
Exploring the Gamut of Receptor Tyrosine Kinases for Their Promise in the Management of Non-Alcoholic Fatty Liver Disease


Abstract
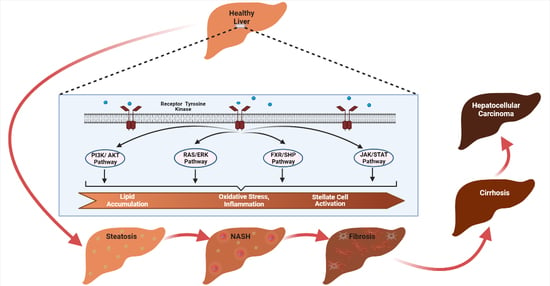
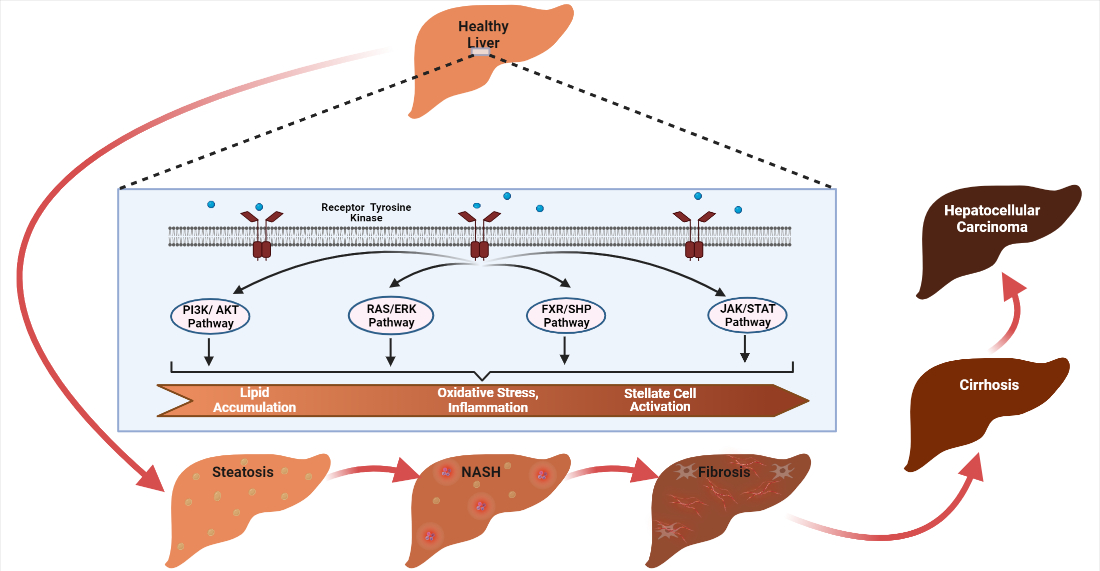
:
1. Introduction
2. Receptor Tyrosine Kinases (RTKs) and Its Role in NAFLD
2.1. Epithelial Growth Factor Receptor (EGFR)
2.2. Hepatocyte Growth Factor Receptor
2.3. TAM (Tyro3, AXL, MERTK) Receptor
2.4. Fibroblast Growth Factor Receptor
2.5. Vascular Endothelial Growth Factor Receptor (VEGFR)
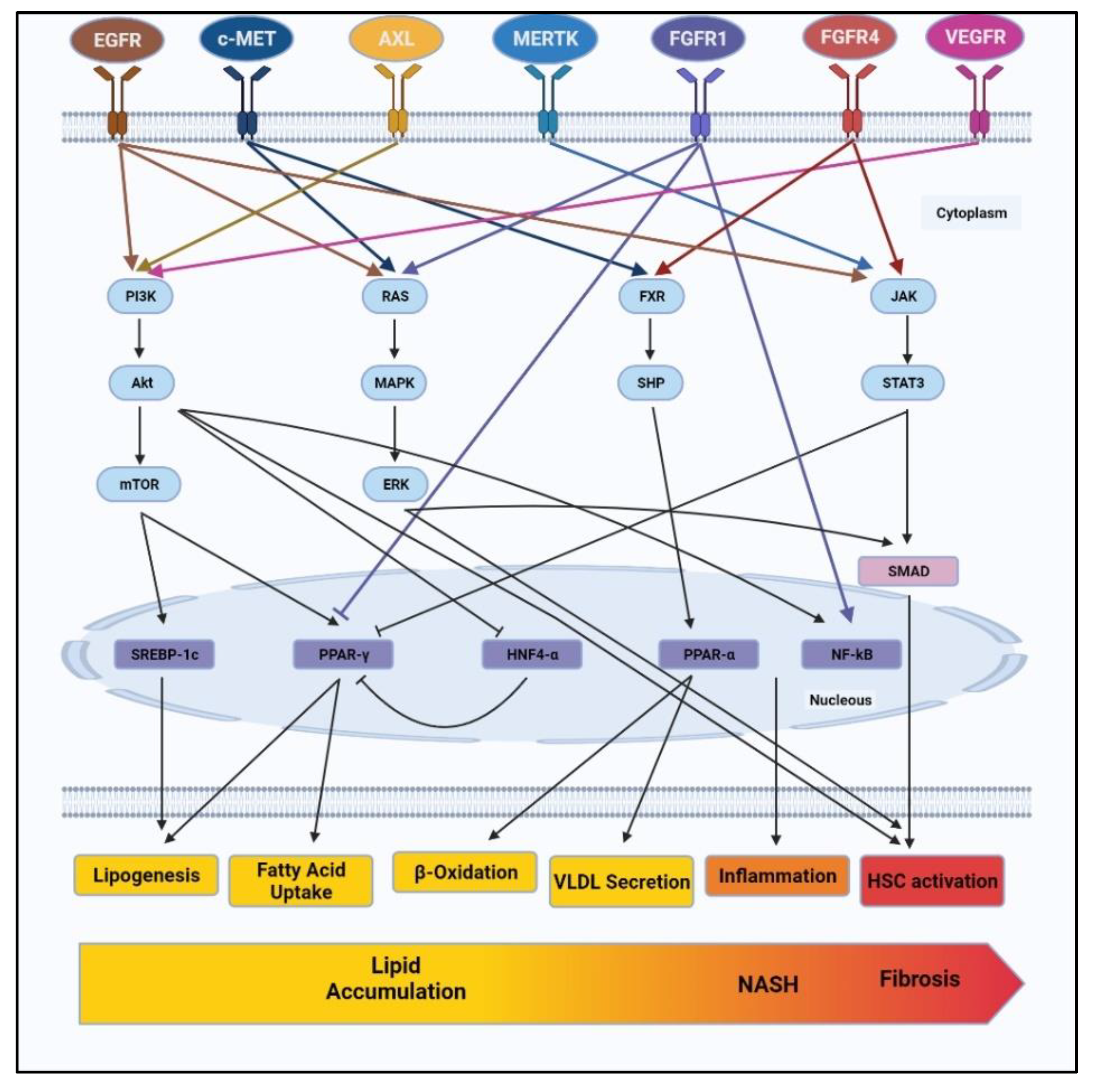
3. Downstream Pathways through Which RTKs Regulate NAFLD
4. Strategies to Target RTKs
4.1. Small Molecule Inhibitors
4.2. Therapeutic Antibodies
4.3. Natural Products
4.4. Nanoparticles
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mantovani, A.; Dalbeni, A. Treatments for NAFLD: State of Art. Int. J. Mol. Sci. 2021, 22, 2350. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2015, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Loomba, R.; Rinella, M.E.; Bugianesi, E.; Marchesini, G.; Neuschwander-Tetri, B.A.; Serfaty, L.; Negro, F.; Caldwell, S.H.; Ratziu, V.; et al. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2018, 68, 361–371. [Google Scholar] [CrossRef]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Rosato, V.; Masarone, M.; Dallio, M.; Federico, A.; Aglitti, A.; Persico, M. NAFLD and Extra-Hepatic Comorbidities: Current Evidence on a Multi-Organ Metabolic Syndrome. Int. J. Environ. Res. Public Health 2019, 16, 3415. [Google Scholar] [CrossRef] [Green Version]
- Targher, G.; Mantovani, A.; Byrne, C.D.; Wang, X.-B.; Yan, H.-D.; Sun, Q.-F.; Pan, K.-H.; Zheng, K.I.; Chen, Y.-P.; Eslam, M.; et al. Risk of severe illness from COVID-19 in patients with metabolic dysfunction-associated fatty liver disease and increased fibrosis scores. Gut 2020, 69, 1545–1547. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, S.; Khandait, H.; Kopel, J.; Aloysius, M.M.; Desai, R.; Goyal, H. NAFLD and COVID-19: A Pooled Analysis. SN Compr. Clin. Med. 2020, 2, 2726–2729. [Google Scholar] [CrossRef] [PubMed]
- BasuRay, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526. [Google Scholar] [CrossRef] [Green Version]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; Hooft, F.V. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [Green Version]
- Smagris, E.; Gilyard, S.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J. Biol. Chem. 2016, 291, 10659–10676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef] [Green Version]
- Di Sessa, A.; Umano, G.R.; Cirillo, G.; Passaro, A.P.; Verde, V.; Cozzolino, D.; Guarino, S.; Marzuillo, P.; del Giudice, E.M. The Membrane-bound O-Acyltransferase7 rs641738 Variant in Pediatric Nonalcoholic Fatty Liver Disease. J. Pediatr. Gastroenterol. Nutr. 2018, 67, 69–74. [Google Scholar] [CrossRef]
- Pafili, K.; Roden, M. Nonalcoholic fatty liver disease (NAFLD) from pathogenesis to treatment concepts in humans. Mol. Metab. 2021, 50, 101122. [Google Scholar] [CrossRef]
- Zheng, S.; Yang, Y.; Wen, C.; Liu, W.; Cao, L.; Feng, X.; Chen, J.; Wang, H.; Tang, Y.; Tian, L.; et al. Effects of environmental contaminants in water resources on nonalcoholic fatty liver disease. Environ. Int. 2021, 154, 106555. [Google Scholar] [CrossRef]
- Satapathy, S.; Kuwajima, V.; Nadelson, J.; Atiq, O.S.A. Drug-induced fatty liver disease: An overview of pathogenesis and management Sanjaya. Ann. Hepatol. 2015, 14, 789–806. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.L.; Dong, M.H.; Loomba, R. Insulin Sensitizers in Nonalcoholic Fatty Liver Disease and Steatohepatitis: Current Status. Adv. Ther. 2019, 26, 893–907. [Google Scholar] [CrossRef]
