Flavolignans from Silymarin as Nrf2 Bioactivators and Their Therapeutic Applications

 ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
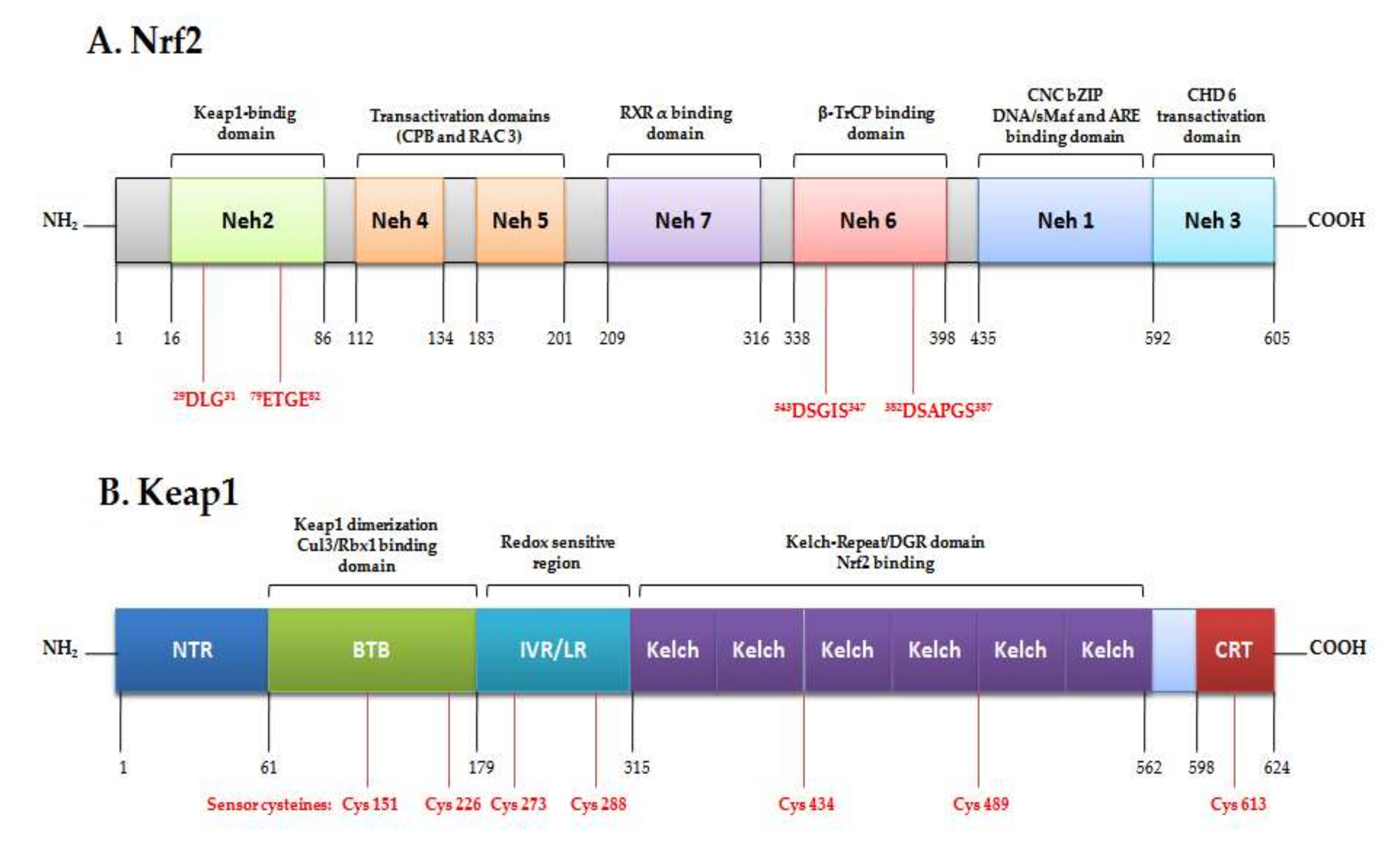
2. The Signaling Pathway Nrf2/Keap1/ARE as Cytoprotector System
Transcription, Regulation and the Antioxidant Response
3. Nrf2 as a Target for Therapeutic Models
3.1. Nrf2 in Aging and Muscular Dysfunction
3.2. Nrf2 and Inflammation
3.3. Nrf2 and Chronic Diseases
3.3.1. Nrf2 in Cancer
3.3.2. Nrf2 in Diabetes
3.3.3. Nrf2 in Neurodegenerative Diseases
3.3.4. Nrf2 in Liver Injury
4. Nfr2 Bioactivators
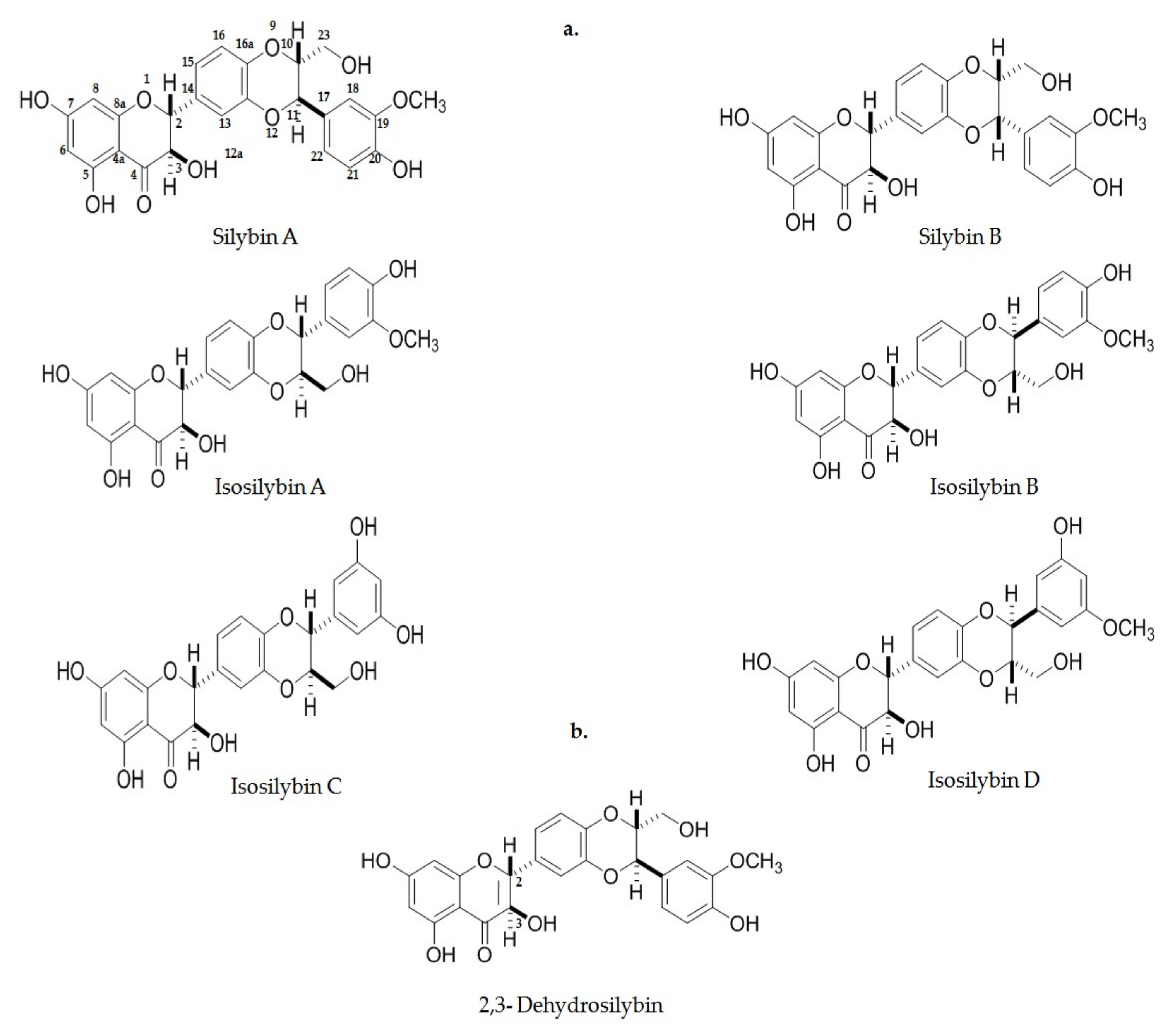
5. Silymarin and Flavolignans as Potent Nrf2 Inducers
5.1. Silymarin and Flavolignans Description
5.2. Silymarin and Flavolignans Pharmacokinetics
Bioavailability and Metabolism
5.3. Nrf2 Activated by Silymarin and Flavolignans: Promising Therapeutic Model
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. CB 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [Green Version]
- Truong, T.H.; Carroll, K.S. Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry 2012, 51, 9954–9965. [Google Scholar] [CrossRef] [Green Version]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. [Google Scholar] [CrossRef]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Surh, Y.J.; Kundu, J.K.; Na, H.K. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008, 74, 1526–1539. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Mendoza, N.; Morales-Gonzalez, A.; Madrigal-Santillan, E.O.; Madrigal-Bujaidar, E.; Alvarez-Gonzalez, I.; Garcia-Melo, L.F.; Anguiano-Robledo, L.; Fregoso-Aguilar, T.; Morales-Gonzalez, J.A. Antioxidant and adaptative response mediated by Nrf2 during physical exercise. Antioxidants 2019. [Google Scholar] [CrossRef] [Green Version]
- Bruns, D.R.; Drake, J.C.; Biela, L.M.; Peelor, F.F., 3rd; Miller, B.F.; Hamilton, K.L. Nrf2 signaling and the slowed aging phenotype: Evidence from long-lived models. Oxidative Med. Cell. Longev. 2015, 2015, 732596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surai, P.F. Silymarin as a natural antioxidant: An overview of the current evidence and perspectives. Antioxidants 2015, 4, 204–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Venugopal, R.; Jaiswal, A.K. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 1998, 17, 3145–3156. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [Green Version]
- Mohler, J.; Vani, K.; Leung, S.; Epstein, A. Segmentally restricted, cephalic expression of a leucine zipper gene during Drosophila embryogenesis. Mech. Dev. 1991, 34, 3–9. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Namani, A.; Li, Y.; Wang, X.J.; Tang, X. Modulation of NRF2 signaling pathway by nuclear receptors: Implications for cancer. Biochim. Biophys. Acta 2014, 1843, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Xiang, M.; Namani, A.; Wu, S.; Wang, X. Nrf2: Bane or blessing in cancer? J. Cancer Res. Clin. Oncol. 2014, 140, 1251–1259. [Google Scholar] [CrossRef]
- Bai, X.; Chen, Y.; Hou, X.; Huang, M.; Jin, J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab. Rev. 2016, 48, 541–567. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Eggler, A.L.; Small, E.; Hannink, M.; Mesecar, A.D. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem. J. 2009, 422, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Eggler, A.L.; Savinov, S.N. Chemical and biological mechanisms of phytochemical activation of Nrf2 and importance in disease prevention. Recent Adv. Phytochem. 2013, 43, 121–155. [Google Scholar]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar]
- Rushmore, T.H.; Pickett, C.B. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J. Biol. Chem. 1990, 265, 1464–14653. [Google Scholar]
- Sun, Z.; Zhang, S.; Chan, J.Y.; Zhang, D.D. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell. Biol. 2007, 27, 6334–6349. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Bjorkoy, G.; Lamark, T.; Johansen, T. p62/SQSTM1: A missing link between protein aggregates and the autophagy machinery. Autophagy 2006, 2, 138–139. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzym. Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Vomund, S.; Schafer, A.; Parnham, M.J.; Brune, B.; von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017. [Google Scholar] [CrossRef] [Green Version]
- Shen, G.; Kong, A.N. Nrf2 plays an important role in coordinated regulation of phase II drug metabolism enzymes and Phase III drug transporters. Biopharm. Drug Dispos. 2009, 30, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, H.J.; Karlenius, T.C.; Tonissen, K.F. Regulation of the human thioredoxin gene promoter and its key substrates: A study of functional and putative regulatory elements. Biochim. Biophys. Acta 2014, 1840, 303–314. [Google Scholar] [CrossRef]
