
Macroautophagy and Mitophagy in Neurodegenerative Disorders: Focus on Therapeutic Interventions

 , and
, and
Abstract
:1. Overview of Autophagy
1.1. Macroautophagy
1.2. Function of Autophagy in Neurons
2. Mitophagy
3. Autophagy and Neurodegenerative Diseases
3.1. Alzheimer’s Disease
3.1.1. Autophagy Impairment in AD
3.1.2. Mitophagy in AD
3.2. Parkinson’s Disease
3.2.1. Autophagy in PD
3.2.2. Mitophagy in PD
3.3. Huntington’s Disease
3.3.1. Autophagy in HD
3.3.2. Mitophagy in HD
4. Therapeutic Strategies
4.1. Targeting Macroautophagy and Mitophagy in AD
4.2. Targeting Macroautophagy and Mitophagy in PD
4.3. Targeting Macroautophagy and Mitophagy in HD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Baehrecke, E.H. Eaten alive: Novel insights into autophagy from multicellular model systems. Trends Cell Biol. 2015, 25, 376–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Ahlberg, J.; Marzella, L.; Glaumann, H. Uptake and degradation of proteins by isolated rat liver lysosomes. Suggestion of a microautophagic pathway of proteolysis. Lab. Investig. J. Tech. Methods Pathol. 1982, 47, 523–532. [Google Scholar]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, C.A.; Kaliappan, A.; Dennis, P.B. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009, 5, 649–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Karanasios, E.; Walker, S.A.; Okkenhaug, H.; Manifava, M.; Hummel, E.; Zimmermann, H.; Ahmed, Q.; Domart, M.C.; Collinson, L.; Ktistakis, N.T. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat. Commun. 2016, 7, 12420. [Google Scholar] [CrossRef]
- Puri, C.; Renna, M.; Bento, C.F.; Moreau, K.; Rubinsztein, D.C. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013, 154, 1285–1299. [Google Scholar] [CrossRef] [Green Version]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef] [Green Version]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Popovic, D.; Dikic, I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep. 2014, 15, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Mack, H.I.; Zheng, B.; Asara, J.M.; Thomas, S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012, 8, 1197–1214. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2013, 2, e00947. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Simonsen, A.; Birkeland, H.C.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 2004, 117, 4239–4251. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Melia, T.J. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr. Opin. Cell Biol. 2017, 47, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Schmidt, O.; Angelova, M.; Faserl, K.; Weys, S.; Kremser, L.; Pfaffenwimmer, T.; Dalik, T.; Kraft, C.; Trajanoski, Z.; et al. The coordinated action of the MVB pathway and autophagy ensures cell survival during starvation. eLife 2015, 4, e07736. [Google Scholar] [CrossRef]
- Liou, W.; Geuze, H.J.; Geelen, M.J.; Slot, J.W. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 1997, 136, 61–70. [Google Scholar] [CrossRef]
- Taguchi-Atarashi, N.; Hamasaki, M.; Matsunaga, K.; Omori, H.; Ktistakis, N.T.; Yoshimori, T.; Noda, T. Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic 2010, 11, 468–478. [Google Scholar] [CrossRef]
- Goo, M.S.; Sancho, L.; Slepak, N.; Boassa, D.; Deerinck, T.J.; Ellisman, M.H.; Bloodgood, B.L.; Patrick, G.N. Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J. Cell Sci. 2017, 216, 2499–2513. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Saegusa, K.; Sato, K.; Hara, T.; Harada, A.; Sato, K. Caenorhabditis elegans SNAP-29 is required for organellar integrity of the endomembrane system and general exocytosis in intestinal epithelial cells. Mol. Biol. Cell 2011, 22, 2579–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takáts, S.; Nagy, P.; Varga, Á.; Pircs, K.; Kárpáti, M.; Varga, K.; Kovács, A.L.; Hegedűs, K.; Juhász, G. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J. Cell Biol. 2013, 201, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, M.G.; Munafó, D.B.; Berón, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef]
- Marshansky, V.; Futai, M. The V-type H+-ATPase in vesicular trafficking: Targeting, regulation and function. Curr. Opin. Cell Biol. 2008, 20, 415–426. [Google Scholar] [CrossRef]
- Xie, Y.; Kang, R.; Sun, X.; Zhong, M.; Huang, J.; Klionsky, D.J.; Tang, D. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 2015, 11, 28–45. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Yue, Z. Autophagy and its normal and pathogenic states in the brain. Ann. Rev. Neurosci. 2014, 37, 55–78. [Google Scholar] [CrossRef] [Green Version]
- Dörrbaum, A.R.; Kochen, L.; Langer, J.D.; Schuman, E.M. Local and global influences on protein turnover in neurons and glia. eLife 2018, 7, e34202. [Google Scholar] [CrossRef]
- Cajigas, I.J.; Will, T.; Schuman, E.M. Protein homeostasis and synaptic plasticity. EMBO J. 2010, 29, 2746–2752. [Google Scholar] [CrossRef]
- Maday, S. Mechanisms of neuronal homeostasis: Autophagy in the axon. Brain Res. 2016, 1649, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, V.; Verstreken, P. Autophagy in the presynaptic compartment in health and disease. J. Cell Sci. 2017, 216, 1895–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.N.; Zhang, L.H.; Wei, E.Q.; Yang, Y. Autophagy in synaptic development, function, and pathology. Neurosci. Bull. 2015, 31, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Soukup, S.F.; Kuenen, S.; Vanhauwaert, R.; Manetsberger, J.; Hernández-Díaz, S.; Swerts, J.; Schoovaerts, N.; Vilain, S.; Gounko, N.V.; Vints, K.; et al. A LRRK2-dependent endophilina phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 2016, 92, 829–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 2017, 36, 1392–1411. [Google Scholar] [CrossRef] [PubMed]
- Okerlund, N.D.; Schneider, K.; Leal-Ortiz, S.; Montenegro-Venegas, C.; Kim, S.A.; Garner, L.C.; Waites, C.L.; Gundelfinger, E.D.; Reimer, R.J.; Garner, C.C. Bassoon controls presynaptic autophagy through Atg5. Neuron 2017, 93, 897–913.e7. [Google Scholar] [CrossRef] [Green Version]
- Shehata, M.; Matsumura, H.; Okubo-Suzuki, R.; Ohkawa, N.; Inokuchi, K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J. Neurosci. 2012, 32, 10413–10422. [Google Scholar] [CrossRef] [Green Version]
- Nikoletopoulou, V.; Sidiropoulou, K.; Kallergi, E.; Dalezios, Y.; Tavernarakis, N. Modulation of autophagy by BDNF underlies synaptic plasticity. Cell Metab. 2017, 26, 230–242.e5. [Google Scholar] [CrossRef] [Green Version]
- Nikoletopoulou, V.; Papandreou, M.E.; Tavernarakis, N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015, 22, 398–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Maday, S.; Holzbaur, E.L. Compartment-Specific Regulation of Autophagy in Primary Neurons. J. Neurosci. 2016, 36, 5933–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [Green Version]
- Kannan, M.; Bayam, E.; Wagner, C.; Rinaldi, B.; Kretz, P.F.; Tilly, P.; Roos, M.; McGillewie, L.; Bär, S.; Minocha, S.; et al. WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc. Natl. Acad. Sci. USA 2017, 114, E9308–E9317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, J.; Suzuki, C.; Nanao, T.; Kakuta, S.; Ozawa, K.; Tanida, I.; Saitoh, T.; Sunabori, T.; Komatsu, M.; Tanaka, K.; et al. Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy 2018, 14, 764–777. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Cherra, S.J., 3rd; Chu, C.T. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008, 3, 309–323. [Google Scholar] [CrossRef]