- Mclntyre, H.D.; Paterson, C.A.; Ma, A.; Ravenscroft, P.J.; Bird, D.M.; Cameron, D.P. Metformin increases insulin sensitivity and basal glucose clearance in Type 2 (non-insulin dependent) diabetes mellitus. Aust. N. Z. J. Med. 1991, 21, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2012, 1, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Ozturk, Z.A.; Kadayifci, A. Insulin sensitizers for the treatment of non-alcoholic fatty liver disease. World J. Hepatol. 2014, 6, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Polimeni, L.; Baratta, F.; Pani, A.; Del Ben, M.; Angelico, F. The efficacy and safety of statins for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2015, 47, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Nseir, W.; Mograbi, J.; Ghali, M. Lipid-Lowering Agents in Nonalcoholic Fatty Liver Disease and Steatohepatitis: Human Studies. Dig. Dis. Sci. 2012, 57, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Sigler, M.A.; Congdon, L.; Edwards, K.L. An Evidence-Based Review of Statin Use in Patients with Nonalcoholic Fatty Liver Disease. Clin. Med. Insights Gastroenterol. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Sugimoto, K.; Inui, H.; Fukusato, T. Current pharmacological therapies for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2015, 21, 3777–3785. [Google Scholar] [CrossRef]
- Tarantino, G.; Citro, V.; Capone, D. Nonalcoholic Fatty Liver Diaease: A Challenge from Mechanisms to Therapy. J. Clin. Med. 2020, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and Prevention of Hepatic Steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Ramachandran, P.; Iredale, J.P. Reversibility of liver fibrosis. Ann. Hepatol. 2009, 8, 283–291. [Google Scholar] [CrossRef]
- Turchinovich, A.; Baranova, A.; Drapkina, O.; Tonevitsky, A. Cell-Free Circulating Nucleic Acids as Early Biomarkers for NAFLD and NAFLD-Associated Disorders. Front. Physiol. 2018, 9, 1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Chen, S.; You, H. Regression of liver fi brosis: Evidence and challenges. Chin. Med. J. (Engl.) 2020, 133, 1696–1702. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, D.; An, T.; Park, H.-J.; Kim, W.; Bae, K.-H.; Oh, K.-J. Metabolic Spectrum of Liver Failure in Type 2 Diabetes and Obesity: From NAFLD to NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4495. [Google Scholar] [CrossRef] [PubMed]
- Montor, W.R.; Ronaldo, A.; Silva, O. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: The current arsenal of inhibitors. Mol. Cancer 2018, 17, 55. [Google Scholar] [CrossRef]
- Cordover, E.; Minden, A. Signaling pathways downstream to receptor tyrosine kinases: Targets for cancer treatment. J. Cancer Metastasis Treat. 2020, 2020. [Google Scholar] [CrossRef]
- Choung, S.; Kim, J.M.; Joung, K.H.; Lee, E.S.; Kim, H.J.; Ku, B.J. Epidermal growth factor receptor inhibition attenuates non-alcoholic fatty liver disease in diet-induced obese mice. PLoS ONE 2019, 14, e0210828. [Google Scholar] [CrossRef]
- Bhushan, B.; Banerjee, S.; Paranjpe, S.; Koral, K.; Mars, W.M.; Stoops, J.W.; Orr, A.; Bowen, W.C.; Locker, J.; Michalopoulos, G.K. Pharmacologic Inhibition of Epidermal Growth Factor Receptor Suppresses Nonalcoholic Fatty Liver Disease in a Murine Fast-Food Diet Model. Hepatology 2019, 70, 1546–1563. [Google Scholar] [CrossRef]
- Kroy, D.C.; Schumacher, F.; Ramadori, P.; Hatting, M.; Bergheim, I.; Gassler, N.; Boekschoten, M.V.; Müller, M.; Streetz, K.L.; Trautwein, C. Hepatocyte specific deletion of c-Met leads to the development of severe non-alcoholic steatohepatitis in mice. J. Hepatol. 2014, 61, 883–890. [Google Scholar] [CrossRef]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.-S.; Lindberg, R.A.; et al. Fibroblast Growth Factor 21 Reverses Hepatic Steatosis, Increases Energy Expenditure, and Improves Insulin Sensitivity in Diet-Induced Obese Mice. Diabetes 2008, 58, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Guo1, Z.; Chen, M.; He, X. The Intestinal-Liver Axis of FGF15-FGFR4-PPARα Signal Control Liver Ischemia Reperfusion Injury Through Regulating Autophagy. Transplantation 2018, 102, s167. [Google Scholar] [CrossRef]
- Carver, R.S.; Stevenson, M.C.; Scheving, L.A.; Russell, W.E. Diverse expression of ErbB receptor proteins during rat liver development and regeneration. Gastroenterology 2002, 123, 2017–2027. [Google Scholar] [CrossRef]
