Doxazosin, a Classic Alpha 1-Adrenoceptor Antagonist, Overcomes Osimertinib Resistance in Cancer Cells via the Upregulation of Autophagy as Drug Repurposing


 ,
,
Abstract
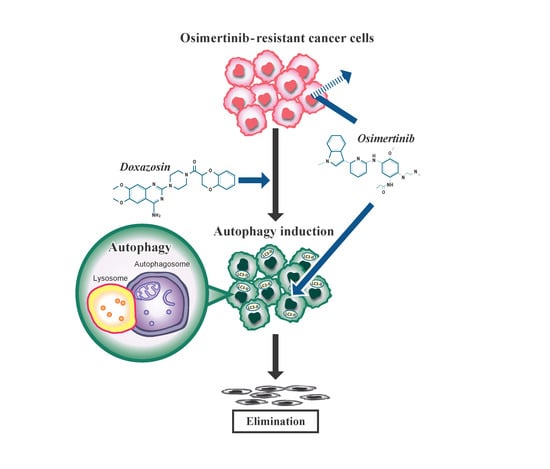
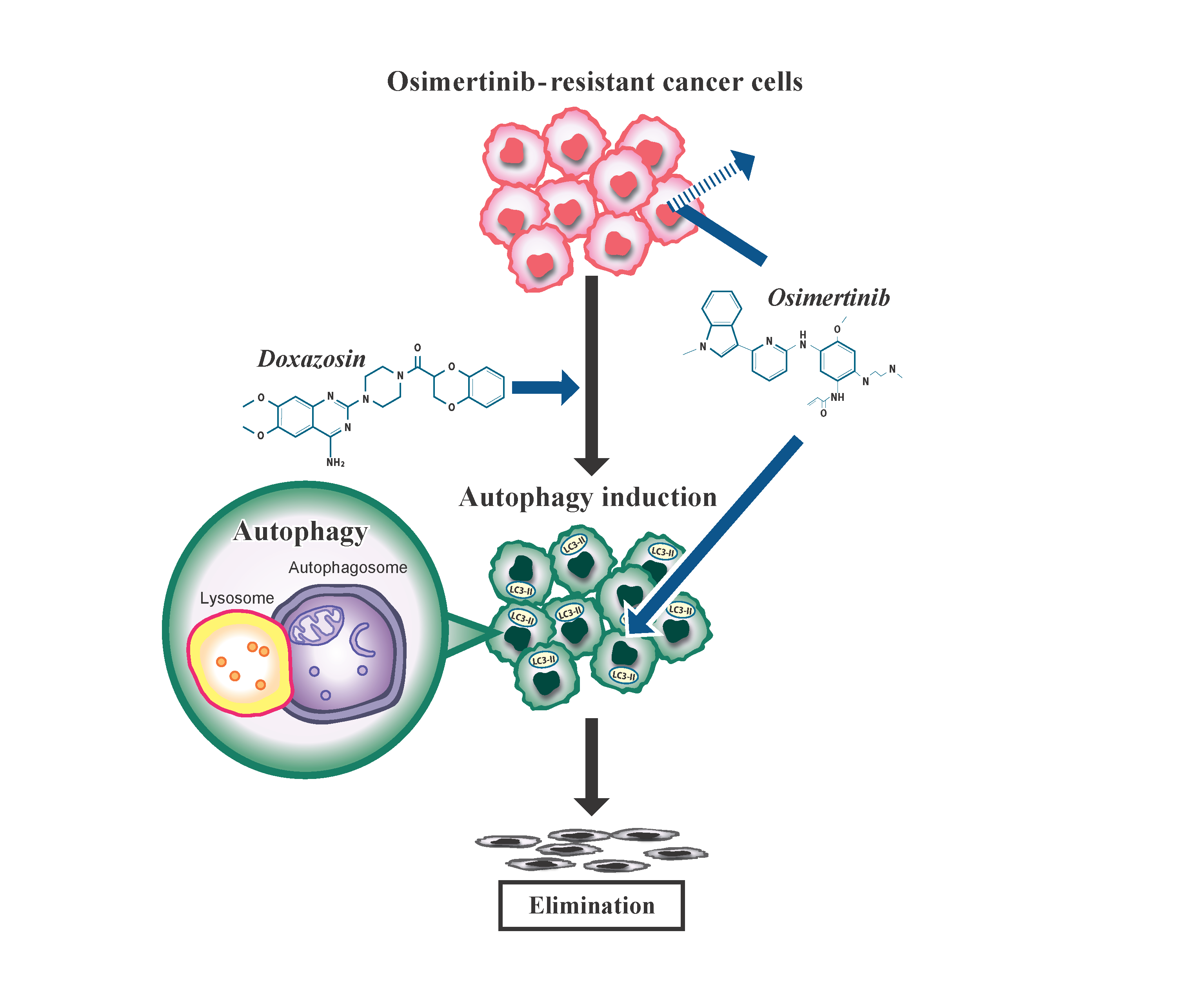
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Cultures
2.3. Cell Viability Assay
2.4. Immunoblot Analysis
2.5. Immunofluorescence Analysis
2.6. Statistical Analysis
3. Results
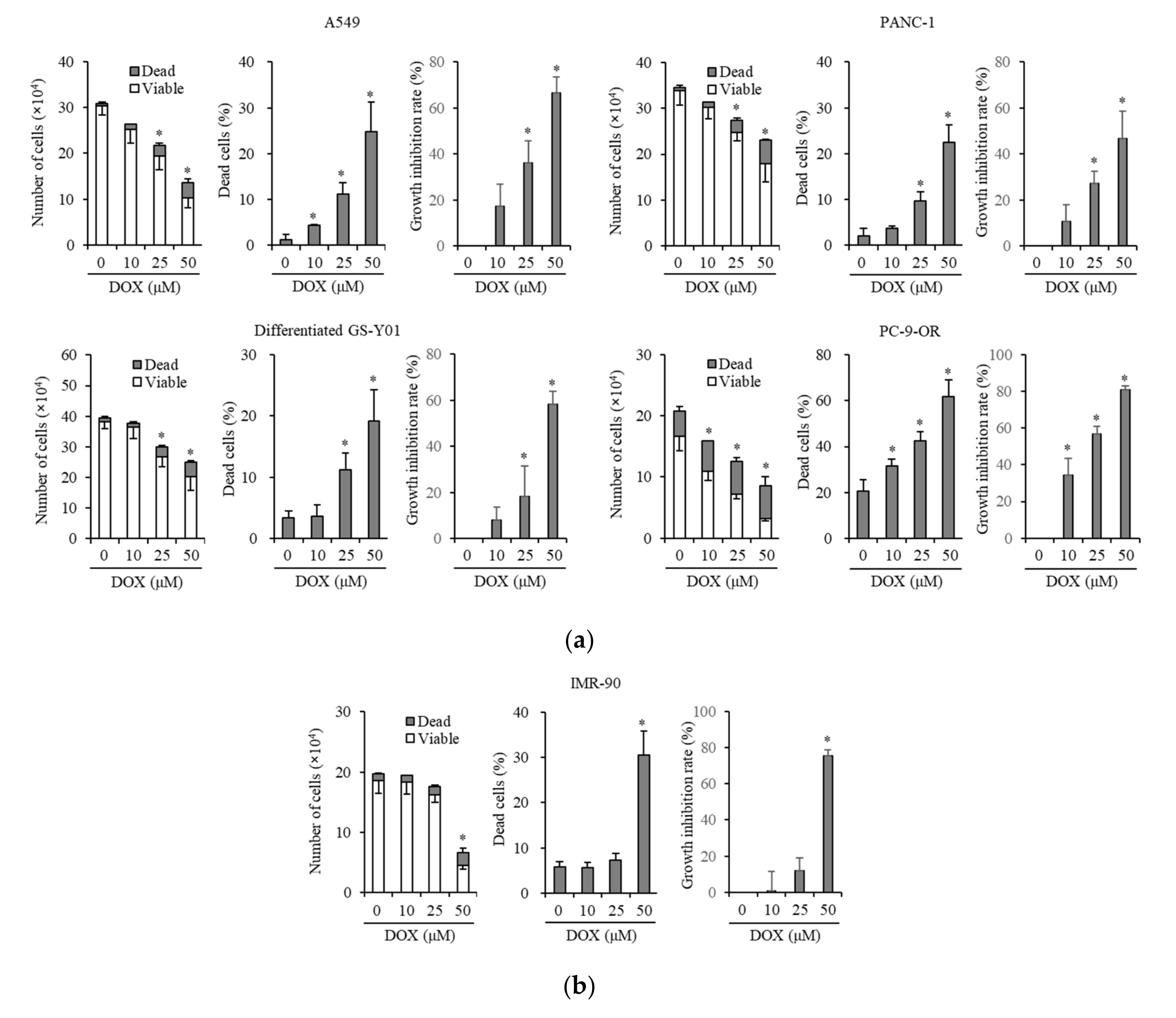
3.1. Doxazosin Inhibits Cancer Cell Growth and Is Cytotoxic to Cancer Cells and Cancer Stem Cells, but not to Non-Cancer Cells
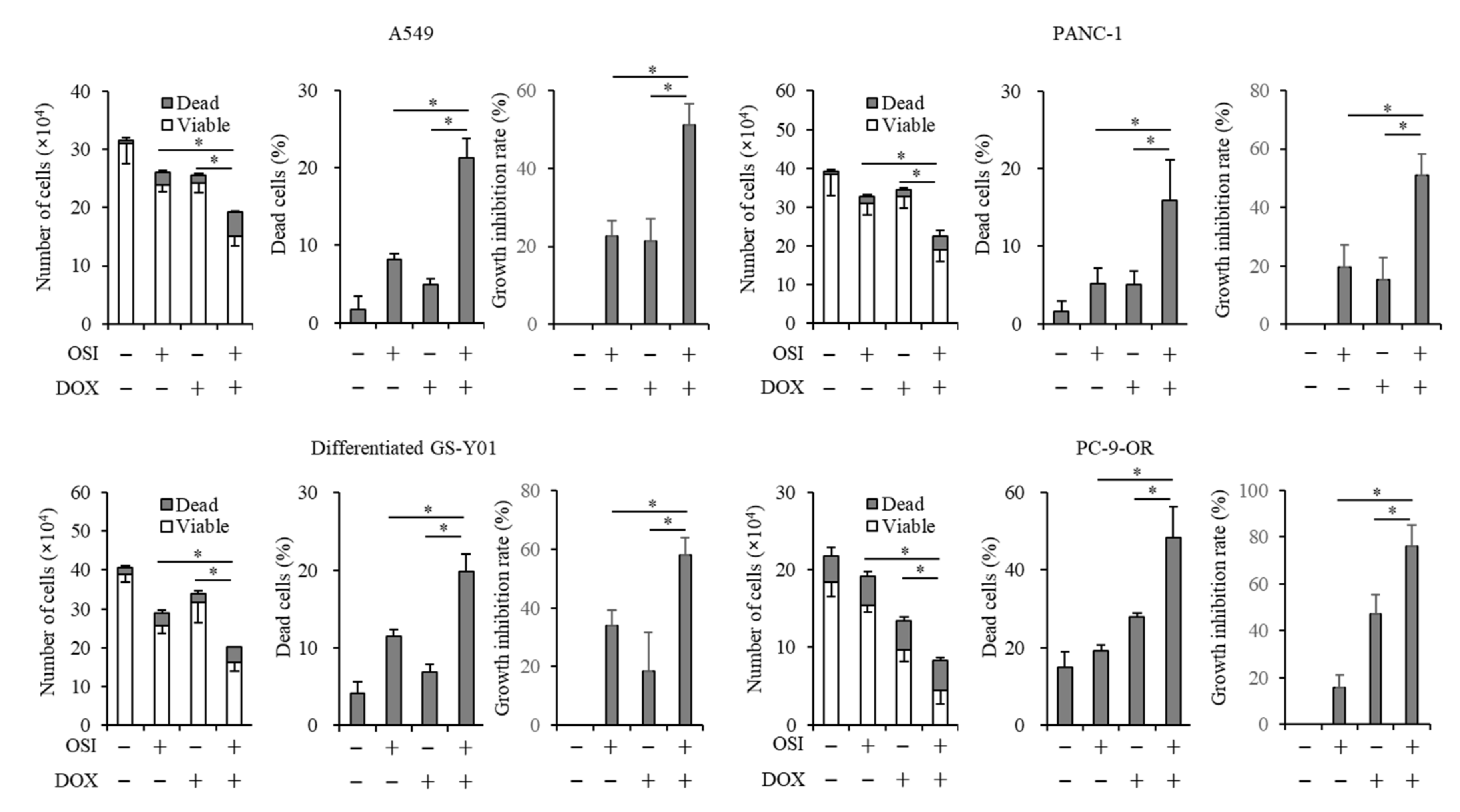
3.2. Doxazosin Enhances Anticancer Effects of Osimertinib in Cancer Cells and CSCs
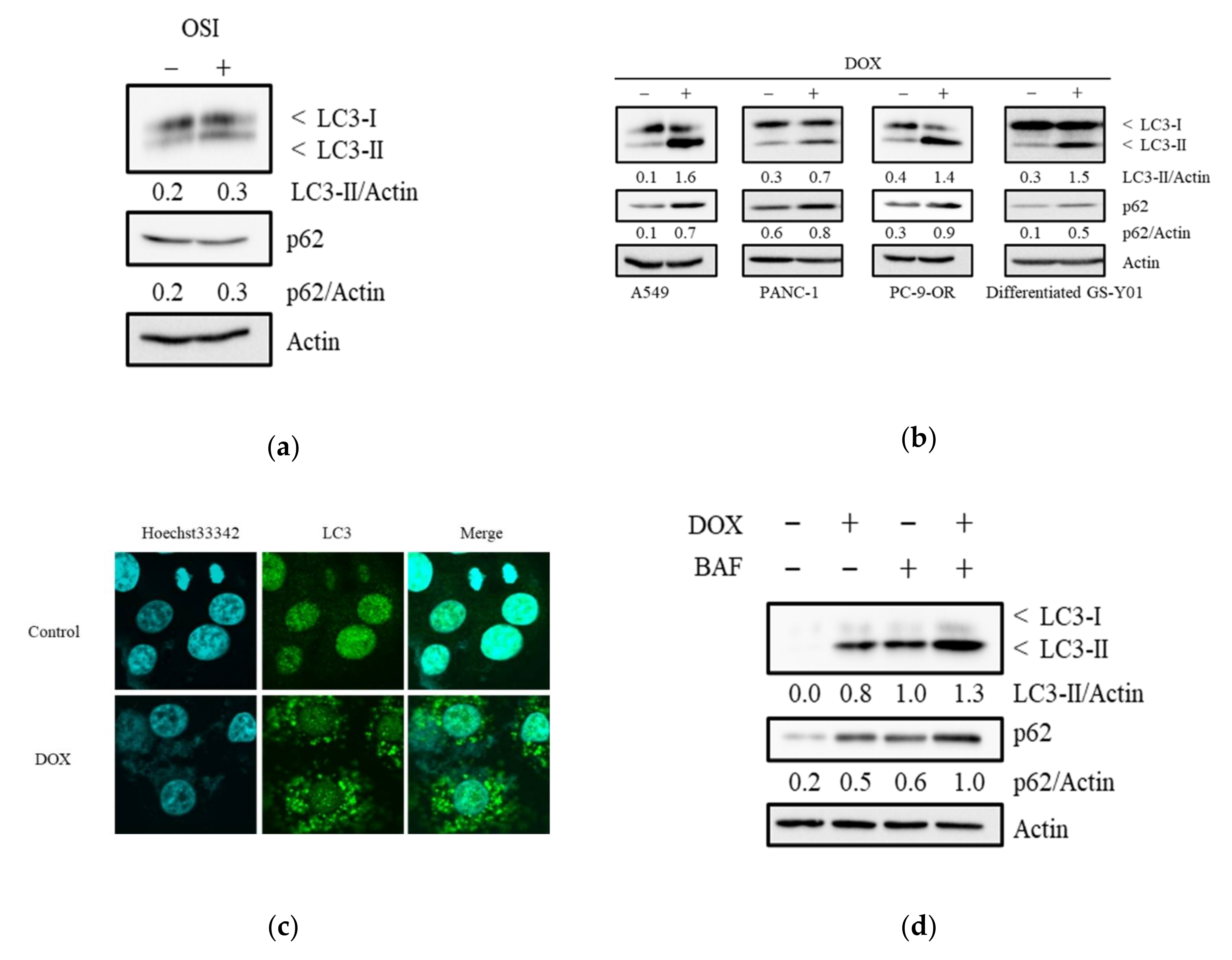
3.3. Doxazosin Induces the Activation of Autophagy
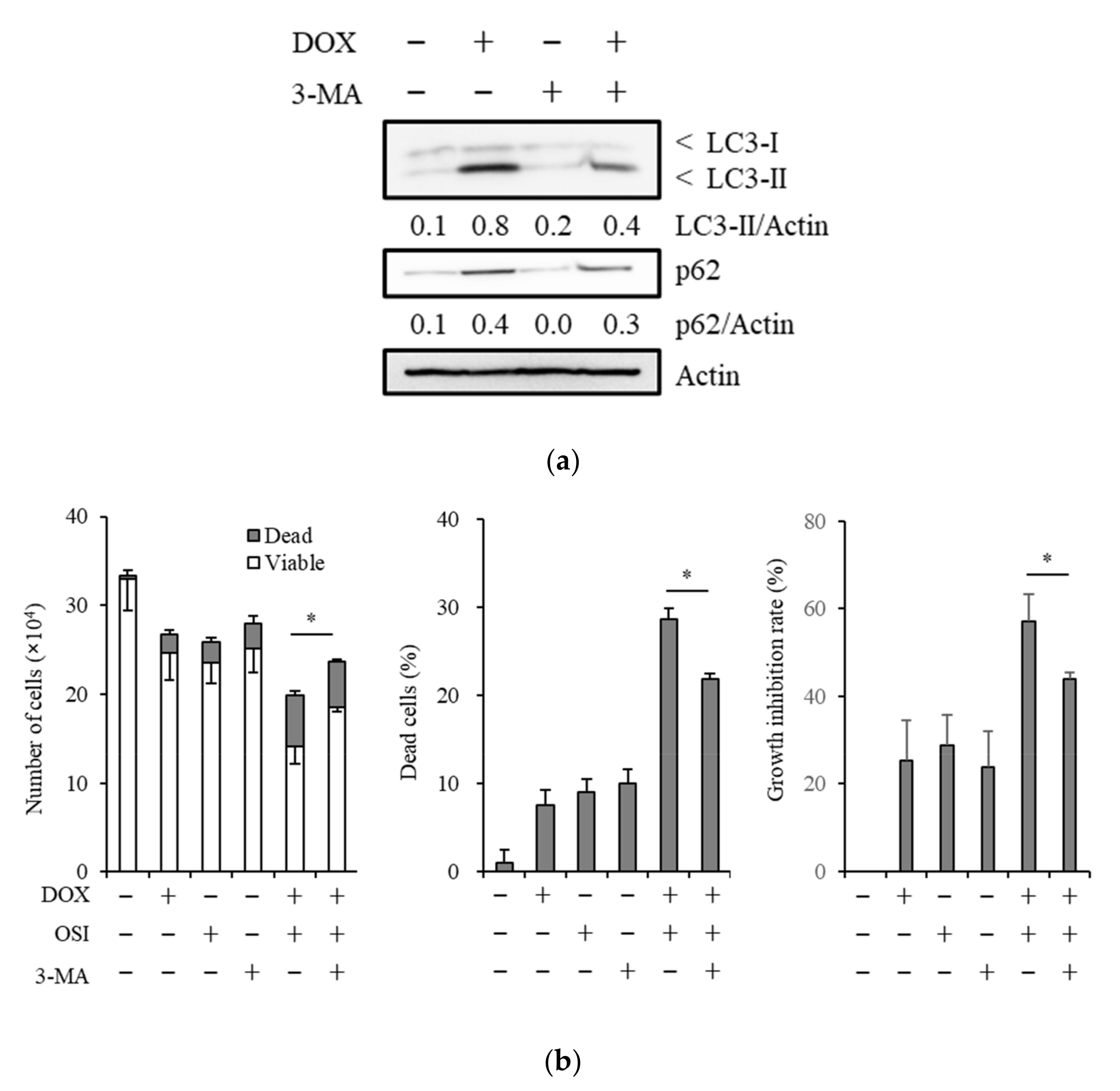
3.4. The Autophagy Inhibitor, 3-Methyladenine, Suppresses Sensitizing Effects of Doxazosin to Osimertinib
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in UntreatedEGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2016, 376, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minari, R.; Bordi, P.; Tiseo, M. Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: Review on emerged mechanisms of resistance. Transl. Lung Cancer Res. 2016, 5, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.-H.; Lu, J.-J. Osimertinib resistance in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Lett. 2018, 420, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Puri, S.; Negrão, M.V.; Nilsson, M.B.; Robichaux, J.P.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.B.