- Chu, C.T. Autophagic stress in neuronal injury and disease. J. Neuropathol. Exp. Neurol. 2006, 65, 423–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.M.; Mizushima, N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 2014, 39, 61–71. [Google Scholar] [CrossRef]
- Arduíno, D.M.; Esteves, A.R.; Cortes, L.; Silva, D.F.; Patel, B.; Grazina, M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial metabolism in Parkinson’s disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum. Mol. Genet. 2012, 21, 4680–4702. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.F.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial Metabolism Power SIRT2-Dependent Deficient Traffic Causing Alzheimer’s-Disease Related Pathology. Mol. Neurobiol. 2017, 54, 4021–4040. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-i.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.L. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 2010, 285, 3499–3509. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Ganetzky, B. Autophagy promotes synapse development in Drosophila. J. Cell Biol. 2009, 187, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Birgisdottir, Å.B.; Lamark, T.; Johansen, T. The LIR motif—crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [Green Version]
- Esteban-Martínez, L.; Boya, P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy 2018, 14, 915–917. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef]
- Koentjoro, B.; Park, J.S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen. Res. 2019, 14, 749–756. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weil, R.; Laplantine, E.; Curic, S.; Génin, P. Role of Optineurin in the Mitochondrial Dysfunction: Potential Implications in Neurodegenerative Diseases and Cancer. Front. Immunol. 2018, 9, 1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, D.L.; Sisodia, S.S. Mutant genes in familial Alzheimer’s disease and transgenic models. Ann. Rev. Neurosci. 1998, 21, 479–505. [Google Scholar] [CrossRef] [PubMed]
- Irvine, G.B.; El-Agnaf, O.M.; Shankar, G.M.; Walsh, D.M. Protein aggregation in the brain: The molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Apelt, J.; Bigl, M.; Wunderlich, P.; Schliebs, R. Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic Tg2576 mice with Alzheimer-like pathology. Int. J. Dev. Neurosci. 2004, 22, 475–484. [Google Scholar] [CrossRef]
- Kinoshita, A.; Fukumoto, H.; Shah, T.; Whelan, C.M.; Irizarry, M.C.; Hyman, B.T. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell Sci. 2003, 116, 3339–3346. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Neurodegeneration: Amyloid-β pathology induced in humans. Nature 2015, 525, 193–194. [Google Scholar] [CrossRef] [Green Version]
- Kins, S.; Crameri, A.; Evans, D.R.; Hemmings, B.A.; Nitsch, R.M.; Gotz, J. Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J. Biol. Chem. 2001, 276, 38193–38200. [Google Scholar] [CrossRef]
- Khlistunova, I.; Biernat, J.; Wang, Y.; Pickhardt, M.; von Bergen, M.; Gazova, Z.; Mandelkow, E.; Mandelkow, E.M. Inducible expression of Tau repeat domain in cell models of tauopathy: Aggregation is toxic to cells but can be reversed by inhibitor drugs. J. Biol. Chem. 2006, 281, 1205–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelkow, E.; von Bergen, M.; Biernat, J.; Mandelkow, E.M. Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol. 2007, 17, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Basurto-Islas, G.; Luna-Muñoz, J.; Guillozet-Bongaarts, A.L.; Binder, L.I.; Mena, R.; García-Sierra, F. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J. NeuroPathol. Exp. Neurol. 2008, 67, 470–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Eckert, A.; Hauptmann, S.; Scherping, I.; Rhein, V.; Müller-Spahn, F.; Götz, J.; Müller, W.E. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neuro-Degener. Dis. 2008, 5, 157–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, I.; Zierz, S.; Möller, H.J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, P.A.; Pickford, F.; Sun, C.-H.; Lucin, K.M.; Masliah, E.; Wyss-Coray, T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS ONE 2010, 5, e11102. [Google Scholar] [CrossRef]
- Khandelwal, P.J.; Herman, A.M.; Hoe, H.S.; Rebeck, G.W.; Moussa, C.E. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum. Mol. Genet. 2011, 20, 2091–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; van Laar, T.; Huang, H.; Zhang, L. APP and APLP1 are degraded through autophagy in response to proteasome inhibition in neuronal cells. Prot. Cell 2011, 2, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Chang, J.C.; Greengard, P.; Flajolet, M. The convergence of endosomal and autophagosomal pathways: Implications for APP-CTF degradation. Autophagy 2014, 10, 694–696. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldo, A.M.; Hamilton, D.J.; Nixon, R.A. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res. 1994, 640, 68–80. [Google Scholar] [CrossRef]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Varo, R.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; De Castro, V.; Jimenez, S.; Ruano, D.; Vizuete, M.; et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 2012, 123, 53–70. [Google Scholar] [CrossRef] [Green Version]
- Cataldo, A.M.; Peterhoff, C.M.; Schmidt, S.D.; Terio, N.B.; Duff, K.; Beard, M.; Mathews, P.M.; Nixon, R.A. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Esteves, A.R.; Cardoso, S.M. Differential protein expression in diverse brain areas of Parkinson’s and Alzheimer’s disease patients. Sci. Rep. 2020, 10, 13149. [Google Scholar] [CrossRef] [PubMed]
- Bieri, G.; Lucin, K.M.; O’Brien, C.E.; Zhang, H.; Villeda, S.A.; Wyss-Coray, T. Proteolytic cleavage of Beclin 1 exacerbates neurodegeneration. Mol. Neurodegener. 2018, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Omata, Y.; Lim, Y.M.; Akao, Y.; Tsuda, L. Age-induced reduction of autophagy-related gene expression is associated with onset of Alzheimer’s disease. Am. J. Neurodegener. Dis. 2014, 3, 134–142. [Google Scholar] [PubMed]
- Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, P.; Sekiguchi, M.; Akagi, T.; Izumi, S.; Komori, T.; Hui, K.; Sörgjerd, K.; Tanaka, M.; Saito, T.; Iwata, N.; et al. Autophagy-related protein 7 deficiency in amyloid β (Aβ) precursor protein transgenic mice decreases Aβ in the multivesicular bodies and induces Aβ accumulation in the Golgi. Am. J. Pathol. 2015, 185, 305–313. [Google Scholar] [CrossRef]