- Berasain, C.; Avila, M.A. The EGFR signalling system in the liver: From hepatoprotection to hepatocarcinogenesis. J. Gastroenterol. 2013, 49, 9–23. [Google Scholar] [CrossRef]
- Komposch, K.; Sibilia, M. EGFR Signaling in Liver Diseases. Int. J. Mol. Sci. 2015, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Michalopoulos, G.K. Role of epidermal growth factor receptor in liver injury and lipid metabolism: Emerging new roles for an old receptor. Chem. Interact. 2020, 324, 109090. [Google Scholar] [CrossRef] [PubMed]
- López-Luque, J.; Caballero-Díaz, D.; Martinez-Palacián, A.; Roncero, C.; Moreno-Càceres, J.; García-Bravo, M.; Grueso, E.; Fernández, A.; Crosas-Molist, E.; García-Álvaro, M.; et al. Dissecting the role of epidermal growth factor receptor catalytic activity during liver regeneration and hepatocarcinogenesis. Hepatology 2016, 63, 604–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paranjpe, S.; Bowen, W.C.; Mars, W.M.; Orr, A.; Haynes, M.M.; DeFrances, M.C.; Liu, S.; Tseng, G.C.; Tsagianni, A.; Michalopoulos, G.K. Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology 2016, 64, 1711–1724. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR Signaling Through an Akt-SREBP-1–Dependent, Rapamycin-Resistant Pathway Sensitizes Glioblastomas to Antilipogenic Therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.; Wang, J.; Tang, H.; Li, L.; Lv, J.; Xia, L.; Han, C.; Xu, F.; He, H.; Xu, H.; et al. Effects of palmitic acid on lipid metabolism homeostasis and apoptosis in goose primary hepatocytes. Mol. Cell. Biochem. 2010, 350, 39–46. [Google Scholar] [CrossRef]
- Ru, P.; Hu, P.; Geng, F.; Mo, X.; Cheng, C.; Yoo, J.Y.; Cheng, X.; Wu, X.; Guo, J.Y.; Nakano, I.; et al. Feedback Loop Regulation of SCAP/SREBP-1 by miR-29 Modulates EGFR Signaling-Driven Glioblastoma Growth. Cell Rep. 2016, 16, 1527–1535. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Lee, Y.K.; Park, J.E.; Lee, M.; Hardwick, J.P. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res. 2018, 2, 209–215. [Google Scholar] [CrossRef]
- Kim, S.; Graham, M.; Lee, R.; Yang, L.; Subramanian, V.; Layne, J.; Cai, L.; Temel, R.; Shih, D.; Lusis, A.; et al. Heparin-binding EGF-like growth factor (HB-EGF) antisense oligonucleotide protected against hyperlipidemia-associated atherosclerosis. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Chen, H.; Zhao, L.; Zhang, W.; Hu, J.; Liu, Z.; Zhong, P.; Wang, W.; Wang, J.; Liang, G. Inhibition of EGFR attenuates fibrosis and stellate cell activation in diet-induced model of nonalcoholic fatty liver disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 133–142. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.-C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR signaling promotes TGFβ-dependent renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 215–224. [Google Scholar]
- Cai, X.; Li, Z.; Zhang, Q.; Qu, Y.; Xu, M.; Wan, X.; Lu, L. CXCL6-EGFR-induced kupffer cells secrete TGF-β1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C-MYC/EZH2 pathway in liver fibrosis. J. Cell. Mol. Med. 2018, 22, 5050–5061. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, Q.; Chen, W.-D.; Wang, Y.-D. HGF/c-MET: A Promising Therapeutic Target in the Digestive System Cancers. Int. J. Mol. Sci. 2018, 19, 3295. [Google Scholar] [CrossRef] [Green Version]
- Shao, Q.; Arakaki, N.; Ohnishi, T.; Nakamura, O.; Daikuhara, Y. Effect of Hepatocyte Growth Factor/Scatter Factor on Lipogenesis in Adult Rat Hepatocytes in Primary Culture. J. Biochem. 1996, 119, 940–946. [Google Scholar] [CrossRef]
- Kaibori, M.; Kwon, A.-H.; Oda, M.; Kamiyama, Y.; Kitamura, N.; Okumura, T. Hepatocyte growth factor stimulates synthesis of lipids and secretion of lipoproteins in rat hepatocytes. Hepatology 1998, 27, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Kosone, T.; Takagi, H.; Horiguchi, N.; Ariyama, Y.; Otsuka, T.; Sohara, N.; Kakizaki, S.; Sato, K.; Mori, M. HGF ameliorates a high-fat diet-induced fatty liver. Am. J. Physiol. Liver Physiol. 2007, 293, G204–G210. [Google Scholar] [CrossRef]