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurie, S.A.; Goss, G.D. Role of Epidermal Growth Factor Receptor Inhibitors in Epidermal Growth Factor Receptor Wild-Type Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2013, 31, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.L.; Burdett, S.; Fisher, D.J.; Navani, N.; Parmar, M.K.; Copas, A.; Tierney, J.F. Should Tyrosine Kinase Inhibitors Be Considered for Advanced Non-Small-Cell Lung Cancer Patients With Wild Type EGFR? Two Systematic Reviews and Meta-Analyses of Randomized Trials. Clin. Lung Cancer 2014, 16, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Pan, G.-Z.; Hou, X.; Liu, T.-H.; Chen, J.; Yanaihara, C.; Yanaihara, N. Epidermal Growth Factor and Its Receptors in Human Pancreatic Carcinoma. Pancreas 1990, 5, 278–283. [Google Scholar] [CrossRef]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.M.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985, 313, 144–147. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Brown, P.D.; Krishnan, S.; Sarkaria, J.N.; Wu, W.; Jaeckle, K.A.; Uhm, J.H.; Geoffroy, F.J.; Arusell, R.; Kitange, G.; Jenkins, R.B.; et al. Phase I/II Trial of Erlotinib and Temozolomide With Radiation Therapy in the Treatment of Newly Diagnosed Glioblastoma Multiforme: North Central Cancer Treatment Group Study N0177. J. Clin. Oncol. 2008, 26, 5603–5609. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M. Brexpiprazole, a Serotonin-Dopamine Activity Modulator, Can Sensitize Glioma Stem Cells to Osimertinib, a Third-Generation EGFR-TKI, via Survivin Reduction. Cancers 2019, 11, 947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Okada, M.; Takeda, H.; Kuramoto, K.; Sanomachi, T.; Togashi, K.; Seino, S.; Yamamoto, M.; Yoshioka, T.; Kitanaka, C. Involvement of GLUT1-mediated glucose transport and metabolism in gefitinib resistance of non-small-cell lung cancer cells. Oncotarget 2018, 9, 32667–32679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhu, G.