- Shultz, C.A.; Quinn, A.M.; Park, J.H.; Harvey, R.G.; Bolton, J.L.; Maser, E.; Penning, T.M. Specificity of human aldo-keto reductases, NAD(P)H: Quinone oxidoreductase, and carbonyl reductases to redox-cycle polycyclic aromatic hydrocarbon diones and 4-hydroxyequilenin-o-quinone. Chem. Res. Toxicol. 2011, 24, 2153–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, L.G.; Kelleher, M.O.; Eggleston, I.M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol. Appl. Pharmacol. 2009, 237, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.M.; Kleeberger, S.R.; Cho, H.Y.; Yamamoto, M.; Kensler, T.W.; Biswal, S.; Reddy, S.P. Deficiency in Nrf2-GSH signaling impairs type II cell growth and enhances sensitivity to oxidants. Am. J. Respir. Cell Mol. Biol. 2007, 37, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Kan, H.; Cai, L.; Ma, Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J. Mol. Cell. Cardiol. 2009, 46, 47–58. [Google Scholar] [CrossRef]
- Patil, N.K.; Saba, H.; MacMillan-Crow, L.A. Effect of S-nitrosoglutathione on renal mitochondrial function: A new mechanism for reversible regulation of manganese superoxide dismutase activity? Free Radic. Biol. Med. 2013, 56, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.M.; Kleeberger, S.R.; Kensler, T.W.; Yamamoto, M.; Hassoun, P.M.; Reddy, S.P. Disruption of Nrf2 impairs the resolution of hyperoxia-induced acute lung injury and inflammation in mice. J. Immunol. 2009, 182, 7264–7271. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.J.; Dudakov, J.A.; Takahashi, K.; Shieh, J.H.; Velardi, E.; Holland, A.M.; Singer, N.V.; West, M.L.; Smith, O.M.; Young, L.F.; et al. Nrf2 regulates haematopoietic stem cell function. Nat. Cell Biol. 2013, 15, 309–316. [Google Scholar] [CrossRef]
- Gomes, M.J.; Martinez, P.F.; Pagan, L.U.; Damatto, R.L.; Cezar, M.D.M.; Lima, A.R.R.; Okoshi, K.; Okoshi, M.P. Skeletal muscle aging: Influence of oxidative stress and physical exercise. Oncotarget 2017, 8, 20428–20440. [Google Scholar] [CrossRef] [Green Version]
- St-Onge, M.P.; Gallagher, D. Body composition changes with aging: The cause or the result of alterations in metabolic rate and macronutrient oxidation? Nutrition 2010, 26, 152–155. [Google Scholar] [CrossRef] [Green Version]
- Safdar, A.; de Beer, J.; Tarnopolsky, M.A. Dysfunctional Nrf2-Keap1 redox signaling in skeletal muscle of the sedentary old. Free Radic. Biol. Med. 2010, 49, 1487–1493. [Google Scholar] [CrossRef]
- Crane, J.D.; Devries, M.C.; Safdar, A.; Hamadeh, M.J.; Tarnopolsky, M.A. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2010, 65, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Tweedie, C.; Romestaing, C.; Burelle, Y.; Safdar, A.; Tarnopolsky, M.A.; Seadon, S.; Britton, S.L.; Koch, L.G.; Hepple, R.T. Lower oxidative DNA damage despite greater ROS production in muscles from rats selectively bred for high running capacity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R544–R553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coen, P.M.; Jubrias, S.A.; Distefano, G.; Amati, F.; Mackey, D.C.; Glynn, N.W.; Manini, T.M.; Wohlgemuth, S.E.; Leeuwenburgh, C.; Cummings, S.R.; et al. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2013, 68, 447–455. [Google Scholar] [CrossRef]
- Calvani, R.; Joseph, A.M.; Adhihetty, P.J.; Miccheli, A.; Bossola, M.; Leeuwenburgh, C.; Bernabei, R.; Marzetti, E. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol. Chem. 2013, 394, 393–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.T.; Epstein, C.J.; Roberts, L.J., II; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Muller, F.L.; Liu, Y.; Ng, R.; Faulkner, J.; Hamilton, M.; Richardson, A.; Huang, T.T.; Epstein, C.J.; Van Remmen, H. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech. Ageing Dev. 2006, 127, 298–306. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Jackson, M.J.; Vasilaki, A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 2014, 48, 12–29. [Google Scholar] [CrossRef]
- Kyle, U.G.; Zhang, F.F.; Morabia, A.; Pichard, C. Longitudinal study of body composition changes associated with weight change and physical activity. Nutrition 2006, 22, 1103–1111. [Google Scholar] [CrossRef]
- Ahn, B.; Ranjit, R.; Premkumar, P.; Pharaoh, G.; Piekarz, K.M.; Matsuzaki, S.; Claflin, D.R.; Riddle, K.; Judge, J.; Bhaskaran, S.; et al. Mitochondrial oxidative stress impairs contractile function but paradoxically increases muscle mass via fibre branching. J. Cachexia Sarcopenia Muscle 2019, 10, 411–428. [Google Scholar] [CrossRef] [Green Version]
- Kitaoka, Y.; Tamura, Y.; Takahashi, K.; Takeda, K.; Takemasa, T.; Hatta, H. Effects of Nrf2 deficiency on mitochondrial oxidative stress in aged skeletal muscle. Physiol. Rep. 2019, 7, e13998. [Google Scholar] [CrossRef]
- Joseph, A.M.; Adhihetty, P.J.; Leeuwenburgh, C. Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J. Physiol. 2016, 594, 5105–5123. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.; Pharaoh, G.; Premkumar, P.; Huseman, K.; Ranjit, R.; Kinter, M.; Szweda, L.; Kiss, T.; Fulop, G.; Tarantini, S.; et al. Nrf2 deficiency exacerbates age-related contractile dysfunction and loss of skeletal muscle mass. Redox Biol. 2018, 17, 47–58. [Google Scholar] [CrossRef]