- Jing, Y.; Sun, Q.; Xiong, X.; Meng, R.; Tang, S.; Cao, S.; Bi, Y.; Zhu, D. Hepatocyte growth factor alleviates hepatic insulin resistance and lipid accumulation in high-fat diet-fed mice. J. Diabetes Investig. 2018, 10, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Dou, Z.; Liu, J.; Chai, B.; Li, Y.; An, X.; Chu, P.; Zhang, X. Therapeutic Effect of HGF on NASH Mice Through HGF/c-Met and JAK2-STAT3 Signalling Pathway. Ann. Hepatol. 2018, 17, 501–510. [Google Scholar] [CrossRef]
- Yang, Y.M.; Fukui, M.; Wang, Z.; Miao, F.; Karriker, M.J.; Seki, E. Interventional Potential of Recombinant Feline Hepatocyte Growth Factor in a Mouse Model of Non-alcoholic Steatohepatitis. Front. Endocrinol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Azuma, T.; Kitamura, N.; Nishida, J.; Tamiya, G.; Oka, A.; Inokuchi, S.; Nishimura, T.; Suematsu, M.; Ishii, H. Pioglitazone prevents alcohol-induced fatty liver in rats through up-regulation of c-Met. Gastroenterology 2004, 126, 873–885. [Google Scholar] [CrossRef]
- Muratsu, J.; Iwabayashi, M.; Sanada, F.; Taniyama, Y.; Otsu, R.; Rakugi, H.; Morishita, R. Hepatocyte Growth Factor Prevented High-Fat Diet-Induced Obesity and Improved Insulin Resistance in Mice. Sci. Rep. 2017, 7, 130. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, J.; Seo, D.; Gómez-Quiroz, L.E.; Uchida, K.; Gillen, M.C.; Kitade, M.; Kaposi-Novak, P.; Conner, E.A.; Factor, V.M.; Thorgeirsson, S.S. Loss of c-Met accelerates development of liver fibrosis in response to CCl4 exposure through deregulation of multiple molecular pathways. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 942–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyama, S.; Yamada, T.; Iwata, H.; Sekino, T.; Matsuo, H.; Yoshida, N.; Miyahara, T.; Umeda, Y.; Matsuno, Y.; Kimura, M.; et al. Reduction of fibrosis in a rat model of non-alcoholic steatohepatitis cirrhosis by human HGF gene transfection using electroporation. J. Gastroenterol. Hepatol. 2008, 23, e471–e476. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rao, B.; Lou, J.; Li, J.; Liu, Z.; Li, A.; Cui, G.; Ren, Z.; Yu, Z. The Function of the HGF/c-Met Axis in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2020, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Fourcot, A.; Couchie, D.; Chobert, M.-N.; Zafrani, E.-S.; Mavier, P.; Laperche, Y.; Brouillet, A. Gas6 deficiency prevents liver inflammation, steatohepatitis, and fibrosis in mice. Am. J. Physiol. Liver Physiol. 2011, 300, G1043–G1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirne, C.; Rigamonti, C.; De Benedittis, C.; Sainaghi, P.P.; Bellan, M.; Burlone, M.E.; Castello, L.M.; Avanzi, G.C. Gas6/TAM Signaling Components as Novel Biomarkers of Liver Fibrosis. Dis. Markers 2019, 2019, 2304931. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.K.; Wilhelm, A.; Antoniades, C.G. TAM receptor tyrosine kinase function and the immunopathology of liver disease. Am. J. Physiol. Liver Physiol. 2016, 310, G899–G905. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wong, W.; Chua, S.; Wee, H.L.; Lim, S.G.; Chua, B.T.; Ho, H.K. Overexpression of Tyro3 and its implications on hepatocellular carcinoma progression. Int. J. Oncol. 2015, 48, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristóbal, H.; Aresté, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 349–368. [Google Scholar] [CrossRef]
- Lafdil, F.; Chobert, M.N.; Couchie, D.; Brouillet, A.; Zafrani, E.S.; Mavier, P.; Laperche, Y. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology 2006, 44, 228–239. [Google Scholar] [CrossRef]
- Bárcena, C.; Stefanovic, M.; Tutusaus, A.; Joannas, L.; Menéndez, A.; García-Ruiz, C.; Sancho-Bru, P.; Marí, M.; Caballeria, J.; Rothlin, C.V.; et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. Eur. Assoc. Study Liver 2015, 63, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Flem-Karlsen, K.; Nyakas, M.; Farstad, I.N.; McFadden, E.; Wernhoff, P.; Jacobsen, K.D.; Flørenes, V.A.; Mælandsmo, G.M. Soluble AXL as a marker of disease progression and survival in melanoma. PLoS ONE 2020, 15, e0227187. [Google Scholar] [CrossRef] [PubMed]