-Y.; Cao, D.; Pan, H.; Li, Y.-W. Gossypol overcomes EGFR-TKIs resistance in non-small cell lung cancer cells by targeting YAP/TAZ and EGFRL858R/T790M. Biomed. Pharmacother. 2019, 115, 108860. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Jiao, L.; Lin, C.; Lu, C.; Zhang, K.; Hu, C.; Ye, J.; Zhang, D.; Wu, H.; et al. Protective autophagy decreases osimertinib cytotoxicity through regulation of stem cell-like properties in lung cancer. Cancer Lett. 2019, 452, 191–202. [Google Scholar] [CrossRef]
- Sakuma, Y.; Matsukuma, S.; Nakamura, Y.; Yoshihara, M.; Koizume, S.; Sekiguchi, H.; Saito, H.; Nakayama, H.; Kameda, Y.; Yokose, T.; et al. Enhanced autophagy is required for survival in EGFR-independent EGFR-mutant lung adenocarcinoma cells. Lab. Investig. 2013, 93, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Sooro, M.A.; Zhang, N.; Zhang, P. Targeting EGFR-mediated autophagy as a potential strategy for cancer therapy. Int. J. Cancer 2018, 143, 2116–2125. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.; Elliott, H.; Meredith, P.; Reid, J. Doxazosin, an alpha 1-adrenoceptor antagonist: Pharmacokinetics and concentration-effect relationships in man. Br. J. Clin. Pharmacol. 1983, 15, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Kirby, R.; Pool, J. Alpha adrenoceptor blockade in the treatment of benign prostatic hyperplasia: Past, present and future. Br. J. Urol. 1997, 80, 521–532. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Barlogie, B.; Tricot, G.; Anaissie, E.; Shaughnessy, J.; Rasmussen, E.; Van Rhee, F.; Fassas, A.; Zangari, M.; Hollmig, K.; Pineda-Roman, M.; et al. Thalidomide and Hematopoietic-Cell Transplantation for Multiple Myeloma. N. Engl. J. Med. 2006, 354, 