- Du, Y.; Wooten, M.C.; Gearing, M.; Wooten, M.W. Age-associated oxidative damage to the p62 promoter: Implications for Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease—Locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. eLife 2017, 6, e21776. [Google Scholar] [CrossRef] [Green Version]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy 2010, 6, 182–183. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2005, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Majid, T.; Ali, Y.O.; Venkitaramani, D.V.; Jang, M.-K.; Lu, H.-C.; Pautler, R.G. In vivo axonal transport deficits in a mouse model of fronto-temporal dementia. NeuroImage Clin. 2014, 4, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.W.; Bandyopadhyay, U.; Jiang, Y.; et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 2011, 7, 788–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditaranto, K.; Tekirian, T.L.; Yang, A.J. Lysosomal membrane damage in soluble Abeta-mediated cell death in Alzheimer’s disease. Neurobiol. Dis. 2001, 8, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.J.; Chandswangbhuvana, D.; Margol, L.; Glabe, C.G. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J. Neurosci. Res. 1998, 52, 691–698. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Schuur, M.; Ikram, M.A.; van Swieten, J.C.; Isaacs, A.; Vergeer-Drop, J.M.; Hofman, A.; Oostra, B.A.; Breteler, M.M.; van Duijn, C.M. Cathepsin D gene and the risk of Alzheimer’s disease: A population-based study and meta-analysis. Neurobiol. Aging 2011, 32, 1607–1614. [Google Scholar] [CrossRef]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, A.; Magrì, A.; Medina, D.X.; Wisely, E.V.; López-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Ioja, E.; Bereczki, E.; Hultenby, K.; Li, C.; Guan, Z.; Winblad, B.; Pei, J.J. mTor mediates tau localization and secretion: Implication for Alzheimer’s disease. Biochim. Biophys. Acta 2015, 1853, 1646–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Zhang, B.; Luan, P.; Gu, B.; Wan, Q.; Huang, X.; Liao, W.; Liu, J. PI3K/AKT/mTOR/p70S6K pathway is involved in Aβ25-35-induced autophagy. BioMed Res. Int. 2015, 2015, 161020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, T.D.; Economou, N.J.; Chamas, A.; Buratto, S.K.; Shea, J.-E.; Bowers, M.T. Interactions between Amyloid-β and Tau fragments promote aberrant aggregates: Implications for amyloid toxicity. J. Phys. Chem. B 2014, 118, 11220–11230. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, R.; Xu, S.; Lakshmana, M.K. Transcription Factor EB Is Selectively Reduced in the Nuclear Fractions of Alzheimer’s and Amyotrophic Lateral Sclerosis Brains. Neurosci. J. 2016, 2016, 4732837. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.A.; Ballabio, A.; Dobrowolski, R. Dysregulation of nutrient sensing and CLEARance in presenilin deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.D.; Zhao, J.J. TFEB participates in the Aβ-induced pathogenesis of alzheimer’s disease by regulating the autophagy-lysosome pathway. DNA Cell Biol. 2015, 34, 661–668. [Google Scholar] [CrossRef]
- Zhang, X.; Garbett, K.; Veeraraghavalu, K.; Wilburn, B.; Gilmore, R.; Mirnics, K.; Sisodia, S.S. A role for presenilins in autophagy revisited: Normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J. Neurosci. 2012, 32, 8633–8648. [Google Scholar] [CrossRef]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.Q.; Bien-Ly, N.; Puoliväli, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Mattson, M.P.; Cheng, A. Permeability transition pore-mediated mitochondrial superoxide flashes regulate cortical neural progenitor differentiation. PLoS ONE 2013, 8, e76721. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Santos, S.; Swerdlow, R.H.; Oliveira, C.R. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001, 15, 1439–1441. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.L.; Ferreiro, E.; Schmidt, J.; Cardoso, J.M.; Pereira, C.M.; Carvalho, A.L.; Oliveira, C.R.; Rego, A.C. Aβ and NMDAR activation cause mitochondrial dysfunction involving ER calcium release. Neurobiol. Aging 2015, 36, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.N.; Pang, Z.; Geddes, J.W.; Begley, J.G.; Germeyer, A.; Waeg, G.; Mattson, M.P. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: Role of the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 1997, 69, 273–284. [Google Scholar] [CrossRef]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Dröse, S.; Brandt, U.; Müller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’Aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: Possible implications for neurodegeneration. JAD 2010, 21, 445–470. [Google Scholar] [CrossRef] [Green Version]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; García, E.; Simón, D.; Avila, J.; García-Escudero, V. Mitophagy Failure in APP and Tau Overexpression Model of Alzheimer’s Disease. JAD 2019, 70, 525–540. [Google Scholar] [CrossRef]
- Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Ciotti, M.T.; Natale, F.; Della Valle, F.; Borreca, A.; Manca, A.; Meli, G.; et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: Implications in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 3058–3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhang, T.; Ge, X.; Chen, J.; Zhao, Y.; Fu, J. Parkin overexpression attenuates Aβ-induced mitochondrial dysfunction in HEK293 cells by restoring impaired mitophagy. Life Sci. 2020, 244, 117322. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Zou, Y.; Zhang, M.; Zhao, N.; Tian, Q.; Gu, M.; Liu, W.; Shi, R.; Lü, Y.; Yu, W. Mitochondrial Sirt3 Expression is Decreased in APP/PS1 Double Transgenic Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2015, 40, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Fuso, A.; Mosca, L.; Forte, E.; Correani, V.; Fontana, M.; Scarpa, S.; Maras, B.; d’Erme, M. Bioenergetic impairment in animal and cellular models of Alzheimer’s disease: PARP-1 inhibition rescues metabolic dysfunctions. JAD 2016, 54, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Burré, J. The synaptic function of α-synuclein. J. Parkinson’s Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Periquet, M.; Fulga, T.; Myllykangas, L.; Schlossmacher, M.G.; Feany, M.B. Aggregated α-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 2007, 27, 3338–3346. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Bennett, M.C.; Bishop, J.F.; Leng, Y.; Chock, P.B.; Chase, T.N.; Mouradian, M.M. Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 1999, 274, 33855–33858. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Björklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [Green Version]
- Higashi, S.; Moore, D.J.; Minegishi, M.; Kasanuki, K.; Fujishiro, H.; Kabuta, T.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Localization of MAP1-LC3 in vulnerable neurons and Lewy bodies in brains of patients with dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 2011, 70, 264–280. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Y.; Yuan, Y.; Chen, N. A new perspective in Parkinson’s disease, chaperone-mediated autophagy. Parkinsonism Relat. Disord. 2011, 17, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H. Glucocerebrosidase and Parkinson disease: Recent advances. Mol. Cell. Neurosci. 2015, 66, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Goker-Alpan, O.; Stubblefield, B.K.; Giasson, B.I.; Sidransky, E. Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010, 120, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, H.J.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. er stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.W.; Leung, C.T.; Liu, H.; Pang, S.Y.; Lam, C.S.; Xian, J.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 2020, 16, 347–370. [Google Scholar] [CrossRef]