- Holstein, E.; Binder, M.; Mikulits, W. Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics. Int. J. Mol. Sci. 2018, 19, 4111. [Google Scholar] [CrossRef] [Green Version]
- Llacuna, L.; Bárcena, C.; Bellido-Martín, L.; Fernández, L.; Stefanovic, M.; Marí, M.; García-Ruiz, C.; Fernández-Checa, J.C.; de Frutos, P.G.; Morales, A. Growth arrest-specific protein 6 is hepatoprotective against murine ischemia/reperfusion injury. Hepatology 2010, 52, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, M.; Pan, G.; Nord, H.; Arzt, E.W.; Wallerman, O.; Wadelius, C. Genetic prevention of hepatitis C virus-induced liver fibrosis by allele-specific downregulation of MERTK. Hepatol. Res. 2017, 47, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 406–421.e7. [Google Scholar] [CrossRef]
- Yang, C.; Wang, C.; Ye, M.; Jin, C.; He, W.; Wang, F.; McKeehan, W.L.; Luo, Y. Control of lipid metabolism by adipocyte FGFR1-mediated adipohepatic communication during hepatic stress. Nutr. Metab. 2012, 9, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Liu, H.-X.; Jena, P.K.; Sheng, L.; Ali, M.R.; Wan, Y.-J.Y. miR-22 inhibition reduces hepatic steatosis via FGF21 and FGFR1 induction. JHEP Rep. 2020, 2, 100093. [Google Scholar] [CrossRef] [Green Version]
- Lin, N.; Chen, S.; Pan, W.; Xu, L.; Hu, K.; Xu, R. NP603, a novel and potent inhibitor of FGFR1 tyrosine kinase, inhibits hepatic stellate cell proliferation and ameliorates hepatic fibrosis in rats. Am. J. Physiol. Physiol. 2011, 301, C469–C477. [Google Scholar] [CrossRef]
- Lou, D.; Han, J.; Zhou, L.; Ma, H.; Xv, J.; Shou, J.; Xu, Z.; Jiang, L.; Qian, Y. Fibroblast growth factor receptor 1 antagonism attenuates lipopolysaccharide-induced activation of hepatic stellate cells via suppressing inflammation. Exp. Ther. Med. 2018, 16, 2909–2916. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Li, H.; Zhang, Y.; Ying, Z.; Wang, X.; Zhang, T.; Zhang, W.; Fan, Z.; Li, X.; et al. Disruption of FGF Signaling Ameliorates Inflammatory Response in Hepatic Stellate Cells. Front. Cell Dev. Biol. 2020, 8, 601. [Google Scholar] [CrossRef]
- Wu, X.; Ge, H.; Baribault, H.; Gupte, J.; Weiszmann, J.; Lemon, B.; Gardner, J.; Fordstrom, P.; Tang, J.; Zhou, M.; et al. Dual actions of fibroblast growth factor 19 on lipid metabolism. J. Lipid Res. 2013, 54, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Yang, C.; Luo, Y.; Jin, C.; Wang, F.; McKeehan, W.L. FGFR4 Prevents Hyperlipidemia and Insulin Resistance but Underlies High-Fat Diet Induced Fatty Liver. Diabetes 2007, 56, 2501–2510. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Jiang, Y.; An, Y.; Zhao, N.; Zhao, Y.; Yu, C. Soluble FGFR4 extracellular domain inhibits FGF19-induced activation of FGFR4 signaling and prevents nonalcoholic fatty liver disease. Biochem. Biophys. Res. Commun. 2011, 409, 651–656. [Google Scholar] [CrossRef]
- Yu, X.X.; Watts, L.M.; Manchem, V.P.; Chakravarty, K.; Monia, B.P.; McCaleb, M.L.; Bhanot, S. Peripheral Reduction of FGFR4 with Antisense Oligonucleotides Increases Metabolic Rate and Lowers Adiposity in Diet-Induced Obese Mice. PLoS ONE 2013, 8, e66923. [Google Scholar]
- Tarantino, G.; Conca, P.; Pasanisi, F.; Ariello, M.; Mastrolia, M.; Arena, A. Vecchione, R. Could inflammatory markers help diagnose nonalcoholic steatohepatitis? Eur. J. Gastroenterol. Hepatol. 2009, 21, 504–511. [Google Scholar] [CrossRef]
- Coulon, S.; Francque, S.; Colle, I.; Verrijken, A.; Blomme, B.; Heindryckx, F.; De Munter, S.; Prawitt, J.; Caron, S.; Staels, B.; et al. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine 2012, 59, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Coulon, S.; Legry, V.; Heindryckx, F.; Van Steenkiste, C.; Casteleyn, C.; Olievier, K.; Libbrecht, L.; Carmeliet, P.; Jonckx, B.; Stassen, J.-M.; et al. Role of vascular endothelial growth factor in the pathophysiology of nonalcoholic steatohepatitis in two rodent models. Hepatology 2013, 57, 1793–1805. [Google Scholar] [CrossRef]