- Miller, C.J.; Gounder, S.S.; Kannan, S.; Goutam, K.; Muthusamy, V.R.; Firpo, M.A.; Symons, J.D.; Paine, R., III; Hoidal, J.R.; Rajasekaran, N.S. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochim. Biophys. Acta 2012, 1822, 1038–1050. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Gladwell, W.; Wang, X.; Chorley, B.; Bell, D.; Reddy, S.P.; Kleeberger, S.R. Nrf2-regulated PPAR{γ} expression is critical to protection against acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2010, 182, 170–182. [Google Scholar] [CrossRef]
- Storz, P.; Toker, A. NF-κB signaling—An alternate pathway for oxidative stress responses. Cell Cycle 2003, 2, 9–10. [Google Scholar] [CrossRef]
- Storz, P.; Doppler, H.; Toker, A. Protein kinase Cδ selectively regulates protein kinase D-dependent activation of NF-κB in oxidative stress signaling. Mol. Cell. Biol. 2004, 24, 2614–2626. [Google Scholar] [CrossRef] [Green Version]
- Storz, P.; Toker, A. Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J. 2003, 22, 109–120. [Google Scholar] [CrossRef]
- Lee, I.T.; Luo, S.F.; Lee, C.W.; Wang, S.W.; Lin, C.C.; Chang, C.C.; Chen, Y.L.; Chau, L.Y.; Yang, C.M. Overexpression of HO-1 protects against TNF-alpha-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am. J. Pathol. 2009, 175, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide-induced expression of NAD(P)H: Quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008, 181, 6730–6737. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Nadeem, A.; Siddiqui, N.; Al-Harbi, N.O.; Al-Harbi, M.M.; Ahmad, S.F. TLR-7 agonist attenuates airway reactivity and inflammation through Nrf2-mediated antioxidant protection in a murine model of allergic asthma. Int. J. Biochem. Cell Biol. 2016, 73, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Baumgart-Vogt, E.; Naidu, S.; Qian, G.; Immenschuh, S. Bruton’s tyrosine kinase is required for TLR-dependent heme oxygenase-1 gene activation via Nrf2 in macrophages. J. Immunol. 2011, 187, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [Green Version]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Potential applications of NRF2 inhibitors in cancer therapy. Oxidative Med. Cell. Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, D.A.; McCabe, M.T.; Arnold, R.S.; Day, M.L. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 2008, 27, 4353–4362. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Reprint of: Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2014, 66, 45–57. [Google Scholar] [CrossRef]
- Reddy, N.M.; Kleeberger, S.R.; Bream, J.H.; Fallon, P.G.; Kensler, T.W.; Yamamoto, M.; Reddy, S.P. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene 2008, 27, 5821–5832. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsby, R.; Kitteringham, N.R.; Goldring, C.E.; Lovatt, C.A.; Chamberlain, M.; Henderson, C.J.; Wolf, C.R.; Park, B.K. Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J. Biol. Chem. 2003, 278, 22243–22249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanpain, C.; Mohrin, M.; Sotiropoulou, P.A.; Passegue, E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 2011, 8, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, M.; Halabian, R.; Gharehbaghian, A.; Amirizadeh, N.; Jahanian-Najafabadi, A.; Roushandeh, A.M.; Roudkenar, M.H. Nrf-2 overexpression in mesenchymal stem cells reduces oxidative stress-induced apoptosis and cytotoxicity. Cell Stress Chaperones 2012, 17, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Suzuki, T.; Mitsuishi, Y.; Miki, Y.; Suzuki, S.; Sugawara, S.; Watanabe, M.; Sakurada, A.; Endo, C.; Uruno, A.; et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012, 103, 760–766. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Wei, Y.; Gong, J.; Cho, H.; Park, J.K.; Sung, E.R.; Huang, H.; Wu, L.; Eberhart, C.; Handa, J.T.; et al. NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia 2014, 57, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. Novel directions for diabetes mellitus drug discovery. Expert Opin. Drug Discov. 2013, 8, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.J.; Yang, Y.M.; Kim, J.W.; Kim, S.G. Identification of a novel class of dithiolethiones that prevent hepatic insulin resistance via the adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway. Hepatology 2007, 46, 730–739. [Google Scholar] [CrossRef]
- Uruno, A.; Yagishita, Y.; Yamamoto, M. The Keap1-Nrf2 system and diabetes mellitus. Arch. Biochem. Biophys. 2015, 566, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wu, W.; Song, H.; Wang, F.; Li, H.; Chen, L.; Lai, Y.; Janicki, J.S.; Ward, K.W.; Meyer, C.J.; et al. Targeting Nrf2 by dihydro-CDDO-trifluoroethyl amide enhances autophagic clearance and viability of beta-cells in a setting of oxidative stress. FEBS Lett. 2014, 588, 2115–2124. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Hur, E.G.; Ryoo, I.G.; Jung, K.A.; Kwak, J.; Kwak, M.K. Involvement of the Nrf2-proteasome pathway in the endoplasmic reticulum stress response in pancreatic beta-cells. Toxicol. Appl. Pharmacol. 2012, 264, 431–438. [Google Scholar] [CrossRef]