- Esteves, A.R.; Swerdlow, R.H.; Cardoso, S.M. LRRK2, a puzzling protein: Insights into Parkinson’s disease pathogenesis. Exp. Neurol. 2014, 261, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef] [Green Version]
- Plowey, E.D.; Cherra, S.J., 3rd; Liu, Y.J.; Chu, C.T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 2008, 105, 1048–1056. [Google Scholar] [CrossRef] [Green Version]
- Ramonet, D.; Daher, J.P.; Lin, B.M.; Stafa, K.; Kim, J.; Banerjee, R.; Westerlund, M.; Pletnikova, O.; Glauser, L.; Yang, L.; et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE 2011, 6, e18568. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Mamais, A.; Roosen, D.A.; Dihanich, S.; Soutar, M.P.; Plun-Favreau, H.; Bandopadhyay, R.; Hardy, J.; Tooze, S.A.; Cookson, M.R.; et al. mTOR independent regulation of macroautophagy by leucine rich repeat kinase 2 via beclin-1. Sci. Rep. 2016, 6, 35106. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [Green Version]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Z.; Cao, G.; Wei, G. A30P mutant α-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 2019, 10, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miki, Y.; Shimoyama, S.; Kon, T.; Ueno, T.; Hayakari, R.; Tanji, K.; Matsumiya, T.; Tsushima, E.; Mori, F.; Wakabayashi, K.; et al. Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol. Aging 2018, 63, 33–43. [Google Scholar] [CrossRef]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 8, 2813. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, I.; Liang, Y.; Schools, S.; Dawson, V.L.; Dawson, T.M.; Savitt, J.M. Development and characterization of a new Parkinson’s disease model resulting from impaired autophagy. J. Neurosci. 2012, 32, 16503–16509. [Google Scholar] [CrossRef]
- Arduíno, D.M.; Esteves, A.R.; Cardoso, S.M. Mitochondrial fusion/fission, transport and autophagy in Parkinson′s disease: When mitochondria get nasty. Parkinson′s Dis. 2011, 2011, 767230. [Google Scholar] [CrossRef] [Green Version]
- Flønes, I.H.; Fernandez-Vizarra, E.; Lykouri, M.; Brakedal, B.; Skeie, G.O.; Miletic, H.; Lilleng, P.K.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; et al. Neuronal complex I deficiency occurs throughout the Parkinson’s disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol. 2018, 135, 409–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Abramov, A.Y. Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci. Lett. 2018, 663, 86–90. [Google Scholar] [CrossRef]
- Perfeito, R.; Ribeiro, M.; Rego, A.C. Alpha-synuclein-induced oxidative stress correlates with altered superoxide dismutase and glutathione synthesis in human neuroblastoma SH-SY5Y cells. Arch. Toxicol. 2017, 91, 1245–1259. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Perfeito, R.; Lázaro, D.F.; Outeiro, T.F.; Rego, A.C. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell. Neurosci. 2014, 62, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.X.; Tsoi, B.; Li, Y.F.; Kurihara, H.; He, R.R. Cardiolipin and its different properties in mitophagy and apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, S.; Wakabayashi, K.; Ishikawa, A.; Nagai, H.; Saito, M.; Maruyama, M.; Takahashi, T.; Ozawa, T.; Tsuji, S.; Takahashi, H. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov. Disord. 2000, 15, 884–888. [Google Scholar] [CrossRef]
- Buhlman, L.; Damiano, M.; Bertolin, G.; Ferrando-Miguel, R.; Lombès, A.; Brice, A.; Corti, O. Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim. Biophys. Acta 2014, 1843, 2012–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollville, E.; Carroll, R.; Cullen, S.; Martin, S. Bcl-2 Family Proteins Participate in Mitochondrial Quality Control by Regulating Parkin/PINK1-Dependent Mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [Green Version]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.S.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cell. Devoted Mol. Cell. Mech. 2010, 15, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.J.; Corti, O.; Araujo, F.; Hampe, C.; Jacquier, S.; Lücking, C.B.; Abbas, N.; Duyckaerts, C.; Rooney, T.; Pradier, L.; et al. The C289G and C418R missense mutations cause rapid sequestration of human Parkin into insoluble aggregates. Neurobiol. Dis. 2003, 14, 357–364. [Google Scholar] [CrossRef]
- Van Rompuy, A.S.; Oliveras-Salvá, M.; Van der Perren, A.; Corti, O.; Van den Haute, C.; Baekelandt, V. Nigral overexpression of alpha-synuclein in the absence of parkin enhances alpha-synuclein phosphorylation but does not modulate dopaminergic neurodegeneration. Mol. Neurodegener. 2015, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.; Nissanka, N.; Moraes, C.T. Lack of Parkin anticipates the phenotype and affects mitochondrial morphology and mtDNA levels in a mouse model of Parkinson’s disease. J. Neurosci. 2018, 38, 1042–1053. [Google Scholar] [CrossRef]
- Khandelwal, P.J.; Dumanis, S.B.; Feng, L.R.; Maguire-Zeiss, K.; Rebeck, G.; Lashuel, H.A.; Moussa, C.E. Parkinson-related parkin reduces α-Synuclein phosphorylation in a gene transfer model. Mol. Neurodegener. 2010, 5, 47. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lämmermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinson’s Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransom, B.R.; Kunis, D.M.; Irwin, I.; Langston, J.W. Astrocytes convert the parkinsonism inducing neurotoxin, MPTP, to its active metabolite, MPP+. Neurosci. Lett. 1987, 75, 323–328. [Google Scholar] [CrossRef]
- Kitayama, S.; Wang, J.B.; Uhl, G.R. Dopamine transporter mutants selectively enhance MPP+ transport. Synapse 1993, 15, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Liu, W.; He, X.; Gao, Y.; Castellani, R.J.; Perry, G.; Smith, M.A.; Zhu, X. DLP1-dependent mitochondrial fragmentation mediates 1-methyl-4-phenylpyridinium toxicity in neurons: Implications for Parkinson’s disease. Aging Cell 2011, 10, 807–823. [Google Scholar] [CrossRef] [Green Version]
- Cartelli, D.; Ronchi, C.; Maggioni, M.; Rodighiero, S.; Giavini, E.; Cappelletti, G. Microtubule dysfunction precedes transport impairment and mitochondria damage in MPP+-induced neurodegeneration. J. Neurochem. 2010, 115, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Xu, H.D.; Guan, J.J.; Hou, Y.S.; Gu, J.H.; Zhen, X.C.; Qin, Z.H. Rotenone impairs autophagic flux and lysosomal functions in Parkinson’s disease. Neuroscience 2015, 284, 900–911. [Google Scholar] [CrossRef]
- Ren, Y.; Jiang, H.; Yang, F.; Nakaso, K.; Feng, J. Parkin protects dopaminergic neurons against microtubule-depolymerizing toxins by attenuating microtubule-associated protein kinase activation. J. Biol. Chem. 2009, 284, 4009–4017. [Google Scholar] [CrossRef] [Green Version]