- Hong, W.; Li, S.; Wu, L.; He, B.; Jiang, J.; Chen, Z. Prediction of VEGF-C as a Key Target of Pure Total Flavonoids From Citrus Against NAFLD in Mice via Network Pharmacology. Front. Pharmacol. 2019, 10, 582. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Zakharia, K.; Banini, B.A.; Mikhail, D.S.; Kim, T.H.; Yang, J.D.; Moser, C.D.; Shaleh, H.M.; Thornburgh, S.R.; Walters, I.; et al. Brivanib Attenuates Hepatic Fibrosis In Vivo and Stellate Cell Activation In Vitro by Inhibition of FGF, VEGF and PDGF Signaling. PLoS ONE 2014, 9, e92273. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Qu, K.; Zhang, J.; Huang, Q.; Qu, P.; Xu, X.; Yuan, P.; Huang, X.; Shao, Y.; Liu, C.; et al. CD147 promotes liver fibrosis progression via VEGF-A/VEGFR2 signalling-mediated cross-talk between hepatocytes and sinusoidal endothelial cells. Clin. Sci. 2015, 129, 699–710. [Google Scholar] [CrossRef]
- Taniguchi, E.; Sakisaka, S.; Matsuo, K.; Tanikawa, K.; Sata, M. Expression and role of vascular endothelial growth factor in liver regeneration after partial hepatectomy in rats. J. Histochem. Cytochem. 2001, 49, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Lui, E.L.H.; Friedman, S.L.; Li, L.; Ye, T.; Chen, Y.; Poon, R.T.; Wo, J.; Kok, T.W.; Fan, S.T. PTK787/ZK22258 attenuates stellate cell activation and hepatic fibrosis in vivo by inhibiting VEGF signaling. Lab. Investig. 2009, 89, 209–221. [Google Scholar] [CrossRef]
- Chen, H. Nutrient mTORC1 signaling contributes to hepatic lipid metabolism in the pathogenesis of non-alcoholic fatty liver disease. Liver Res. 2020, 4, 15–22. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting Autophagy and Lipid Metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, Y. mTORC1 signaling in hepatic lipid metabolism. Protein Cell 2018, 9, 145–151. [Google Scholar] [CrossRef]
- Matsuda, S.; Kobayashi, M.; Kitagishi, Y. Roles for PI3K/AKT/PTEN Pathway in Cell Signaling of Nonalcoholic Fatty Liver Disease. ISRN Endocrinol. 2013, 2013, 472432. [Google Scholar] [CrossRef]
- Kong, X.; Horiguchi, N.; Mori, M.; Gao, B. Cytokines and STATs in Liver Fibrosis. Front. Physiol. 2012, 3, 69. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Song, J.; Bian, H.; Bo, J.; Lv, S.; Pan, W.; Lv, X. Apelin promotes hepatic fibrosis through ERK signaling in LX-2 cells. Mol. Cell. Biochem. 2019, 460, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foglia, B.; Cannito, S.; Bocca, C.; Parola, M.; Novo, E. ERK Pathway in Activated, Myofibroblast-Like, Hepatic Stellate Cells: A Critical Signaling Crossroad Sustaining Liver Fibrosis. Int. J. Mol. Sci. 2019, 20, 2700. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Qi, Y.-F.; Yu, Y.-R. STAT3: A key regulator in liver fibrosis. Ann. Hepatol. 2021, 21, 100224. [Google Scholar] [CrossRef]
- Neuzillet, C.; De Gramont, A.; Tijeras-Raballand, A.; de Mestier, L.; Cros, J.; Faivre, S.; Raymond, E. Perspectives of TGF-β inhibition in pancreatic and hepatocellular carcinomas. Oncotarget 2013, 5, 78–94. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Huang, X.; Zhu, X.; Liu, L.; Mo, S.; Wang, H.; Wei, X.; Lu, S.; Bai, F.; Wang, D.; et al. HBOA ameliorates CCl 4 -incuded liver fibrosis through inhibiting TGF-β1/Smads, NF-κB and ERK signaling pathways. Biomed. Pharmacother. 2019, 115, 108901. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Hoshida, Y.; Fujii, T.; Wei, L.; Yamada, S.; Lauwers, G.Y.; McGinn, C.M.; DePeralta, D.K.; Chen, X.; Kuroda, T.; et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014, 59, 1577–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svegliati-Baroni, G.; Ridolfi, F.; Hannivoort, R.; Saccomanno, S.; Homan, M.; de Minicis, S.; Jansen, P.L.; Candelaresi, C.; Benedetti, A.; Moshage, H. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology 2005, 128, 1042–1055. [Google Scholar] [CrossRef]