1021–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batty, M.; Pugh, R.; Rathinam, I.; Simmonds, J.; Walker, E.; Forbes, A.; Anoopkumar-Dukie, S.; McDermott, C.; Spencer, B.; Christie, D.R.; et al. The Role of α1-Adrenoceptor Antagonists in the Treatment of Prostate and Other Cancers. Int. J. Mol. Sci. 2016, 17, 1339. [Google Scholar] [CrossRef]
- Partin, J.V.; Anglin, I.E.; Kyprianou, N. Quinazoline-based α1-adrenoceptor antagonists induce prostate cancer cell apoptosis via TGF-β signalling and IκBα induction. Br. J. Cancer 2003, 88, 1615–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, H.-X.; Fernando, M.A.; Heaney, A.P. The α1-adrenergic receptor antagonist doxazosin inhibits EGFR and NF-κB signalling to induce breast cancer cell apoptosis. Eur. J. Cancer 2008, 44, 160–166. [Google Scholar] [CrossRef]
- Lin, S.-C.; Chueht, S.-C.; Hsiao, C.-J.; Li, T.-K.; Chen, T.-H.; Liao, C.-H.; Lyu, P.-C.; Guh, J.-H. Prazosin Displays Anticancer Activity against Human Prostate Cancers: Targeting DNA, Cell Cycle. Neoplasia 2007, 9, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Arencibia, J.M.; Del Rio, M.; Bonnin, A.; Lopes, R.; Lemoine, N.R.; López-Barahona, M. Doxazosin induces apoptosis in LNCaP prostate cancer cell line through DNA binding and DNA-dependent protein kinase down-regulation. Int. J. Oncol. 2005, 27, 1617–1623. [Google Scholar] [PubMed]
- Shaw, Y.-J.; Yang, Y.-T.; Garrison, J.B.; Kyprianou, N.; Chen, C.-S. Pharmacological Exploitation of the α1-Adrenoreceptor Antagonist Doxazosin to Develop a Novel Class of Antitumor Agents That Block Intracellular Protein Kinase B/Akt Activation. J. Med. Chem. 2004, 47, 4453–4462. [Google Scholar] [CrossRef] [PubMed]
- Garrison, J.B.; Shaw, Y.-J.; Chen, C.-S.; Kyprianou, N. Novel quinazoline-based compounds impair prostate tumorigenesis by targeting tumor vascularity. Cancer Res. 2007, 67, 11344–11352. [Google Scholar] [CrossRef] [Green Version]
