Therapeutic Targeting of Autoreactive B Cells: Why, How, and When?


Abstract
:1. Introduction: The “Why?”
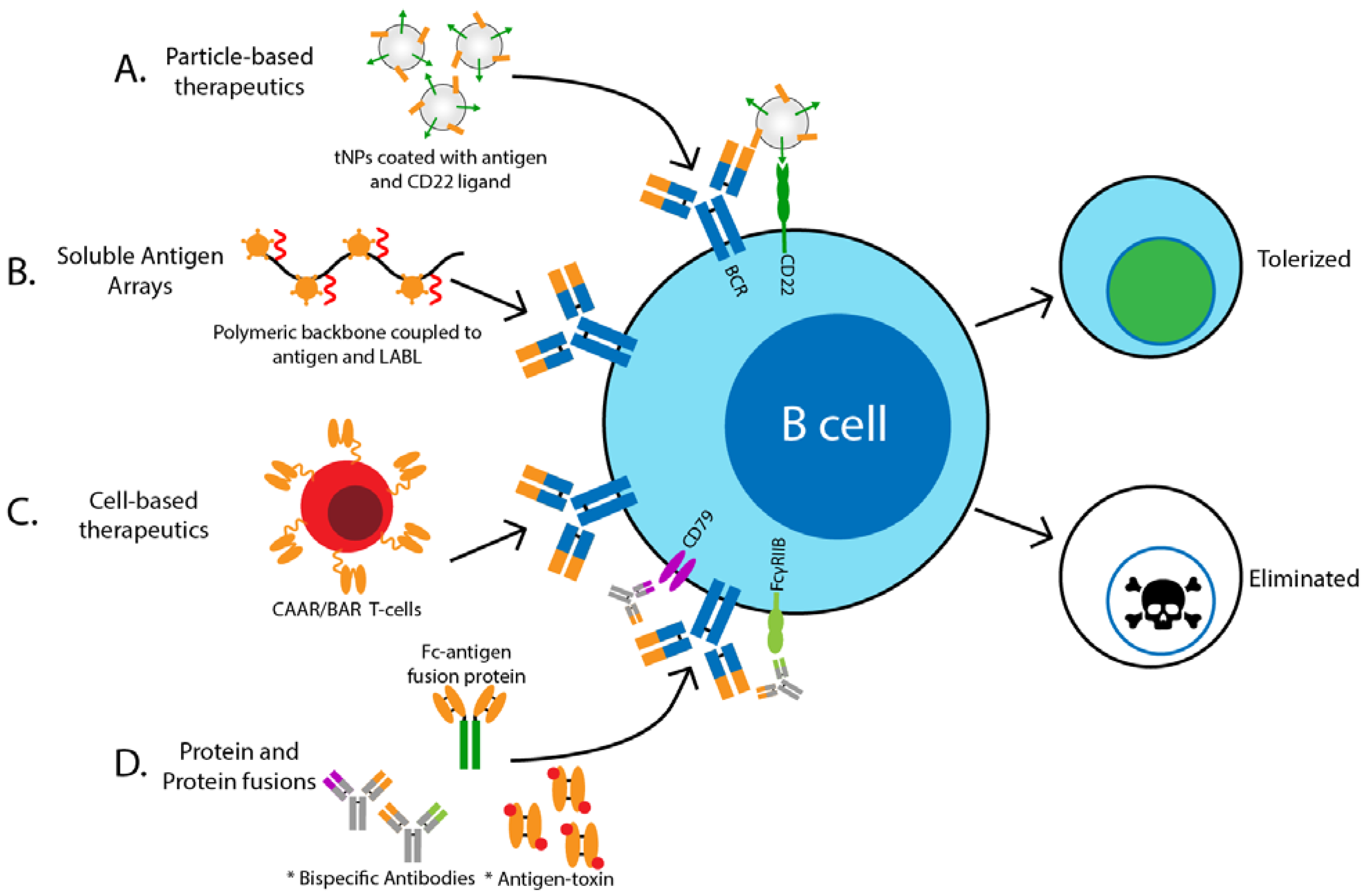
2. Methods to Target Antigen-Specific B Cells: The “How?”
2.1. Particle-Based Therapeutics
2.2. Soluble Antigen Arrays
2.3. Cell-Based Therapeutics
2.4. Protein and Protein Fusions
3. Challenges to Antigen-Specific B Cell Therapy: The “When?”
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SLE | systemic lupus erythematosus |
| T1D | type 1 diabetes |
| MS | multiple sclerosis |
| RA | rheumatoid arthritis |
| BCR | B cell receptor |
| tNPs | tolerogenic nanoparticles |
| APC | antigen-presenting cell |
| MHC | major histocompatibility complex |
| EAE | experimental autoimmune encephalomyelitis |
| FVIII | Factor VIII |
| CAR | chimeric antigen receptor |
| ITAM | immunoreceptor tyrosine-based activation motif |
| PV | pemphigus vulgaris |
| Dsg3 | desmoglein 3 |
| NOD | non-obese diabetic |
| BAR | B-cell-targeting Antibody Receptor |
| CAAR | chimeric auto-antibody receptor |
| Treg | T regulatory cell |
| Teff | T effector cell |
| NK | natural killer cell |
| ADCC | antibody-dependent cell-mediated cytotoxicity |
| ADC | antibody-drug conjugate |
References
- Cross, A.H.; Stark, J.L.; Lauber, J.; Ramsbottom, M.J.; Lyons, J.-A. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J. Neuroimmunol. 2006, 180, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly, P.; Mouquet, H.; Roujeau, J.-C.; D’Incan, M.; Gilbert, D.; Jacquot, S.; Gougeon, M.-L.; Bedane, C.; Muller, R.; Dreno, B.; et al. A single cycle of rituximab for the treatment of severe pemphigus. N. Engl. J. Med. 2007, 357, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Popa, C.; Leandro, M.J.; Cambridge, G.; Edwards, J.C.W. Repeated B lymphocyte depletion with rituximab in rheumatoid arthritis over 7 yrs. Rheumatology 2007, 46, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Rubbert-Roth, A.; Tak, P.P.; Zerbini, C.; Tremblay, J.-L.; Carreño, L.; Armstrong, G.; Collinson, N.; Shaw, T.M.; on behalf of the MIRROR Trial Investigators. Efficacy and safety of various repeat treatment dosing regimens of rituximab in patients with active rheumatoid arthritis: Results of a Phase III randomized study (MIRROR). Rheumatology 2010, 49, 1683–1693. [Google Scholar] [CrossRef] [Green Version]
- Tandan, R.; Hehir, M.K.; Waheed, W.; Howard, D.B. Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve 2017, 56, 185–196. [Google Scholar] [CrossRef]
- Smith, K.G.C.; Jones, R.B.; Burns, S.M.; Jayne, D.R.W. Long-term comparison of rituximab treatment for refractory systemic lupus erythematosus and vasculitis: Remission, relapse, and re-treatment. Arthritis Rheum. 2006, 54, 2970–2982. [Google Scholar] [CrossRef]