- Liu, Y.; Wen, X.M.; Lui, E.L.H.; Friedman, S.L.; Cui, W.; Ho, N.P.S.; Li, L.; Ye, T.; Fan, S.T.; Zhang, H. Therapeutic targeting of the PDGF and TGF-Β-signaling pathways in hepatic stellate cells by PTK787/ZK22258. Lab. Investig. 2009, 89, 1152–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tugues, S.; Fernandez-Varo, G.; Muñoz-Luque, J.; Ros, J.; Arroyo, V.; Rodés, J.; Friedman, S.L.; Carmeliet, P.; Jiménez, W.; Morales-Ruiz, M. Antiangiogenic treatment with Sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology 2007, 46, 1919–1926. [Google Scholar] [CrossRef]
- Majumder, S.; Piguet, A.-C.; Dufour, J.-F.; Chatterjee, S. Study of the cellular mechanism of Sunitinib mediated inactivation of activated hepatic stellate cells and its implications in angiogenesis. Eur. J. Pharmacol. 2013, 705, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kato, C.; Kato, A. Therapeutic antibodies: Their mechanisms of action and the pathological findings they induce in toxicity studies. J. Toxicol. Pathol. 2015, 28, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.D. Unlocking the potential of natural products in drug discovery. Microb. Biotechnol. 2019, 12, 55–57. [Google Scholar] [CrossRef] [Green Version]
- Yin, B.; Fang, D.-M.; Zhou, X.-L.; Gao, F. Natural products as important tyrosine kinase inhibitors. Eur. J. Med. Chem. 2019, 182, 111664. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, Z.; Ji, G. Herbal Extracts and Natural Products in Alleviating Non-alcoholic Fatty Liver Disease via Activating Autophagy. Front. Pharmacol. 2018, 9, 1459. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.A.; Rushworth, S.A. Curcumin: Potential for hepatic fibrosis therapy? Br. J. Pharmacol. 2008, 153, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Yan, N.; Wang, P.; Xia, Y.; Hao, H.; Wang, G.; Gonzalez, F.J. Herbal drug discovery for the treatment of nonalcoholic fatty liver disease. Acta Pharm. Sin. B 2020, 10, 3–18. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Xu, W.; Wang, S.; Hu, Z.; Zhang, Q.; Deng, X.; Wang, J.; Zhang, J.; Guo, C. Antifibrotic effects of luteolin on hepatic stellate cells and liver fibrosis by targeting AKT/mTOR/p70S6K and TGFβ/Smad signalling pathways. Liver Int. 2014, 35, 1222–1233. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Gao, L.; Lin, H.; Wu, Y.; Han, X.; Zhu, Y.; Li, J. Luteolin improves non-alcoholic fatty liver disease in db/db mice by inhibition of liver X receptor activation to down-regulate expression of sterol regulatory element binding protein 1c. Biochem. Biophys. Res. Commun. 2017, 482, 720–726. [Google Scholar] [CrossRef]
- Yin, H.-Q.; Kim, Y.-C.; Chung, Y.-S.; Shin, Y.-K.; Lee, B.-H. Honokiol reverses alcoholic fatty liver by inhibiting the maturation of sterol regulatory element binding protein-1c and the expression of its downstream lipogenesis genes. Toxicol. Appl. Pharmacol. 2009, 236, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.L.; Spohn, W.H.; Hung, M.C. Curcumin inhibits tyrosine kinase activity of p185neu and also depletes p185neu. Clin. Cancer Res. 1999, 5, 1884–1891. [Google Scholar] [PubMed]
- Kim, K.-C.; Baek, S.-H.; Lee, C. Curcumin-induced downregulation of Axl receptor tyrosine kinase inhibits cell proliferation and circumvents chemoresistance in non-small lung cancer cells. Int. J. Oncol. 2015, 47, 2296–2303. [Google Scholar] [CrossRef] [Green Version]
- Golonko, A.; Lewandowska, H.; Świsłocka, R.; Jasińska, U.; Priebe, W.; Lewandowski, W. Curcumin as tyrosine kinase inhibitor in cancer treatment. Eur. J. Med. Chem. 2019, 181, 111512. [Google Scholar] [CrossRef]
- Leeman-Neill, R.J.; Cai, Q.; Joyce, S.C.; Thomas, S.M.; Bhola, N.E.; Neill, D.B.; Arbiser, J.L.; Grandis, J.R. Honokiol inhibits EGFR signaling and enhances the antitumor effects of EGFR inhibitors. Clin. Cancer Res. 2011, 16, 2571–2579. [Google Scholar] [CrossRef] [Green Version]
- Okuda, K.; Umemura, A.; Umemura, S.; Kataoka, S.; Taketani, H.; Seko, Y.; Nishikawa, T.; Yamaguchi, K.; Moriguchi, M.; Kanbara, Y.; et al. Honokiol Prevents Non-Alcoholic Steatohepatitis-Induced Liver Cancer via EGFR Degradation through the Glucocorticoid Receptor—MIG6 Axis. Cancers 2021, 13, 1515. [Google Scholar] [CrossRef]