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-mediated regulation of skeletal muscle glycogen Metabolism. Mol. Cell. Biol. 2016, 36, 1655–1672. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [Green Version]
- Castro-Sanchez, S.; Garcia-Yague, A.J.; Lopez-Royo, T.; Casarejos, M.; Lanciego, J.L.; Lastres-Becker, I. Cx3cr1-deficiency exacerbates alpha-synuclein-A53T induced neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Glia 2018, 66, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Xu, M.; Wang, Y.; Xie, F.; Zhang, G.; Qin, X. Nrf2-a promising therapeutic target for defensing against oxidative stress in stroke. Mol. Neurobiol. 2017, 54, 6006–6017. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Gold, R.; Linker, R.A. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: Therapeutic modulation via fumaric acid esters. Int. J. Mol. Sci. 2012, 13, 11783–11803. [Google Scholar] [CrossRef] [Green Version]
- Post-White, J.; Ladas, E.J.; Kelly, K.M. Advances in the use of milk thistle (Silybum marianum). Integr. Cancer Ther. 2007, 6, 104–109. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Shin, S.; Slocum, S.L.; Agoston, E.S.; Wakabayashi, J.; Kwak, M.K.; Misra, V.; Biswal, S.; Yamamoto, M.; Kensler, T.W. Regulation of notch1 signaling by nrf2: Implications for tissue regeneration. Sci. Signal. 2010. [Google Scholar] [CrossRef] [Green Version]
- Zuber, R.; Modriansky, M.; Dvorak, Z.; Rohovsky, P.; Ulrichova, J.; Simanek, V.; Anzenbacher, P. Effect of silybin and its congeners on human liver microsomal cytochrome P450 activities. Phytother. Res. PTR 2002, 16, 632–638. [Google Scholar] [CrossRef]
- Zuber, R.; Anzenbacherova, E.; Anzenbacher, P. Cytochromes P450 and experimental models of drug metabolism. J. Cell. Mol. Med. 2002, 6, 189–198. [Google Scholar] [CrossRef]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of NRF2: A review of their potential for clinical development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of three major cysteine sensors of Keap1 in stress response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Lleres, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Dinkova-Kostova, A.T. Diffusion dynamics of the Keap1-cullin3 interaction in single live cells. Biochem. Biophys. Res. Commun. 2013, 433, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Palliyaguru, D.L.; Kensler, T.W. Frugal chemoprevention: Targeting Nrf2 with foods rich in sulforaphane. Semin. Oncol. 2016, 43, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R.; Gupta, S.; Maru, G.B. Dietary curcumin modulates transcriptional regulators of phase I and phase II enzymes in benzo[a]pyrene-treated mice: Mechanism of its anti-initiating action. Carcinogenesis 2008, 29, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Hassan, F.U.; Rehman, M.S.; Khan, M.S.; Ali, M.A.; Javed, A.; Nawaz, A.; Yang, C. Curcumin as an alternative epigenetic modulator: Mechanism of action and potential effects. Front. Genet. 2019, 10, 514. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Jang, J.H.; Li, M.H.; Surh, Y.J. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 331, 993–1000. [Google Scholar] [CrossRef]
- Su, X.; Jiang, X.; Meng, L.; Dong, X.; Shen, Y.; Xin, Y. Anticancer activity of sulforaphane: The epigenetic mechanisms and the Nrf2 signaling pathway. Oxidative Med. Cell. Longev. 2018, 2018, 5438179. [Google Scholar] [CrossRef]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane: Translational research from laboratory bench to clinic. Nutr. Rev. 2013, 71, 709–726. [Google Scholar] [CrossRef]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and other nutrigenomic Nrf2 activators: Can the clinician’s expectation be matched by the reality? Oxidative Med. Cell. Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef] [Green Version]
- Botti, M.G.; Taylor, M.G.; Botting, N.P. Studies on the mechanism of myrosinase. Investigation of the effect of glycosyl acceptors on enzyme activity. J. Biol. Chem. 1995, 270, 20530–20535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nicola, G.R.; Bagatta, M.; Pagnotta, E.; Angelino, D.; Gennari, L.; Ninfali, P.; Rollin, P.; Iori, R. Comparison of bioactive phytochemical content and release of isothiocyanates in selected brassica sprouts. Food Chem. 2013, 141, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Klomparens, E.A.; Ding, Y. The neuroprotective mechanisms and effects of sulforaphane. Brain Circ. 2019, 5, 74–83. [Google Scholar] [PubMed]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci. CMLS 2008, 65, 1631–1652. [Google Scholar] [CrossRef]