- Smith, R.M.; Jones, R.B.; Guerry, M.-J.; Laurino, S.; Catapano, F.; Chaudhry, A.; Smith, K.G.C.; Jayne, D.R.W. Rituximab for remission maintenance in relapsing antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2012, 64, 3760–3769. [Google Scholar] [CrossRef]
- Venhoff, N.; Niessen, L.; Kreuzaler, M.; Rolink, A.G.; Hässler, F.; Rizzi, M.; Voll, R.E.; Thiel, J. Reconstitution of the peripheral B lymphocyte compartment in patients with ANCA-associated vasculitides treated with rituximab for relapsing or refractory disease. Autoimmunity 2014, 47, 401–408. [Google Scholar] [CrossRef]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The lupus nephritis assessment with rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef]
- Fabris, M.; De Vita, S.; Blasone, N.; Visentini, D.; Pezzarini, E.; Pontarini, E.; Fabro, C.; Quartuccio, L.; Mazzolini, S.; Curcio, F.; et al. Serum levels of anti-CCP antibodies, anti-MCV antibodies and RF IgA in the follow-up of patients with rheumatoid arthritis treated with rituximab. Autoimmun. Highlights 2010, 1, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, R.A.; Lamothe, R.A.; Ferrari, J.D.; Zhang, A.-H.; Rossi, R.J.; Kolte, P.N.; Griset, A.P.; O’Neil, C.P.; Altreuter, D.H.; Browning, E.A.; et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, E156–E165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeste, A.; Nadeau, M.; Burns, E.J.; Weiner, H.L.; Quintana, F.J. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2012, 109, 11270–11275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getts, D.R.; Martin, A.J.; McCarthy, D.P.; Terry, R.L.; Hunter, Z.N.; Yap, W.T.; Getts, M.T.; Pleiss, M.A.; Luo, X.; King, N.J.C.; et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 2012, 30, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Casares, X.; Blanco, J.; Ambalavanan, P.; Yamanouchi, J.; Singha, S.; Fandos, C.; Tsai, S.; Wang, J.; Garabatos, N.; Izquierdo, C.; et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nat. Cell Biol. 2016, 530, 434–440. [Google Scholar] [CrossRef]
- Tsai, S.; Shameli, A.; Yamanouchi, J.; Clemente-Casares, X.; Wang, J.; Serra, P.; Yang, Y.; Medarova, Z.; Moore, A.; Santamaria, P. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity 2010, 32, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Temchura, V.; Kozlova, D.; Sokolova, V.; Überla, K.; Epple, M. Targeting and activation of antigen-specific B-cells by calcium phosphate nanoparticles loaded with protein antigen. Biomaterials 2014, 35, 6098–6105. [Google Scholar] [CrossRef]
- Hong, S.; Zhang, Z.; Liu, H.; Tian, M.; Zhu, X.; Zhang, Z.; Wang, W.; Zhou, X.; Zhang, F.; Ge, Q.; et al. B cells are the dominant antigen-presenting cells that activate naive CD4+ T cells upon immunization with a virus-derived nanoparticle antigen. Immunity 2018, 49, 695–708.e4. [Google Scholar] [CrossRef] [Green Version]