- Boey, A.; Ho, H.K. All Roads Lead to the Liver: Metal Nanoparticles and Their Implications for Liver Health. Small 2020, 16, e2000153. [Google Scholar] [CrossRef] [PubMed]
- Tee, J.K.; Peng, F.; Ho, H.K. Effects of inorganic nanoparticles on liver fibrosis: Optimizing a double-edged sword for therapeutics. Biochem. Pharmacol. 2019, 160, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Tee, J.K.; Setyawati, M.I.; Ding, X.; Yeo, H.L.A.; Tan, Y.L.; Leong, D.T.; Ho, H.K. Inorganic Nanomaterials as Highly Efficient Inhibitors of Cellular Hepatic Fibrosis. ACS Appl. Mater. Interfaces 2018, 10, 31938–31946. [Google Scholar] [CrossRef]
- Saeed, B.A.; Lim, V.; Yusof, N.A.; Khor, K.Z.; Rahman, H.; Samad, N.A. Antiangiogenic properties of nanoparticles: A systematic review. Int. J. Nanomed. 2019, 14, 5135–5146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darweesh, R.S.; Ayoub, N.M.; Nazzal, S. Gold nanoparticles and angiogenesis: Molecular mechanisms and biomedical applications. Int. J. Nanomed. 2019, 14, 7643–7663. [Google Scholar] [CrossRef] [Green Version]
- Otlewski, J.; Szlachcic, A.; Pala, K.; Zakrzewska, M.; Jakimowicz, P.; Wiedlocha, A. FGF1-gold nanoparticle conjugates targeting FGFR efficiently decrease cell viability upon NIR irradiation. Int. J. Nanomed. 2012, 7, 5915–5927. [Google Scholar] [CrossRef] [Green Version]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal Models of Nonalcoholic Fatty Liver Disease—A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, A. The Flaws and Human Harms of Animal Experimentation. Camb. Q. Healthc. Ethics 2015, 24, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, A.; Beli, A.; Zanger, U.M.; Rozman, D. Molecular interactions between NAFLD and xenobiotic metabolism. Front. Genet. 2013, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| RTK | Inhibitor | Study | Reference |
|---|---|---|---|
| EGFR | Gefitinib | Gefitinib attenuated palmitic acid induced lipid accumulation in Huh7 cells by inhibiting lipogenic genes | [32] |
| Erlotinib | Erlotinib attenuated HSC activation and fibrosis after liver injury in mice treated with C 1Cl4 and bile duct ligation. | [40,104] | |
| AG1478 | AG1478 reduced diet-induced fat accumulation as well as HSC activation and proliferation | [49,105] | |
| PD153035 | PD153035 controlled lipid accumulation in high fat fed mice by downregulating lipogenic genes | [32] | |
| AXL | Bemcentinib (BGB324) | Bemcentinib inhibits AXL and reduces liver inflammation and fibrosis in diet induced mouse model by inactivation of AXL/AKT phosphorylation and blocking of successive HSC activation | [68,70] |
| FGFR4 | Soluble FGFR4 extracellular domain fragment | Blocking FGFR4 by soluble extracellular domain leads to decrease in steatosis. | [83] |
| VEGFR | DC101 | Treatment with anti VEGFR-2 antibodies (DC101) reduced steatosis, inflammation as well as fibrosis in mice fed on MCD diet. | [87] |
| PTK787/ZK222584 (PTK/ZK) | Inhibits HSC activation by attenuating HSC proliferation, migration, and collagen synthesis through the VEGF pathway | [92,106] | |
| Sunitinib | Treatment with Sunitinib resulted in decrease in inflammatory infiltrates as well as fibrotic markers such as α-SMA and collagen through VEGF pathway | [107,108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhave, S.; Ho, H.K. Exploring the Gamut of Receptor Tyrosine Kinases for Their Promise in the Management of Non-Alcoholic Fatty Liver Disease. Biomedicines 2021, 9, 1776. https://doi.org/10.3390/biomedicines9121776
Bhave S, Ho HK. Exploring the Gamut of Receptor Tyrosine Kinases for Their Promise in the Management of Non-Alcoholic Fatty Liver Disease. Biomedicines. 2021; 9(12):1776. https://doi.org/10.3390/biomedicines9121776
Chicago/Turabian StyleBhave, Sayali, and Han Kiat Ho. 2021. "Exploring the Gamut of Receptor Tyrosine Kinases for Their Promise in the Management of Non-Alcoholic Fatty Liver Disease" Biomedicines 9, no. 12: 1776. https://doi.org/10.3390/biomedicines9121776