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.; Liu, L.; Bennett, J.P., Jr. Chronic, low-dose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol. Neurodegener. 2008, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.R.; Zhang, X.N.; Chen, Z. Mitochondrial transport serves as a mitochondrial quality control strategy in axons: Implications for central nervous system disorders. CNS Neurosci. Ther. 2019, 25, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Morreale, M.K. Huntington’s disease: Looking beyond the movement disorder. Adv. Psychosom. Med. 2015, 34, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Sieradzan, K.A.; Mann, D.M. The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 1–21. [Google Scholar] [CrossRef]
- Waldvogel, H.J.; Kim, E.H.; Tippett, L.J.; Vonsattel, J.P.; Faull, R.L. The Neuropathology of Huntington’s Disease. Curr. Topics Behav. Neurosci. 2015, 22, 33–80. [Google Scholar] [CrossRef]
- Ayala-Peña, S. Role of oxidative DNA damage in mitochondrial dysfunction and Huntington’s disease pathogenesis. Free Radic. Biol. Med. 2013, 62, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; Wanker, E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef] [Green Version]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Sapp, E.; Schwarz, C.; Chase, K.; Bhide, P.G.; Young, A.B.; Penney, J.; Vonsattel, J.P.; Aronin, N.; DiFiglia, M. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann. Neurol. 1997, 42, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Li, S. Proteasomal dysfunction in aging and Huntington disease. Neurobiol. Dis. 2011, 43, 4–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Zhang, X.D.; Wu, J.C.; Lin, F.; Wang, J.; DiFiglia, M.; Qin, Z.H. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE 2012, 7, e46834. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Arias, E.; Kaushik, S.; Sulzer, D.; Cuervo, A.M. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J. Neurosci. 2011, 31, 18492–18505. [Google Scholar] [CrossRef]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vincente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.J.; La Spada, A.R. The many faces of autophagy dysfunction in Huntington’s disease: From mechanism to therapy. Drug Discov. Today 2014, 19, 963–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kegel, K.B.; Kim, M.; Sapp, E.; McIntyre, C.; Castaño, J.G.; Aronin, N.; DiFiglia, M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 2000, 20, 7268–7278. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Tellez-Nagel, I.; Johnson, A.B.; Terry, R.D. Studies on brain biopsies of patients with Huntington’s chorea. J. Neuropathol. Exp. Neurol. 1974, 33, 308–332. [Google Scholar] [CrossRef]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef]
- Wold, M.S.; Lim, J.; Lachance, V.; Deng, Z.; Yue, Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol. Neurodegener. 2016, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Noh, J.Y.; Oh, Y.; Kim, Y.; Chang, J.W.; Chung, C.W.; Lee, S.T.; Kim, M.; Ryu, H.; Jung, Y.K. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 2012, 21, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Nagaoka, U.; Kim, K.; Jana, N.R.; Doi, H.; Maruyama, M.; Mitsui, K.; Oyama, F.; Nukina, N. Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J. Neurochem. 2004, 91, 57–68. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rué, L.; López-Soop, G.; Gelpi, E.; Martínez-Vicente, M.; Alberch, J.; Pérez-Navarro, E. Brain region- and age-dependent dysregulation of p62 and NBR1 in a mouse model of Huntington’s disease. Neurobiol. Dis. 2013, 52, 219–228. [Google Scholar] [CrossRef]
- Huang, N.; Erie, C.; Lu, M.L.; Wei, J. Aberrant subcellular localization of SQSTM1/p62 contributes to increased vulnerability to proteotoxic stress recovery in Huntington’s disease. Mol. Cell. Neurosci. 2018, 88, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, M.; Matsumoto, G.; Kino, Y.; Okuno, M.; Kurosawa-Yamada, M.; Washizu, C.; Taniguchi, H.; Nakaso, K.; Yanagawa, T.; Warabi, E.; et al. Depletion of p62 reduces nuclear inclusions and paradoxically ameliorates disease phenotypes in Huntington’s model mice. Hum. Mol. Genet. 2015, 24, 1092–1105. [Google Scholar] [CrossRef] [Green Version]
- Lamark, T.; Perander, M.; Outzen, H.; Kristiansen, K.; Øvervatn, A.; Michaelsen, E.; Bjørkøy, G.; Johansen, T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J. Biol. Chem. 2003, 278, 34568–34581. [Google Scholar] [CrossRef] [Green Version]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakson, P.; Holland, P.; Simonsen, A. The role of ALFY in selective autophagy. Cell Death Differ. 2013, 20, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Mori, F.; Tanji, K.; Toyoshima, Y.; Yoshida, M.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Optineurin immunoreactivity in neuronal nuclear inclusions of polyglutamine diseases (Huntington’s, DRPLA, SCA2, SCA3) and intranuclear inclusion body disease. Acta Neuropathol. 2012, 123, 747–749. [Google Scholar] [CrossRef]
- Okita, S.; Morigaki, R.; Koizumi, H.; Kaji, R.; Nagahiro, S.; Goto, S. Cell type-specific localization of optineurin in the striatal neurons of mice: Implications for neuronal vulnerability in Huntington’s disease. Neuroscience 2012, 202, 363–370. [Google Scholar] [CrossRef]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Oguro, A.; Kubota, H.; Shimizu, M.; Ishiura, S.; Atomi, Y. Protective role of the ubiquitin binding protein Tollip against the toxicity of polyglutamine-expansion proteins. Neurosci. Lett. 2011, 503, 234–239. [Google Scholar] [CrossRef]
- Doi, H.; Mitsui, K.; Kurosawa, M.; Machida, Y.; Kuroiwa, Y.; Nukina, N. Identification of ubiquitin-interacting proteins in purified polyglutamine aggregates. FEBS Lett. 2004, 571, 171–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, M.; Lu, T.; Furuya, T.; Degterev, A.; Mizushima, N.; Yoshimori, T.; MacDonald, M.; Yankner, B.; Yuan, J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006, 281, 14474–14485. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mealer, R.G.; Murray, A.J.; Shahani, N.; Subramaniam, S.; Snyder, S.H. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef] [Green Version]
- Metzger, S.; Saukko, M.; Van Che, H.; Tong, L.; Puder, Y.; Riess, O.; Nguyen, H.P. Age at onset in Huntington’s disease is modified by the autophagy pathway: Implication of the V471A polymorphism in Atg7. Hum. Genet. 2010, 128, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; La Spada, A.R. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012, 4, 142ra197. [Google Scholar] [CrossRef] [Green Version]
- Saft, C.; Zange, J.; Andrich, J.; Müller, K.; Lindenberg, K.; Landwehrmeyer, B.; Vorgerd, M.; Kraus, P.H.; Przuntek, H.; Schöls, L. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington’s disease. Mov. Disord. 2005, 20, 674–679. [Google Scholar] [CrossRef]
- Naia, L.; Cunha-Oliveira, T.; Rodrigues, J.; Rosenstock, T.R.; Oliveira, A.; Ribeiro, M.; Carmo, C.; Oliveira-Sousa, S.I.; Duarte, A.I.; Hayden, M.R.; et al. Histone deacetylase inhibitors protect against pyruvate dehydrogenase dysfunction in Huntington’s disease. J. Neurosci. 2017, 37, 2776–2794. [Google Scholar] [CrossRef] [Green Version]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laço, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in Huntington’s disease Models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.M.; Jekabsons, M.B.; Chen, S.; Lin, A.; Rego, A.C.; Gonçalves, J.; Ellerby, L.M.; Nicholls, D.G. Mitochondrial dysfunction in Huntington’s disease: The bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem. 2007, 101, 241–249. [Google Scholar] [CrossRef]