- Duong, B.H.; Tian, H.; Ota, T.; Completo, G.; Han, S.; Vela, J.L.; Ota, M.; Kubitz, M.; Bovin, N.; Paulson, J.C.; et al. Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. J. Exp. Med. 2010, 207, 173–187. [Google Scholar] [CrossRef]
- Macauley, M.S.; Pfrengle, F.; Rademacher, C.; Nycholat, C.M.; Gale, A.J.; Von Drygalski, A.; Paulson, J.C. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J. Clin. Investig. 2013, 123, 3074–3083. [Google Scholar] [CrossRef] [Green Version]
- Hartwell, B.L.; Martinez-Becerra, F.J.; Chen, J.; Shinogle, H.; Sarnowski, M.; Moore, D.S.; Berkland, C.J. Antigen-specific binding of multivalent soluble antigen arrays induces receptor clustering and impedes B cell receptor mediated signaling. Biomacromolecules 2016, 17, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Sestak, J.O.; Fakhari, A.; Badawi, A.H.; Siahaan, T.J.; Berkland, C.J. Structure, size, and solubility of antigen arrays determines efficacy in experimental autoimmune encephalomyelitis. AAPS J. 2014, 16, 1185–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwell, B.L.; Pickens, C.J.; Leon, M.; Berkland, C.J. Multivalent soluble antigen arrays exhibit high avidity binding and modulation of B cell receptor-mediated signaling to drive efficacy against experimental autoimmune encephalomyelitis. Biomacromolecules 2017, 18, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Leon, M.A.; Wemlinger, S.M.; Larson, N.R.; Ruffalo, J.K.; Middaugh, C.R.; Cambier, J.C.; Berkland, C.J.; Sestak, J.O. Soluble antigen arrays for selective desensitization of insulin-reactive B cells. Mol. Pharm. 2019, 16, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Beilke, J.N.; Jasinski, J.M.; Kobayashi, M.; Miao, N.; Li, M.; Coulombe, M.G.; Liu, E.; Elliott, J.F.; Gill, R.G.; et al. Priming and effector dependence on insulin B:9–23 peptide in NOD islet autoimmunity. J. Clin. Investig. 2007, 117, 1835–1843. [Google Scholar] [CrossRef]
- Zhang, L.; Crawford, F.; Yu, L.; Michels, A.; Nakayama, M.; Davidson, H.W.; Kappler, J.W.; Eisenbarth, G.S. Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 2656–2661. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Sosinowski, T.; Cox, A.R.; Cepeda, J.R.; Sekhar, N.S.; Hartig, S.M.; Miao, D.; Yu, L.; Pietropaolo, M.; Davidson, H.W. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J. Autoimmun. 2019, 96, 50–58. [Google Scholar] [CrossRef]
- Serreze, D.V.; Fleming, S.A.; Chapman, H.D.; Richard, S.D.; Leiter, E.H.; Tisch, R. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Immunol. 1998, 161, 3912–3918. [Google Scholar]
- Hulbert, C.; Riseili, B.; Rojas, M.; Thomas, J.W. B cell specificity contributes to the outcome of diabetes in nonobese diabetic mice. J. Immunol. 2001, 167, 5535–5538. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.-H.; Yoon, J.; Kim, Y.C.; Scott, D.W. Targeting antigen-specific B cells using antigen-expressing transduced regulatory T cells. J. Immunol. 2018, 201, 1434–1441. [Google Scholar] [CrossRef] [Green Version]