- Forbes, A.; Anoopkumar-Dukie, S.; Chess-Williams, R.; McDermott, C. Relative cytotoxic potencies and cell death mechanisms of α1-adrenoceptor antagonists in prostate cancer cell lines. Prostate 2016, 76, 757–766. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Wu, C.-C.; Chen, W.P.; Chen, Y.-L.; Su, M.-J. Prazosin induces p53-mediated autophagic cell death in H9C2 cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 384, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Sanomachi, T.; Suzuki, S.; Togashi, K.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M.; Yamamoto, M. Brexpiprazole Reduces Survivin and Reverses EGFR Tyrosine Kinase Inhibitor Resistance in Lung and Pancreatic Cancer. Anticancer Res. 2019, 39, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Shibuya, K.; Sato, A.; Seino, S.; Watanabe, E.; Suzuki, S.; Seino, M.; Kitanaka, C. Specific role of JNK in the maintenance of the tumor-initiating capacity of A549 human non-small cell lung cancer cells. Oncol. Rep. 2013, 30, 1957–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Okada, M.; Shibuya, K.; Seino, M.; Sato, A.; Takeda, H.; Seino, S.; Yoshioka, T.; Kitanaka, C. JNK suppression of chemotherapeutic agents-induced ROS confers chemoresistance on pancreatic cancer stem cells. Oncotarget 2014, 6, 458–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, K.-I.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanomachi, T.; Suzuki, S.; Kuramoto, K.; Takeda, H.; Sakaki, H.; Togashi, K.; Seino, S.; Yoshioka, T.; Okada, M.; Kitanaka, C. Olanzapine, an Atypical Antipsychotic, Inhibits Survivin Expression and Sensitizes Cancer Cells to Chemotherapeutic Agents. Anticancer Res. 2017, 37, 6177–6188. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Yamamoto, M.; Togashi, K.; Sanomachi, T.; Sugai, A.