- Naia, L.; Ferreira, I.; Cunha-Oliveira, T.; Duarte, A.; Ribeiro, M.; Rosenstock, T.; Nôro Laço, M.; Ribeiro, M.; Oliveira, C.; Saudou, F.; et al. Activation of IGF-1 and Insulin Signaling Pathways Ameliorate Mitochondrial Function and Energy Metabolism in Huntington’s Disease Human Lymphoblasts. Mol. Neurobiol. 2014, 51, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Sarmento-Ribeiro, A.B.; Januário, C.; Rego, A.C.; Oliveira, C.R. Evidence of apoptosis and mitochondrial abnormalities in peripheral blood cells of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2008, 374, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Cunha-Oliveira, T.; Nascimento, M.; Ribeiro, M.; Proença, M.; Januario, C.; Oliveira, C.; Rego, A.C. Bioenergetic dysfunction in Huntington’s disease human cybrids. Exp. Neurol. 2011, 231, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.L.; Nascimento, M.V.; Ribeiro, M.; Almeida, S.; Cardoso, S.M.; Grazina, M.; Pratas, J.; Santos, M.J.; Januário, C.; Oliveira, C.R.; et al. Mitochondrial-dependent apoptosis in Huntington’s disease human cybrids. Exp. Neurol. 2010, 222, 243–255. [Google Scholar] [CrossRef]
- Milakovic, T.; Quintanilla, R.A.; Johnson, G.V. Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: Functional consequences. J. Biol. Chem. 2006, 281, 34785–34795. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, J.M.; Chen, S.; Almeida, S.; Riley, R.; Gonçalves, J.; Oliveira, C.R.; Hayden, M.R.; Nicholls, D.G.; Ellerby, L.M.; Rego, A.C. Mitochondrial-dependent Ca2+ handling in Huntington’s disease striatal cells: Effect of histone deacetylase inhibitors. J. Neurosci. 2006, 26, 11174–11186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.C.; Almeida, S.; Laço, M.; Duarte, A.I.; Domingues, J.; Oliveira, C.R.; Januário, C.; Rego, A.C. Mitochondrial respiratory chain complex activity and bioenergetic alterations in human platelets derived from pre-symptomatic and symptomatic Huntington’s disease carriers. Mitochondrion 2013, 13, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci 2002, 5, 731–736. [Google Scholar] [CrossRef]
- Reddy, P.H.; Shirendeb, U.P. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease. Biochim. Biophys. Acta 2012, 1822, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Kyles, P.; Tyagi, S.C.; Tyagi, N. Autophagy of mitochondria: A promising therapeutic target for neurodegenerative disease. Cell Biochem. Biophys. 2014, 70, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Costa, V.; Giacomello, M.; Hudec, R.; Lopreiato, R.; Ermak, G.; Lim, D.; Malorni, W.; Davies, K.J.; Carafoli, E.; Scorrano, L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2010, 2, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Liévens, J.C. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef] [Green Version]
- Aladdin, A.; Király, R.; Boto, P.; Regdon, Z.; Tar, K. Juvenile Huntington’s Disease Skin Fibroblasts Respond with Elevated Parkin Level and Increased Proteasome Activity as a Potential Mechanism to Counterbalance the Pathological Consequences of Mutant Huntingtin Protein. Int. J. Mol. Sci. 2019, 20, 5338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedes-Dias, P.; Pinho, B.R.; Soares, T.R.; de Proença, J.; Duchen, M.R.; Oliveira, J.M. Mitochondrial dynamics and quality control in Huntington’s disease. Neurobiol. Dis. 2016, 90, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Hutt, D.M.; Balch, W.E. Expanding proteostasis by membrane trafficking networks. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; De Pinto, V.; Messina, A.; Branca, C.; Oddo, S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J. Neurosci. 2014, 34, 7988–7998. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.L.; Jahrling, J.B.; Zhang, W.; DeRosa, N.; Bakshi, V.; Romero, P.; Galvan, V.; Richardson, A. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2017, 37, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Siman, R.; Cocca, R.; Dong, Y.N. The mTOR inhibitor rapamycin mitigates perforant pathway neurodegeneration and synapse loss in a mouse model of early-stage Alzheimer-type tauopathy. PLoS ONE 2015, 10, e0142340. [Google Scholar] [CrossRef]
- Tian, Y.; Bustos, V.; Flajolet, M.; Greengard, P. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011, 25, 1934–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Ferreira, E.; Branca, C.; Oddo, S. p62 improves AD-like pathology by increasing autophagy. Mol. Psychiatry 2017, 22, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Lemke, M.R. Effect of carbamazepine on agitation in Alzheimer’s inpatients refractory to neuroleptics. J. Clin. Psychiatry 1995, 56, 354–357. [Google Scholar] [PubMed]
- Zhang, L.; Wang, L.; Wang, R.; Gao, Y.; Che, H.; Pan, Y.; Fu, P. Evaluating the Effectiveness of GTM-1, Rapamycin, and Carbamazepine on Autophagy and Alzheimer Disease. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Coria, H.; Green, K.N.; Billings, L.M.; Kitazawa, M.; Albrecht, M.; Rammes, G.; Parsons, C.G.; Gupta, S.; Banerjee, P.; LaFerla, F.M. Memantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic mice. Am. J. Pathol. 2010, 176, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Li, Y.; Lin, L.; Cao, Y. Anti-autophagic and anti-apoptotic effects of memantine in a SH-SY5Y cell model of Alzheimer’s disease via mammalian target of rapamycin-dependent and -independent pathways. Mol. Med. Rep. 2015, 12, 7615–7622. [Google Scholar] [CrossRef] [Green Version]
- Hirano, K.; Fujimaki, M.; Sasazawa, Y.; Yamaguchi, A.; Ishikawa, K.I.; Miyamoto, K.; Souma, S.; Furuya, N.; Imamichi, Y.; Yamada, D.; et al. Neuroprotective effects of memantine via enhancement of autophagy. Biochem. Biophys. Res. Commun. 2019, 518, 161–170. [Google Scholar] [CrossRef]
- Glinz, D.; Gloy, V.L.; Monsch, A.U.; Kressig, R.W.; Patel, C.; McCord, K.A.; Ademi, Z.; Tomonaga, Y.; Schwenkglenks, M.; Bucher, H.C.; et al. Acetylcholinesterase inhibitors combined with memantine for moderate to severe Alzheimer’s disease: A meta-analysis. Swiss Med. Wkly. 2019, 149, w20093. [Google Scholar] [CrossRef] [Green Version]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the insulin sensitizer metformin in Alzheimer disease: Pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis. Assoc. Dis. 2017, 31, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, K.; Wang, R.; Liu, Y.; Kwak, Y.D.; Ma, T.; Thompson, R.C.; Zhao, Y.; Smith, L.; Gasparini, L.; et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc. Natl. Acad. Sci. USA 2009, 106, 3907–3912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.M.; Shin, H.J.; Byun, J.; Kook, S.Y.; Moon, M.; Chang, Y.J.; Mook-Jung, I. Metformin Facilitates Amyloid-β Generation by β- and γ-Secretases via Autophagy Activation. J. Alzheimer’s Dis. 2016, 51, 1197–1208. [Google Scholar] [CrossRef]
- Labuzek, K.; Liber, S.; Gabryel, B.; Adamczyk, J.; Okopień, B. Metformin increases phagocytosis and acidifies lysosomal/endosomal compartments in AMPK-dependent manner in rat primary microglia. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2010, 381, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Köhler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef] [Green Version]