- Pohl, A.D.P.; Venkatesha, S.H.; Zhang, A.-H.; Scott, D.W. Suppression of FVIII-specific memory B cells by chimeric BAR receptor-engineered natural regulatory T cells. Front. Immunol. 2020, 11, 693. [Google Scholar] [CrossRef] [PubMed]
- Parvathaneni, K.; Scott, D.W. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. 2018, 2, 2332–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Lundgren, D.K.; Mao, X.; Manfredo-Vieira, S.; Nunez-Cruz, S.; Williams, E.F.; Assenmacher, C.-A.; Radaelli, E.; Oh, S.; Wang, B.; et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J. Clin. Investig. 2020, 130, 6317–6324. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.A.; Kendall, P.L.; Thomas, J.W. Autoantigen-specific B-cell depletion overcomes failed immune tolerance in type 1 diabetes. Diabetes 2012, 61, 2037–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.I.; Schroer, J.A.; Marcus-Samuels, B.; McElduff, A.; Bender, T.P. Binding of insulin to its receptor impairs recognition by monoclonal anti-insulin antibodies. Diabetes 1984, 33, 778–784. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.; Hippen, K.; Minskoff, S.; Mellman, I.; Pani, G.; Siminovitch, K.; Cambier, J. Recruitment and activation of PTP1C in negative regulation of antigen receptor signaling by Fc gamma RIIB1. Science 1995, 268, 293–297. [Google Scholar] [CrossRef]
- Chu, S.Y.; Yeter, K.C.; Kotha, R.; Pong, E.; Miranda, Y.; Phung, S.; Chen, H.; Lee, S.-H.; Leung, I.; Bonzon, C.; et al. Suppression of rheumatoid arthritis B cells by XmAb5871, an anti-CD19 antibody that coengages B cell antigen receptor complex and fcγ receptor IIb inhibitory receptor. Arthritis Rheumatol. 2014, 66, 1153–1164. [Google Scholar] [CrossRef]
- Horton, H.M.; Chu, S.Y.; Ortiz, E.C.; Pong, E.; Cemerski, S.; Leung, I.W.L.; Jacob, N.; Zalevsky, J.; DesJarlais, J.R.; Stohl, W.; et al. Antibody-mediated coengagement of FcγRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J. Immunol. 2011, 186, 4223–4233. [Google Scholar] [CrossRef]
- Merrill, J.; June, J.; Koumpouras, F.; Machua, W.; Khan, M.F.; Askanase, A.; Khosroshahi, A.; Sheikh, S.; James, J.A.; Guthridge, J.; et al. Phase 2, double-blind, randomized, placebo-controlled study of a reversible B cell inhibitor, Xmab (R) 5871, in systemic lupus erythematosus (Sle). Ann. Rheum. Dis. 2019, 78, 761–762. [Google Scholar] [CrossRef] [Green Version]