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M. In vitro and in vivo anti-tumor effects of brexpiprazole, a newly-developed serotonin-dopamine activity modulator with an improved safety profile. Oncotarget 2019, 10, 3547–3558. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Okada, M.; Kuramoto, K.; Takeda, H.; Sakaki, H.; Watarai, H.; Sanomachi, T.; Seino, S.; Yoshioka, T.; Kitanaka, C. Aripiprazole, an Antipsychotic and Partial Dopamine Agonist, Inhibits Cancer Stem Cells and Reverses Chemoresistance. Anticancer Res. 2016, 36, 5153–5162. [Google Scholar] [CrossRef] [Green Version]
- Seino, M.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, S.; Ohta, T.; Kurachi, H.; Kitanaka, C. Requirement of JNK signaling for self-renewal and tumor-initiating capacity of ovarian cancer stem cells. Anticancer Res. 2014, 34, 4723–4731. [Google Scholar]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, M.; Liu, H.; Yin, W. AZD9291 promotes autophagy and inhibits PI3K/Akt pathway in NSCLC cancer cells. J. Cell. Biochem. 2018, 120, 756–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.-H.; Cao, W.-X.; Su, M.-X.; Chen, X.; Lu, J.-J. Osimertinib induces autophagy and apoptosis via reactive oxygen species generation in non-small cell lung cancer cells. Toxicol. Appl. Pharmacol. 2017, 321, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef]
- Kyprianou, N.; Benning, C.M. Suppression of human prostate cancer cell growth by alpha1-adrenoceptor antagonists doxazosin and terazosin via induction of apoptosis. Cancer Res. 2000, 60, 4550–4555. [Google Scholar]
- Benning, C.M.; Kyprianou, N. Quinazoline-derived alpha1-adrenoceptor antagonists induce prostate cancer cell apoptosis via an alpha1-adrenoceptor-independent action. Cancer Res. 2002, 62, 597–602. [Google Scholar] [PubMed]