- Bijak, M. Silybin, a major bioactive component of milk thistle (Silybum marianum L. Gaernt.)—Chemistry, bioavailability, and metabolism. Molecules 2017. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Mendoza, N.; Madrigal-Santillan, E.; Morales-Gonzalez, A.; Esquivel-Soto, J.; Esquivel-Chirino, C.; Garcia-Luna, Y.G.-R.M.; Gayosso-de-Lucio, J.A.; Morales-Gonzalez, J.A. Hepatoprotective effect of silymarin. World J. Hepatol. 2014, 6, 144–149. [Google Scholar] [CrossRef]
- Madrigal-Santillan, E.; Madrigal-Bujaidar, E.; Alvarez-Gonzalez, I.; Sumaya-Martinez, M.T.; Gutierrez-Salinas, J.; Bautista, M.; Morales-Gonzalez, A.; Garcia-Luna y Gonzalez-Rubio, M.; Aguilar-Faisal, J.L.; Morales-Gonzalez, J.A. Review of natural products with hepatoprotective effects. World J. Gastroenterol. 2014, 20, 14787–14804. [Google Scholar] [CrossRef]
- Trappoliere, M.; Caligiuri, A.; Schmid, M.; Bertolani, C.; Failli, P.; Vizzutti, F.; Novo, E.; di Manzano, C.; Marra, F.; Loguercio, C.; et al. Silybin, a component of sylimarin, exerts anti-inflammatory and anti-fibrogenic effects on human hepatic stellate cells. J. Hepatol. 2009, 50, 1102–1111. [Google Scholar] [CrossRef]
- Li, L.; Zeng, J.; Gao, Y.; He, D. Targeting silibinin in the antiproliferative pathway. Expert Opin. Investig. Drugs 2010, 19, 243–255. [Google Scholar] [CrossRef]
- Biedermann, D.; Vavrikova, E.; Cvak, L.; Kren, V. Chemistry of silybin. Nat. Prod. Rep. 2014, 31, 1138–1157. [Google Scholar] [CrossRef]
- Javed, S.; Kohli, K.; Ali, M. Reassessing bioavailability of silymarin. Altern. Med. Rev. A J. Clin. Ther. 2011, 16, 239–249. [Google Scholar]
- Wu, J.W.; Lin, L.C.; Hung, S.C.; Chi, C.W.; Tsai, T.H. Analysis of silibinin in rat plasma and bile for hepatobiliary excretion and oral bioavailability application. J. Pharm. Biomed. Anal. 2007, 45, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.N.; Zhu, Y.; Wang, L.; Peng, M.; Tong, S.S.; Cao, X.; Qiu, H.; Xu, X.M. Enhancement of oral bioavailability of the poorly water-soluble drug silybin by sodium cholate/phospholipid-mixed micelles. Acta Pharmacol. Sin. 2010, 31, 759–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Sanchez, A.; Cuyas, E.; Ruiz-Torres, V.; Agullo-Chazarra, L.; Verdura, S.; Gonzalez-Alvarez, I.; Bermejo, M.; Joven, J.; Micol, V.; Bosch-Barrera, J.; et al. Intestinal permeability study of clinically relevant formulations of silibinin in CaCo-2 cell monolayers. Int. J. Mol. Sci. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, D.; Lucker, P.W.; Mennicke, W.H.; Wetzelsberger, N. Pharmacokinetic studies with silymarin in human serum and bile. Methods Find. Exp. Clin. Pharmacol. 1984, 6, 655–661. [Google Scholar]
- Zhu, H.J.; Brinda, B.J.; Chavin, K.D.; Bernstein, H.J.; Patrick, K.S.; Markowitz, J.S. An assessment of pharmacokinetics and antioxidant activity of free silymarin flavonolignans in healthy volunteers: A dose escalation study. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 1679–1685. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Kim, E.J.; Lee, E.D.; Kim, J.H.; Jang, S.W.; Kim, Y.G.; Kwon, J.W.; Kim, W.B.; Lee, M.G. Comparative bioavailability of silibinin in healthy male volunteers. Int. J. Clin. Pharmacol. Ther. 2003, 41, 593–596. [Google Scholar] [CrossRef]
- Parveen, R.; Baboota, S.; Ali, J.; Ahuja, A.; Vasudev, S.S.; Ahmad, S. Oil based nanocarrier for improved oral delivery of silymarin: In vitro and in vivo studies. Int. J. Pharm. 2011, 413, 245–253. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Liu, Z.; Liu, G.; Duan, C.; Jia, L.; Feng, F.; Zhang, X.; Shi, Y.; Zhang, Q. In vitro and in vivo evaluation of silybin nanosuspensions for oral and intravenous delivery. Nanotechnology 2010, 21, 155104. [Google Scholar] [CrossRef]
- Theodosiou, E.; Purchartová, K.; Stamatis, H.; Kolisis, F.; Kren, V. Bioavility of silymarin flavonolignans: Drug formulations and biotransformation. Phytochem. Rev. 2014, 12, 1–18. [Google Scholar] [CrossRef]
- Jancova, P.; Siller, M.; Anzenbacherova, E.; Kren, V.; Anzenbacher, P.; Simanek, V. Evidence for differences in regioselective and stereoselective glucuronidation of silybin diastereomers from milk thistle (Silybum marianum) by human UDP-glucuronosyltransferases. Xenobiotica Fate Foreign Compd. Biol. Syst. 2011, 41, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Miranda, S.R.; Lee, J.K.; Brouwer, K.L.; Wen, Z.; Smith, P.C.; Hawke, R.L. Hepatic metabolism and biliary excretion of silymarin flavonolignans in isolated perfused rat livers: Role of multidrug resistance-associated protein 2 (Abcc2). Drug Metab. Dispos. Biol. Fate Chem. 2008, 36, 2219–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, L.; King, C.D.; Whitington, P.F.; Green, M.D.; Roy, S.K.; Tephly, T.R.; Coffman, B.L.; Ratain, M.J. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J. Clin. Investig. 1998, 101, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Wenning, L.A.; Petry, A.S.; Kost, J.T.; Jin, B.; Breidinger, S.A.; DeLepeleire, I.; Carlini, E.J.; Young, S.; Rushmore, T.; Wagner, F.; et al. Pharmacokinetics of raltegravir in individuals with UGT1A1 polymorphisms. Clin. Pharmacol. Ther. 2009, 85, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, J.M.; Goldberg, R.M.; Qu, P.; Ibrahim, J.G.; McLeod, H.L. UGT1A1*28 genotype and irinotecan-induced neutropenia: Dose matters. J. Natl. Cancer Inst. 2007, 99, 1290–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Miranda, S.R.; Hoskins, J.M.; Hawke, R.L. Role of UDP-glucuronosyltransferase 1A1 in the metabolism and pharmacokinetics of silymarin flavonolignans in patients with HCV and NAFLD. Molecules 2017. [Google Scholar] [CrossRef] [Green Version]