- Veri, M.-C.; Burke, S.; Huang, L.; Li, H.; Gorlatov, S.; Tuaillon, N.; Rainey, G.J.; Ciccarone, V.; Zhang, T.; Shah, K.; et al. Therapeutic control of b-cell activation via recruitment of Fcγ receptor IIB (CD32B) inhibitory function with a novel bispecific antibody scaffold. Arthritis Rheum. 2010, 62, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Pandya, N.; Chen, W.; Lohr, J.; Yao, X.-T.; Burns, R.; Li, H.; Muth, J.; Goldwater, R.; Bonvini, E.; Johnson, S.; et al. OP0201 Safety, Tolerability, and Functional Activity of MGD010, A Dart® Molecule Targeting CD32B and CD79B, Following A Single Dose Administration in Healthy Volunteers. Ann. Rheum. Dis. 2016, 75, 132–133. [Google Scholar] [CrossRef]
- Hardy, I.R.; Anceriz, N.; Rousseau, F.; Seefeldt, M.B.; Hatterer, E.; Irla, M.; Buatois, V.; Chatel, L.E.; Getahun, A.; Fletcher, A.; et al. Anti-CD79 antibody induces B cell anergy that protects against autoimmunity. J. Immunol. 2014, 192, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, F.; Putt, M.; Koo, Y.K.; Madaio, M.; Cambier, J.C.; Cohen, P.L.; Eisenberg, R.A. B cell depletion with anti-CD79 mAbs ameliorates autoimmune disease in MRL/lpr Mice1. J. Immunol. 2008, 181, 2961–2972. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.R.W.; Gliddon, L.; Fieles, W.; et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Palanca-Wessels, M.C.A.; Czuczman, M.; Salles, G.; Assouline, S.; Sehn, L.H.; Flinn, I.; Patel, M.R.; Sangha, R.; Hagenbeek, A.; Advani, R.; et al. Safety and activity of the anti-CD79B antibody–drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: A phase 1 study. Lancet Oncol. 2015, 16, 704–715. [Google Scholar] [CrossRef]
- Saunders, L.R.; Bankovich, A.J.; Anderson, W.C.; Aujay, M.A.; Bheddah, S.; Black, K.; Desai, R.; Escarpe, P.A.; Hampl, J.; Laysang, A.; et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 2015, 7, 302ra136. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, J.L.; Niesvizky, R.; Stadtmauer, E.A.; Chanan-Khan, A.; Siegel, D.; Horne, H.; Wegener, W.A.; Goldenberg, D.M. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 163, 478–486. [Google Scholar] [CrossRef]
- Pickens, C.J.; Christopher, M.A.; Leon, M.A.; Pressnall, M.M.; Johnson, S.N.; Thati, S.; Sullivan, B.P.; Berkland, C.J. Antigen-drug conjugates as a novel therapeutic class for the treatment of antigen-specific autoimmune disorders. Mol. Pharm. 2019, 16, 2452–2461. [Google Scholar] [CrossRef]
- Dema, B.; Charles, N. Autoantibodies in SLE: Specificities, isotypes and receptors. Antibodies 2016, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.S.; Hemmer, B.; Cepok, S. The role of antibodies in multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 239–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifacio, E.; Lampasona, V.; Bernasconi, L.; Ziegler, A.-G. Maturation of the humoral autoimmune response to epitopes of GAD in preclinical childhood type 1 diabetes. Diabetes 2000, 49, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, E.B.; Stenberg, P.; Book, C.; Sjöberg, K. Antibodies against transglutaminases, peptidylarginine deiminase and citrulline in rheumatoid arthritis—New pathways to epitope spreading. Clin. Exp. Rheumatol. 2006, 24, 12–18. [Google Scholar] [PubMed]