- Galluzzi, L.; Pedro, J.M.B.-S.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.-K.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12–16. [Google Scholar] [CrossRef]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Xia, B.; White, E. Autophagy-Mediated Tumor Promotion. Cell 2013, 155, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Ling, Y.-H.; Sironi, J.; Schwartz, E.L.; Perez-Soler, R.; Piperdi, B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J. Thorac. Oncol. 2013, 8, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.-G.; Jeong, E.-H.; Kim, S.Y.; Kim, H.-R.; Kim, C.H. The combination of irreversible EGFR TKIs and SAHA induces apoptosis and autophagy-mediated cell death to overcome acquired resistance in EGFRT790M-mutated lung cancer. Int. J. Cancer 2014, 136, 2717–2729. [Google Scholar] [CrossRef]
- Chen, P.; Huang, H.-P.; Wang, Y.; Jin, J.; Long, W.-G.; Chen, K.; Zhao, X.-H.; Chen, C.-G.; Li, J. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J. Exp. Clin. Cancer Res. 2019, 38, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ancker, O.V.; Wehland, M.; Bauer, J.; Infanger, M.; Grimm, D. The Adverse Effect of Hypertension in the Treatment of Thyroid Cancer with Multi-Kinase Inhibitors. Int. J. Mol. Sci. 2017, 18, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahmatzopoulos, A.; Kyprianou, N. Apoptotic impact of alpha1-blockers on prostate cancer growth: A myth or an inviting reality? Prostate 2004, 59, 91–100. [Google Scholar] [CrossRef]
- Park, M.S.; Kim, B.-R.; Kang, S.; Kim, D.-Y.; Rho, S.B. The antihypertension drug doxazosin suppresses JAK/STATs phosphorylation and enhances the effects of IFN-α/γ-induced apoptosis. Genes Cancer 2014, 5, 470. [Google Scholar] [CrossRef] [Green Version]