- Jancova, P.; Anzenbacherova, E.; Papouskova, B.; Lemr, K.; Luzna, P.; Veinlichova, A.; Anzenbacher, P.; Simanek, V. Silybin is metabolized by cytochrome P450 2C8 in vitro. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 2035–2039. [Google Scholar] [CrossRef] [Green Version]
- Gunaratna, C.; Zhang, T. Application of liquid chromatography-electrospray ionization-ion trap mass spectrometry to investigate the metabolism of silibinin in human liver microsomes. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 794, 303–310. [Google Scholar] [CrossRef]
- Doehmer, J.; Weiss, G.; McGregor, G.P.; Appel, K. Assessment of a dry extract from milk thistle (Silybum marianum) for interference with human liver cytochrome-P450 activities. Toxicol. Vitr. Int. J. Publ. Assoc. Bibra 2011, 25, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi-Suzuki, M.; Frye, R.F.; Zhu, H.J.; Brinda, B.J.; Chavin, K.D.; Bernstein, H.J.; Markowitz, J.S. The effects of milk thistle (Silybum marianum) on human cytochrome P450 activity. Drug Metab. Dispos. Biol. Fate Chem. 2014, 42, 1611–1616. [Google Scholar] [CrossRef] [Green Version]
- Roubalova, L.; Dinkova-Kostova, A.T.; Biedermann, D.; Kren, V.; Ulrichova, J.; Vrba, J. Flavonolignan 2,3-dehydrosilydianin activates Nrf2 and upregulates NAD(P)H: Quinone oxidoreductase 1 in Hepa1c1c7 cells. Fitoterapia 2017, 119, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentova, K.; Purchartova, K.; Rydlova, L.; Roubalova, L.; Biedermann, D.; Petraskova, L.; Krenkova, A.; Pelantova, H.; Holeckova-Moravcova, V.; Tesarova, E.; et al. Sulfated metabolites of flavonolignans and 2,3-dehydroflavonolignans: Preparation and properties. Int. J. Mol. Sci. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalthoff, S.; Strassburg, C.P. Contribution of human UDP-glucuronosyltransferases to the antioxidant effects of propolis, artichoke and silymarin. Phytomed. Int. J. Phytother. Phytopharm. 2019, 56, 35–39. [Google Scholar] [CrossRef]
- Abdelsalam, H.M.; Samak, M.A.; Alsemeh, A.E. Synergistic therapeutic effects of Vitis vinifera extract and Silymarin on experimentally induced cardiorenal injury: The pertinent role of Nrf2. Biomed. Pharmacother. 2019, 110, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, Q.; Chen, Y. Effect of silibinin on CFLAR-JNK pathway in oleic acid-treated HepG2 cells. Biomed. Pharmacother. 2018, 108, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Ou, Q.; Weng, Y.; Wang, S.; Zhao, Y.; Zhang, F.; Zhou, J.; Wu, X. Silybin alleviates hepatic steatosis and fibrosis in NASH mice by inhibiting oxidative stress and involvement with the Nf-κB Pathway. Dig. Dis. Sci. 2018, 63, 3398–3408. [Google Scholar] [CrossRef]
- Arafa Keshk, W.; Zahran, S.M.; Katary, M.A.; Abd-Elaziz Ali, D. Modulatory effect of silymarin on nuclear factor-erythroid-2-related factor 2 regulated redox status, nuclear factor-κB mediated inflammation and apoptosis in experimental gastric ulcer. Chem. Biol. Interact. 2017, 273, 266–272. [Google Scholar] [CrossRef]
- Li, L.; Sun, H.Y.; Liu, W.; Zhao, H.Y.; Shao, M.L. Silymarin protects against acrylamide-induced neurotoxicity via Nrf2 signalling in PC12 cells. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2017, 102, 93–101. [Google Scholar] [CrossRef]
- Zhou, J.; Chao, G.; Li, Y.; Wu, M.; Zhong, S.; Feng, Z. Activation of NRF2/ARE by isosilybin alleviates Aβ25–35-induced oxidative stress injury in HT-22 cells. Neurosci. Lett. 2016, 632, 92–97. [Google Scholar] [CrossRef]
- Al-Rasheed, N.; Faddah, L.; Al-Rasheed, N.; Bassiouni, Y.A.; Hasan, I.H.; Mahmoud, A.M.; Mohamad, R.A.; Yacoub, H.I. Protective effects of silymarin, alone or in combination with chlorogenic acid and/or melatonin, against carbon tetrachloride-induced hepatotoxicity. Pharmacogn. Mag. 2016, 12, S337–S345. [Google Scholar]
- Zhao, F.; Shi, D.; Li, T.; Li, L.; Zhao, M. Silymarin attenuates paraquat-induced lung injury via Nrf2-mediated pathway in vivo and in vitro. Clin. Exp. Pharmacol. Physiol. 2015, 42, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Prabu, S.M.; Muthumani, M. Silibinin ameliorates arsenic induced nephrotoxicity by abrogation of oxidative stress, inflammation and apoptosis in rats. Mol. Biol. Rep. 2012, 39, 11201–11216. [Google Scholar] [CrossRef] [PubMed]