- Barini, E.; Antico, O.; Zhao, Y.; Asta, F.; Tucci, V.; Catelani, T.; Marotta, R.; Xu, H.; Gasparini, L. Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy. Mol. Neurodegener. 2016, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.M.; Stephenson, M.D.; de Courten, B.; Chapman, I.; Bellman, S.M.; Aromataris, E. Metformin Use Associated with Reduced Risk of Dementia in Patients with Diabetes: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2018, 65, 1225–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Wu, Z.; Shang, J.; Xie, Z.; Chen, C.; Zhang, C. The effects of metformin on autophagy. Biomed. Pharmacother. 2021, 137, 111286. [Google Scholar] [CrossRef]
- Dasgupta, B.; Milbrandt, J. Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7217–7222. [Google Scholar] [CrossRef] [Green Version]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [Green Version]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int. 2009, 54, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beher, D.; Wu, J.; Cumine, S.; Kim, K.W.; Lu, S.C.; Atangan, L.; Wang, M. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem. Biol. Drug Des. 2009, 74, 619–624. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Sabogal-Guáqueta, A.M.; Muñoz-Manco, J.I.; Ramírez-Pineda, J.R.; Lamprea-Rodriguez, M.; Osorio, E.; Cardona-Gómez, G.P. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.C.; Klein, P.S. Multiple roles for glycogen synthase kinase-3 as a drug target in Alzheimer’s disease. Curr. Drug targets 2006, 7, 1389–1397. [Google Scholar] [CrossRef]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: In vivo and in vitro studies. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef] [Green Version]
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Aβ exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005, 94, 215–225. [Google Scholar] [CrossRef]
- Ma, T.; Hoeffer, C.A.; Capetillo-Zarate, E.; Yu, F.; Wong, H.; Lin, M.T.; Tampellini, D.; Klann, E.; Blitzer, R.D.; Gouras, G.K. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e12845. [Google Scholar] [CrossRef]
- Leyhe, T.; Eschweiler, G.W.; Stransky, E.; Gasser, T.; Annas, P.; Basun, H.; Laske, C. Increase of BDNF serum concentration in lithium treated patients with early Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 16, 649–656. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, S.; Kishi, T.; Annas, P.; Basun, H.; Hampel, H.; Iwata, N. Lithium as a Treatment for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2015, 48, 403–410. [Google Scholar] [CrossRef]
- Mendes, C.T.; Mury, F.B.; de Sá Moreira, E.; Alberto, F.L.; Forlenza, O.V.; Dias-Neto, E.; Gattaz, W.F. Lithium reduces Gsk3b mRNA levels: Implications for Alzheimer Disease. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259, 16–22. [Google Scholar] [CrossRef]
- Leroy, K.; Ando, K.; Héraud, C.; Yilmaz, Z.; Authelet, M.; Boeynaems, J.M.; Buée, L.; De Decker, R.; Brion, J.P. Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J. Alzheimer’s Dis. 2010, 19, 705–719. [Google Scholar] [CrossRef] [Green Version]
- Sofola, O.; Kerr, F.; Rogers, I.; Killick, R.; Augustin, H.; Gandy, C.; Allen, M.J.; Hardy, J.; Lovestone, S.; Partridge, L. Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS Genet. 2010, 6, e1001087. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saiki, S.; O’Kane, C.J.; Rubinsztein, D.C. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum. Mol. Genet. 2008, 17, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Maiuri, M.C.; Tasdemir, E.; Vitale, I.; Fiebig, A.A.; Andrews, D.; Molgó, J.; Díaz, J.; Lavandero, S.; Harper, F.; et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007, 14, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, K.N.; Steffan, J.S.; Martinez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef] [PubMed]
- Phelan, M. Phase II clinical trial of nicotinamide for the treatment of mild to Moderate Alzheimer’s disease. J. Geriatr. Med. Gerontol. 2017, 3, 2469–5858. [Google Scholar] [CrossRef]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef] [Green Version]
- Landel, V.; Baranger, K.; Virard, I.; Loriod, B.; Khrestchatisky, M.; Rivera, S.; Benech, P.; Féron, F. Temporal gene profiling of the 5XFAD transgenic mouse model highlights the importance of microglial activation in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini-Stoica, H.; Cole, A.L.; Swartzlander, D.B.; Chen, F.; Wan, Y.W.; Bajaj, L.; Bader, D.A.; Lee, V.M.Y.; Trojanowski, J.Q.; Liu, Z.; et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 2018, 215, 2355–2377. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, K. Turning up mitophagy in Alzheimer disease. Nat. Rev. Drug Discov. 2019, 18, 252. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.M.; Lee, W.K.; Lee, Y.H.; Kang, E.S.; Cha, B.S.; Lee, B.W. Metformin Restores Parkin-Mediated Mitophagy, Suppressed by Cytosolic p53. Int. J. Mol. Sci. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 2014, 92, 373–386. [Google Scholar] [CrossRef]
- Liu, K.; Shi, N.; Sun, Y.; Zhang, T.; Sun, X. Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem. Res. 2013, 38, 201–207. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with lewy body disease and in models of α-synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef]
- Wong, M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed. J. 2013, 36, 40–50. [Google Scholar] [CrossRef]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.H.; Tan, J.Q.; Durairajan, S.S.; Liu, L.F.; Zhang, Z.H.; Ma, L.; Shen, H.M.; Chan, H.Y.; Li, M. Isorhynchophylline, a natural alkaloid, promotes the degradation of alpha-synuclein in neuronal cells via inducing autophagy. Autophagy 2012, 8, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.L.; Kaasik, A. BECN1 is involved in the initiation of mitophagy: It facilitates PARK2 translocation to mitochondria. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.; Muqit, M.M.K. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 2019, 705, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Hertz, N.T.; Berthet, A.; Sos, M.L.; Thorn, K.S.; Burlingame, A.L.; Nakamura, K.; Shokat, K.M. A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell 2013, 154, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Sawada, T.; Lee, S.; Yu, W.; Silverio, G.; Alapatt, P.; Millan, I.; Shen, A.; Saxton, W.; Kanao, T.; et al. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 2012, 8, e1002537. [Google Scholar] [CrossRef]
- Bian, M.; Liu, J.; Hong, X.; Yu, M.; Huang, Y.; Sheng, Z.; Fei, J.; Huang, F. Overexpression of Parkin Ameliorates Dopaminergic Neurodegeneration Induced by 1- Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine in Mice. PLoS ONE 2012, 7, e39953. [Google Scholar] [CrossRef]
- Koch, J.C.; Tatenhorst, L.; Roser, A.E.; Saal, K.A.; Tönges, L.; Lingor, P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol. Ther. 2018, 189, 1–21. [Google Scholar] [CrossRef]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; Angus McQuibban, G. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain J. Neurol. 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J.M. Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013, 4, e545. [Google Scholar] [CrossRef]