- Syren, K.; Hartmann, U.; Meinck, H.M.; Knip, M.; Akerblom, H.K.; Richter, W.O. Epitope spreading and a varying but not disease-specific GAD65 antibody response in Type I diabetes. Diabetol. 2000, 43, 210–217. [Google Scholar] [CrossRef]
- Tuohy, V.K.; Yu, M.; Weinstock-Guttman, B.; Kinkel, R.P. Diversity and plasticity of self recognition during the development of multiple sclerosis. J. Clin. Investig. 1997, 99, 1682–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Woude, D.; Rantapää-Dahlqvist, S.; Ioan-Facsinay, A.; Onnekink, C.; Schwarte, C.M.; Verpoort, K.N.; Drijfhout, J.W.; Huizinga, T.W.J.; Toes, R.E.M.; Pruijn, G.J.M. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann. Rheum. Dis. 2010, 69, 1554–1561. [Google Scholar] [CrossRef]
- Fisher, D.E.; Reeves, W.H.; Wisniewolski, R.; Lahita, R.G.; Chiorazzi, N. Temporal shifts from Sm to ribonucleoprotein reactivity in systemic lupus erythematosus. Arthritis Rheum. 1985, 28, 1348–1355. [Google Scholar] [CrossRef]


{kind=link}
| Disease Model | Method | Results | References |
|---|---|---|---|
| Hemophilia | tNPs coated with FVIII and CD22 ligands | FVIII-specific B cell deletion | [20] |
| tNP with FVIII encapsulated with rapamycin | FVIII-specific B cell deletion | [12] | |
| FVIII-BAR Treg | FVIII-specific B cell tolerance and antibody suppression | [27] | |
| FVIII-BAR CD8+ T cell | FVIII-specific B cell deletion | [30] | |
| EAE | Soluble antigen array with PLP + LABL | PLP-reactive B cell anergy | [21,23] |
| Pemphigus Vulgaris | Dsg3-CAAR T cell | Dsg3-specific B cell deletion | [33,34] |
| Autoimmune diabetes | Soluble antigen array with insulin + LABL | Insulin-specific B cell anergy ex vivo | [24] |
| 287-CAR CD8+ T cell | I-Ag7-B:9–23 presenting APC deletion | [27] | |
| mab123 | Insulin-binding B cell deletion | [35] | |
| Fc-insulin fusion protein (AKS-107) | Insulin-binding B cell deletion/anergy | unpublished | |
| Insulin-toxin | Insulin-binding B cell deletion | unpublished |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stensland, Z.C.; Cambier, J.C.; Smith, M.J. Therapeutic Targeting of Autoreactive B Cells: Why, How, and When? Biomedicines 2021, 9, 83. https://doi.org/10.3390/biomedicines9010083
Stensland ZC, Cambier JC, Smith MJ. Therapeutic Targeting of Autoreactive B Cells: Why, How, and When? Biomedicines. 2021; 9(1):83. https://doi.org/10.3390/biomedicines9010083
Chicago/Turabian StyleStensland, Zachary C., John C. Cambier, and Mia J. Smith. 2021. "Therapeutic Targeting of Autoreactive B Cells: Why, How, and When?" Biomedicines 9, no. 1: 83. https://doi.org/10.3390/biomedicines9010083