- Au, A.Y.; Hasenwinkel, J.M.; Frondoza, C.G. Hepatoprotective effects of S-adenosylmethionine and silybin on canine hepatocytes in vitro. J. Anim. Physiol. Anim. Nutr. 2013, 97, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Podder, B.; Kim, Y.S.; Zerin, T.; Song, H.Y. Antioxidant effect of silymarin on paraquat-induced human lung adenocarcinoma A549 cell line. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2012, 50, 3206–3214. [Google Scholar] [CrossRef]
- Choi, E.J.; Kim, E.K.; Jeoung, N.H.; Kim, S.H. Effect of silymarin on gluconeogenesis and lactate production in exercising rats. Food Sci. Biotechnol. 2016, 25, 119–124. [Google Scholar] [CrossRef]
- Schmoll, D.; Engel, C.K.; Glombik, H. The Keap1-Nrf2 protein-protein interaction: A suitable target for small molecules. Drug Discov. Today Technol. 2017, 24, 11–17. [Google Scholar] [CrossRef]
- Leung, C.H.; Zhang, J.T.; Yang, G.J.; Liu, H.; Han, Q.B.; Ma, D.L. Emerging screening approaches in the development of Nrf2-Keap1 protein-protein interaction inhibitors. Int. J. Mol. Sci. 2019. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos, N.D.; Talapatra, S.K.; Gatliff, J.; Kozielski, F.; Wells, G. Modified peptide inhibitors of the keap1-Nrf2 protein-protein interaction incorporating unnatural amino acids. Chembiochem. A Eur. J. Chem. Biol. 2018, 19, 1810–1816. [Google Scholar] [CrossRef]
- Li, M.; Huang, W.; Jie, F.; Wang, M.; Zhong, Y.; Chen, Q.; Lu, B. Discovery of Keap1-Nrf2 small-molecule inhibitors from phytochemicals based on molecular docking. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2019, 133, 110758. [Google Scholar] [CrossRef]
- Heightman, T.D.; Callahan, J.F.; Chiarparin, E.; Coyle, J.E.; Griffiths-Jones, C.; Lakdawala, A.S.; McMenamin, R.; Mortenson, P.N.; Norton, D.; Peakman, T.M.; et al. Structure-activity and structure-conformation relationships of aryl propionic acid inhibitors of the kelch-like ECH-associated protein 1/nuclear factor erythroid 2-related factor 2 (KEAP1/NRF2) protein-protein interaction. J. Med. Chem. 2019, 62, 4683–4702. [Google Scholar] [CrossRef]
- Tran, K.T.; Pallesen, J.S.; Solbak, S.M.O.; Narayanan, D.; Baig, A.; Zang, J.; Aguayo-Orozco, A.; Carmona, R.M.C.; Garcia, A.D.; Bach, A. A comparative assessment study of known small-molecule KEAP1-Nrf2 protein-protein interaction inhibitors: Chemical synthesis, binding properties, and cellular activity. J. Med. Chem. 2019, 62, 8028–8052. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Ursua, M.A.; Bello, M.; Hernandez-Martinez, C.F.; Santillan-Torres, I.; Guerrero-Ramirez, R.; Correa-Basurto, J.; Arias-Montano, J.A.; Trujillo-Ferrara, J.G. Cell-based assays and molecular dynamics analysis of a boron-containing agonist with different profiles of binding to human and guinea pig beta2 adrenoceptors. Eur. Biophys. J. EBJ 2019, 48, 83–97. [Google Scholar] [CrossRef] [PubMed]
- De Freitas Silva, M.; Pruccoli, L.; Morroni, F.; Sita, G.; Seghetti, F.; Viegas, C.; Tarozzi, A. The Keap1/Nrf2-ARE pathway as a pharmacological target for chalcones. Molecules 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Protocol | Results | Reference |
|---|---|---|---|
| Human esophangeal squamous cell carcinoma (KYSE70) cells in OS model induced by tBHQ | -tBHQ (100 μM) -SM (100 μM) -Artichoke (1 mg/mL) -Propolis (1%) 48 h of exposition | ↑ UGT1A1 enzyme activity by the AhR and Nrf2 pathway ↑ Induction of UGT1A7 with propolis, artichoke and SM (7.3, 5 and 4.5-fold respectively) ↑ UGT catalytic activity with porpolis, artichoke and SM treatment (21%, 29% and 20% respectively) in 90 min in the presence of substrate ↓ hydrogen peroxide were lower in tBHQ cells treated with phytochemicals | Kalthoff, et al. [153] |
| Rat cardiorenal injury model | Male Wistar albino rats -Doxorubicin via rat tail vein (3 and 2 mg/kg) to induce twice for 2 weeks to induce nephropathy -SM (600 mg/kg) for 4 weeks -Aqueous Vitis vinifera extract (400 mg/kg) for 4 weeks -Ethanolic Vitis vinifera extract (500 mg/kg) for 4 weeks -SM+EVVE (600 mg and 500 mg/kg) for 4 weeks | All the following results were significantly higher in SM+EVVE group: ↑ Nrf2 expression ↓ biochemical markers: BUN, urea, lipid profile ↓ hs-CRP and serum lipid profile ↓ histopathological alterations: improve cytomorphologic structure and restoration of myocardial architecture | Abdelsalam, et al. [154] |
| Hepa1c1c7 cells | Sulfate compounds from SM at 50 μM for 48 h exposition. | ↑ activity of NQO1 compared to control: silybin A 20-O-sulfate1.2-fold, silybin B 20-O-sulfate0.9-fold, 2,3-dehydrosilybin-20-O-sulfate 1.3-fold, 2,3-dehydrosilybin-7,20-di-O-sulfate 1.2 fold, silychristin-19-O-sulfate1.3-fold. 2,3-dehydrosilychristin-19-O-sulfate1.4- fold | Valentová, et al. [152] |
| Oleic acid-treated HepG2 cell as in vitro model of steatosis, OS and IR | Silibinin at 5, 20, 50 and 100 μM for 24 h exposition | ↓ lipid metabolism genes: SREBP-1C, PPAR α, PNPLA3 ↓ intracellular levels of TG and NO ↑ oxidative stress response genes:Nrf2, CYP2E1 and CYP4A ↑ glucose uptake: proteins PI3K and pAKT | Liu, et al. [155] |