- Su, C.; Yang, X.; Lou, J. Geniposide reduces α-synuclein by blocking microRNA-21/lysosome-associated membrane protein 2A interaction in Parkinson disease models. Brain Res. 2016, 1644, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Tecedor, L.; Chen, Y.H.; Monteys, A.M.; Sowada, M.J.; Thompson, L.M.; Davidson, B.L. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron 2015, 85, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, C.; Menzies, F.M.; Renna, M.; Acevedo-Arozena, A.; Corrochano, S.; Sadiq, O.; Brown, S.D.; Rubinsztein, D.C. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 2144–2153. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Estevez, M.A.; Casarejos, M.J.; López Sendon, J.; Garcia Caldentey, J.; Ruiz, C.; Gomez, A.; Perucho, J.; de Yebenes, J.G.; Mena, M.A. Trehalose reverses cell malfunction in fibroblasts from normal and huntington’s disease patients caused by proteosome inhibition. PLoS ONE 2014, 9, e90202. [Google Scholar] [CrossRef]
- Jiang, W.; Wei, W.; Gaertig, M.A.; Li, S.; Li, X.-J. Therapeutic effect of berberine on Huntington’s disease transgenic mouse model. PLoS ONE 2015, 10, e0134142. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Menzies, F.M.; Garcia-Arencibia, M.; Imarisio, S.; O’Sullivan, N.C.; Ricketts, T.; Kent, B.A.; Rao, M.V.; Lam, W.; Green-Thompson, Z.W.; Nixon, R.A.; et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 2015, 22, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Luis-Ravelo, D.; Estévez-Silva, H.; Barroso-Chinea, P.; Afonso-Oramas, D.; Salas-Hernández, J.; Rodríguez-Núñez, J.; Acevedo-Arozena, A.; Marcellino, D.; González-Hernández, T. Pramipexole reduces soluble mutant huntingtin and protects striatal neurons through dopamine D3 receptors in a genetic model of Huntington’s disease. Exp. Neurol. 2018, 299, 137–147. [Google Scholar] [CrossRef]
- Jin, J.; Gu, H.; Anders, N.M.; Ren, T.; Jiang, M.; Tao, M.; Peng, Q.; Rudek, M.A.; Duan, W. Metformin Protects Cells from Mutant Huntingtin Toxicity Through Activation of AMPK and Modulation of Mitochondrial Dynamics. NeuroMol. Med. 2016, 18, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Manrique, R.P.; Farina, F.; Cambon, K.; Dolores Sequedo, M.; Parker, A.J.; Millán, J.M.; Weiss, A.; Déglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, A.S.; Miller, J.; Arrasate, M.; Wong, J.S.; Pleiss, M.A.; Finkbeiner, S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc. Natl. Acad. Sci. USA 2010, 107, 16982–16987. [Google Scholar] [CrossRef] [Green Version]
- Siddiqi, F.H.; Menzies, F.M.; Lopez, A.; Stamatakou, E.; Karabiyik, C.; Ureshino, R.; Ricketts, T.; Jimenez-Sanchez, M.; Esteban, M.A.; Lai, L.; et al. Felodipine induces autophagy in mouse brains with pharmacokinetics amenable to repurposing. Nat. Commun. 2019, 10, 1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Wang, C.; Wang, Z.; Zhu, C.; Li, J.; Sha, T.; Ma, L.; Gao, C.; Yang, Y.; Sun, Y.; et al. Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature 2019, 575, 203–209. [Google Scholar] [CrossRef]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Wacker, J.L.; Huang, S.Y.; Steele, A.D.; Aron, R.; Lotz, G.P.; Nguyen, Q.; Giorgini, F.; Roberson, E.D.; Lindquist, S.; Masliah, E.; et al. Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s disease. J. Neurosci. 2009, 29, 9104–9114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, V.K.; Ali, A.; Dutta, N.; Ghosh, S.; Jana, M.; Ganguli, A.; Komarov, A.; Paul, S.; Dwivedi, V.; Chatterjee, S.; et al. Azadiradione ameliorates polyglutamine expansion disease in Drosophila by potentiating DNA binding activity of heat shock factor 1. Oncotarget 2016, 7, 78281–78296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]







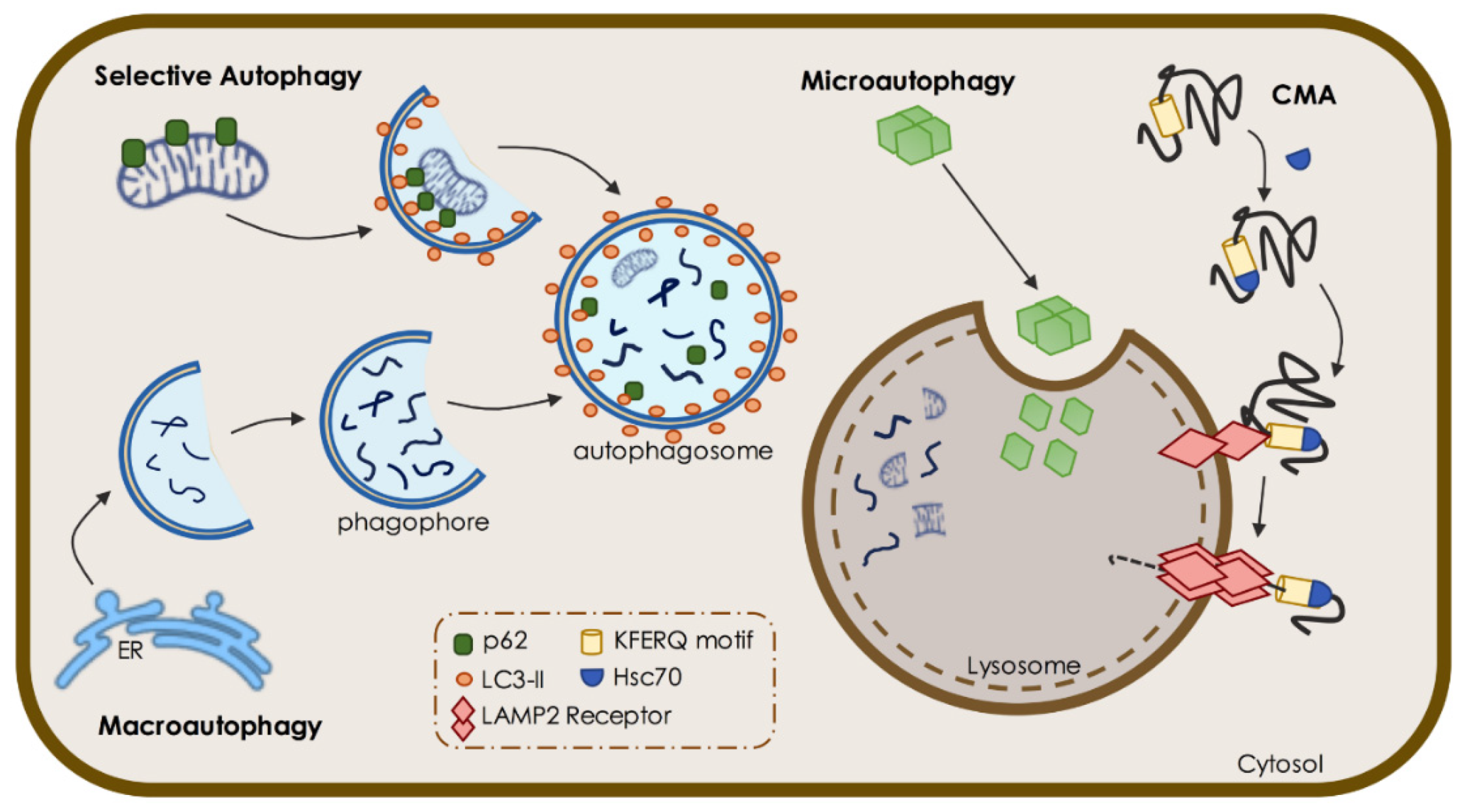{kind=link}
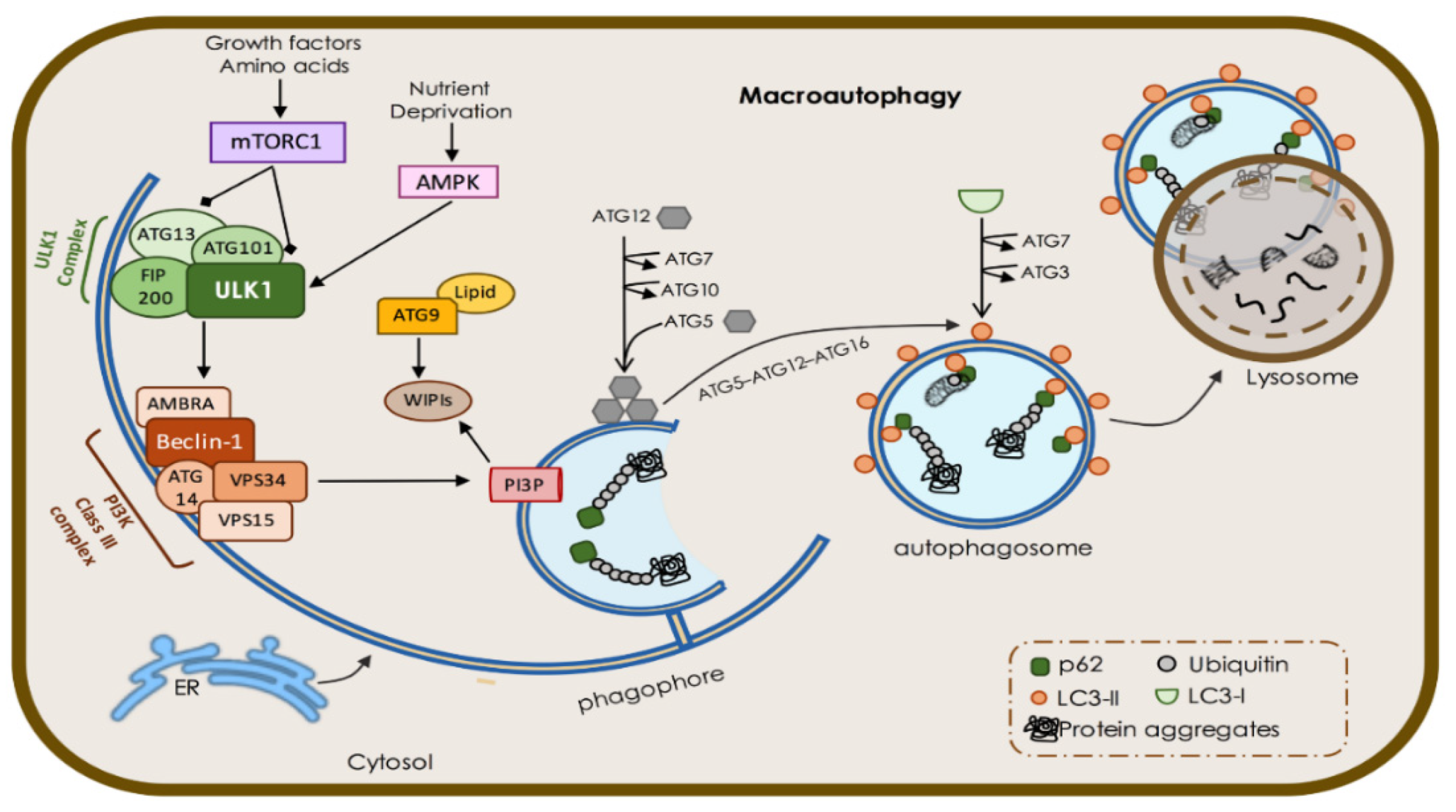{kind=link}
{kind=link}
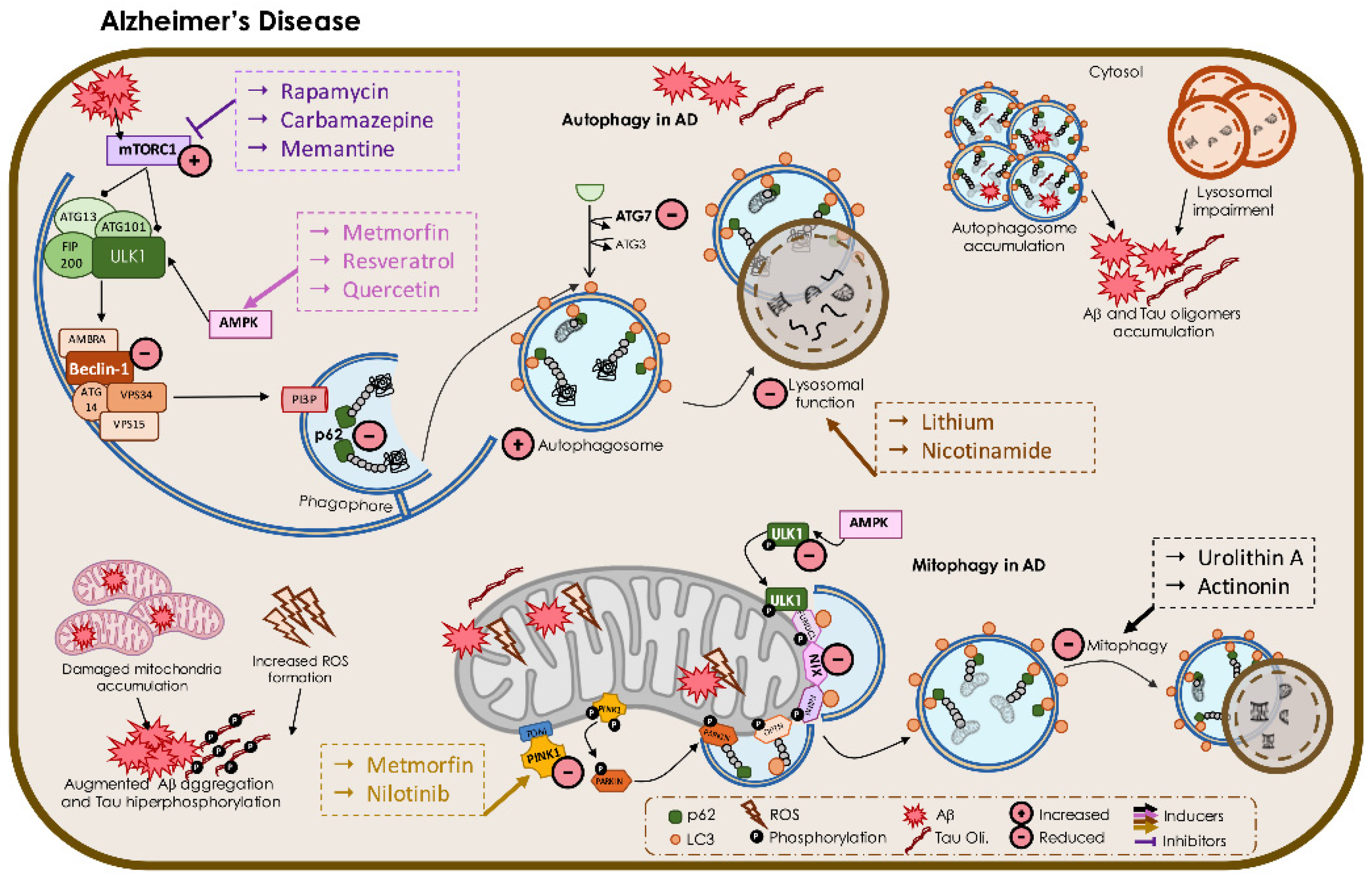{kind=link}
{kind=link}
{kind=link}
| Disease | Protective Compounds-Related Pathways | Compounds | Models | References |
|---|---|---|---|---|
| Alzheimer’s Disease | mTORC1 | Rapamycin | Tg2576 mice; Apolipoprotein E ϵ4 transgenic mice; an adeno-associated viral vector-based AD mice; AD hippocampal primary neurons; 3xTg-AD mice; | [318,319,320,321,322] |
| Carbamazepine | AD patients; | [325] | ||
| Memantine | AD patients; APP695swe expression in SH-SY5Y cells; | [327,328] | ||
| AMPK | Metformin | AD patients; AD rat primary microglia; Tau transgenic mice primary neurons; human tau (P301S) transgenic mouse model; | [331,334,335,336] | |
| Resveratrol | AD primary neurons; Tg19959 mice; AD patients; | [340,341,343] | ||
| Quercetin | 3xTg-AD mice; | [344] | ||
| Lysosomal Function | Lithium | AD patients; Mutant tau transgenic mice; Drosophila AD model | [349,350,352,353] Clinical trial: [NCT03185208] | |
| Nicotinamide | 3xTg-AD model mice; AD patients; | [357,358,359] Clinical [trials: NCT03061474] | ||
| PINK1 | Nilotinib | C57BL/6 injected with Aβ1–42; | [365] | |
| Mitophagy | Urolithin A | APP/PS1 mice; | [163] | |
| Actinonin | APP/PS1 mice; | [163] | ||
| Parkinson’s Disease | mTORC1 | Rapamycin | Dopaminergic neurons from MPTP-induced mice; 6-OHDA-treated mice; overexpressing WT aSyn mice; | [366,367,368] |
| AMPK | Metformin | MPTP-treated mice; | [369] | |
| Beclin-1 | Isorhynchophylline | Dopaminergic neurons with A53T aSyn overexpressing; | [372] | |
| PINK1 | Kinetin triphosphate | Dopaminergic SH-SY5Y cells; | [375] | |
| Parkin | SR3677 | Paraquat-treated fly model; | [378] | |
| Lamp2 | Geniposide | MPTP-induced mice; | [382] | |
| Huntington’s Disease | mTORC1 | Rapamycin | HD fly model; | [271] |
| CCI-779 | HD-N171-N82Q mice; | [383] | ||
| AMPK | Metformin | STHdhQ111/Q111 cells; 109Q/109Q mouse striatal cells; HD patients; | [391,392,393] | |
| Berberine | N171-82Q HD mouse model | [387] | ||
| Hsc70 | Azadiradione | HD fly model; | [403] | |
| Autophagic Function | Rilmenidine | N171-82Q HD mice | [384] | |
| Trehalose | HD R6/2 transgenic mice; HD patient fibroblasts; | [385,386] | ||
| Lithium | Drosophila HD models; | [354] | ||
| Clonidine | HD fly and zebrafish models; | [388] | ||
| Calpain | HD Drosophila model; N171-82Q mice; | [389] | ||
| Pramipexole | R6/1 HD mouse model; | [390] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhães, J.D.; Fão, L.; Vilaça, R.; Cardoso, S.M.; Rego, A.C. Macroautophagy and Mitophagy in Neurodegenerative Disorders: Focus on Therapeutic Interventions. Biomedicines 2021, 9, 1625. https://doi.org/10.3390/biomedicines9111625
Magalhães JD, Fão L, Vilaça R, Cardoso SM, Rego AC. Macroautophagy and Mitophagy in Neurodegenerative Disorders: Focus on Therapeutic Interventions. Biomedicines. 2021; 9(11):1625. https://doi.org/10.3390/biomedicines9111625
Chicago/Turabian StyleMagalhães, João Duarte, Lígia Fão, Rita Vilaça, Sandra Morais Cardoso, and Ana Cristina Rego. 2021. "Macroautophagy and Mitophagy in Neurodegenerative Disorders: Focus on Therapeutic Interventions" Biomedicines 9, no. 11: 1625. https://doi.org/10.3390/biomedicines9111625