- Fernando, M.A.; Heaney, A.P. α1-Adrenergic Receptor Antagonists: Novel Therapy for Pituitary Adenomas. Mol. Endocrinol. 2005, 19, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Harris, A.M.; Warner, B.W.; Wilson, J.M.; Becker, A.; Rowland, R.G.; Conner, W.; Lane, M.; Kimbler, K.; Durbin, E.B.; Baron, A.; et al. Effect of α1-Adrenoceptor Antagonist Exposure on Prostate Cancer Incidence: An Observational Cohort Study. J. Urol. 2007, 178, 2176–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.M.; Harris, A.M.; Rowland, R.G.; Conner, W.; Lane, M.; Durbin, E.; Baron, A.T.; Kyprianou, N. Decreased risk of bladder cancer in men treated with quinazoline-based α1-adrenoceptor antagonists. Gene Ther. Mol. Biol. 2008, 12, 253–258. [Google Scholar] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Okada, M.; Yoshioka, T.; Kitanaka, C. Doxazosin, a Classic Alpha 1-Adrenoceptor Antagonist, Overcomes Osimertinib Resistance in Cancer Cells via the Upregulation of Autophagy as Drug Repurposing. Biomedicines 2020, 8, 273. https://doi.org/10.3390/biomedicines8080273
Suzuki S, Yamamoto M, Sanomachi T, Togashi K, Sugai A, Seino S, Okada M, Yoshioka T, Kitanaka C. Doxazosin, a Classic Alpha 1-Adrenoceptor Antagonist, Overcomes Osimertinib Resistance in Cancer Cells via the Upregulation of Autophagy as Drug Repurposing. Biomedicines. 2020; 8(8):273. https://doi.org/10.3390/biomedicines8080273
Chicago/Turabian StyleSuzuki, Shuhei, Masahiro Yamamoto, Tomomi Sanomachi, Keita Togashi, Asuka Sugai, Shizuka Seino, Masashi Okada, Takashi Yoshioka, and Chifumi Kitanaka. 2020. "Doxazosin, a Classic Alpha 1-Adrenoceptor Antagonist, Overcomes Osimertinib Resistance in Cancer Cells via the Upregulation of Autophagy as Drug Repurposing" Biomedicines 8, no. 8: 273. https://doi.org/10.3390/biomedicines8080273