| Male C57BL/6 mice a NASH model | -DL-methionine (3 g/kg) and choline bitartrate (2 g/kg) diet to induce NASH -orally ingestion of silybin at a dose of 105 mg/kg/day for 8 weeks | ↓ Weight loss ↓ ALT and AST activities ↓ lipid metabolism gene expression ↓ NF-κB signaling pathway ↑ Nrf2 pathway: expression levels of GCLM, GCLC, NQO1, HOX1, GSTM1 ↑ levels of GPx and SOD in hepatic tissue ↓ histological disorders: hepatic steatosis, hepatocellular ballooning with inflammatory cell infiltration and liver fibrosis | Ou, et al. [156] |
| Rat gastric ulcer model | -5 days pre-treatment with ranitidine at 25 mg/kg orally (positive control) -5 days pre-treatment with SM at 50 mg/kg orally -Indometacin-induced gastric injury (25 mg/kg) in the last day | Prevent gastric OS by: ↑ Nrf2 expression, ↑ SOD and GPx activity ↓ gastric inflammation: TNF- α, IL-6 and myeloperoxidase activity, ↑ NF-κB expression Histological changes: protection observed as mild sub-mucosal edema and inflammatory cellular infiltrates and minimal alterations in epithelial surface | Arafa Kesnk, et al. [157] |
| Murine hepatoma Hepa1c1c7 cells | S. marianumflavolingnans and its 2,3- dehiydroderivates (25 μM) And 2,3-dehydrosilydianin (50 μM) 6 h exposition | ↑ expression of Nrf2, Gclm and Gclcat 25 and 50 μM ↑ protein concentration of NOQ1 and GCLM at 50 μM HMOX1 and GCLC did not changed significantly | Roubalová, et al. [151] |
| PC12 cells Acrylamide-induced neurotoxicity model | Cells pre-treated with SM at 12, 24, 48, 96 or 192 μg/mL for 3 h, then cells were exposed to a 5 mM Acrylamide for 24 h | ↑ mRNA and protein expression of Nrf2 in nuclear fractions ↑ translocation of Nrf2 from cytosol into the nucleus ↑ cytoprotective genes: GPx, GCLC, GCLM ↑ intracellular levels of GSH ↓ levels of ROS and MDA | Li, et al. [158] |
| HT-22 hipocampal cells AD model | Cells treated with: -Aβ25-35AT 2 μM for 24 h to induce toxicity in cells -Isosilybin exposition at 2, 4 and 10 Μm | ↑ Nrf2/ARE signaling pathway: HO-1, AKR1C2 and GST ↓ ROS production ↓ cellular OS injury ↑ total antioxidant capacity in cells by ↓release MDA and LDH | Zhou, et al. [159] |
| Male Wistar rats CCl4 damage model | Hepatotoxicity induced with single dose of CCl4 (1 ml/Kg, IP) Orally administration of SM (200 mg/kg) alone or in combination with CA (60 mg/kg) and/or ME (20 mg (kg) for 21 days | ↓ expression of fibrogenic and apoptotic factors ↓ Serum ALT activity ↓ liver caspase-3 activity ↓ hepatic CYP2E1 activity in SM, CA and ME treated group compared with SM alone ↓ oxidative DNA damage in liver: 8-OxodG levels | Al-Rasheed, et al. [160] |
| Male Sprague-Dawley rats Paraquat lung injury model | Paraquat exposition (30 mg/kg) to induce lung injury SM treatment (200 mg/kg) 3 days exposition | ↓ MDA ↑ SOD, Cat, and GPx in lung tissue and serum. ↑ Nrf2, HO-1, and NQO1 expression ↓ Inflammatory cell infiltration and collagen deposition in the alveolar septum ↓ MPO activity and HYP content ↓ NO and iNOS ↓ Proinflammatory mediator levels: TNFα, IL-1β, IL-6 and the TGF-β1 | Zhao, et al. [161] |
| Rat arsenic toxic model | Adult male Wistar albino rats treated with: -Arsenic (5 mg/kg) for 4 weeks silibinin at 75 mg/kg/day for 4 weeks | ↓ lipid peroxidation, NADPH Oxidase, iNOS, NF-kB and TNFα ↓ lipid hydroperoxides, protein carbonyls and TBARS ↑ mRNA expression of Nrf2 and NADPH in renal tissue ↑ activities of enzymatic antioxidants: SOD, CA, GPx, and GST ↑ Non-enzymatic antioxidants: GSH, TSH, Vitamin C and E in kidney tissue | Prabu, et al. [162] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vargas-Mendoza, N.; Morales-González, Á.; Morales-Martínez, M.; Soriano-Ursúa, M.A.; Delgado-Olivares, L.; Sandoval-Gallegos, E.M.; Madrigal-Bujaidar, E.; Álvarez-González, I.; Madrigal-Santillán, E.; Morales-Gonzalez, J.A. Flavolignans from Silymarin as Nrf2 Bioactivators and Their Therapeutic Applications. Biomedicines 2020, 8, 122. https://doi.org/10.3390/biomedicines8050122
Vargas-Mendoza N, Morales-González Á, Morales-Martínez M, Soriano-Ursúa MA, Delgado-Olivares L, Sandoval-Gallegos EM, Madrigal-Bujaidar E, Álvarez-González I, Madrigal-Santillán E, Morales-Gonzalez JA. Flavolignans from Silymarin as Nrf2 Bioactivators and Their Therapeutic Applications. Biomedicines. 2020; 8(5):122. https://doi.org/10.3390/biomedicines8050122
Chicago/Turabian StyleVargas-Mendoza, Nancy, Ángel Morales-González, Mauricio Morales-Martínez, Marvin A. Soriano-Ursúa, Luis Delgado-Olivares, Eli Mireya Sandoval-Gallegos, Eduardo Madrigal-Bujaidar, Isela Álvarez-González, Eduardo Madrigal-Santillán, and José A. Morales-Gonzalez. 2020. "Flavolignans from Silymarin as Nrf2 Bioactivators and Their Therapeutic Applications" Biomedicines 8, no. 5: 122. https://doi.org/10.3390/biomedicines8050122