Exposure to CuO Nanoparticles Mediates NFκB Activation and Enhances Amyloid Precursor Protein Expression



Abstract
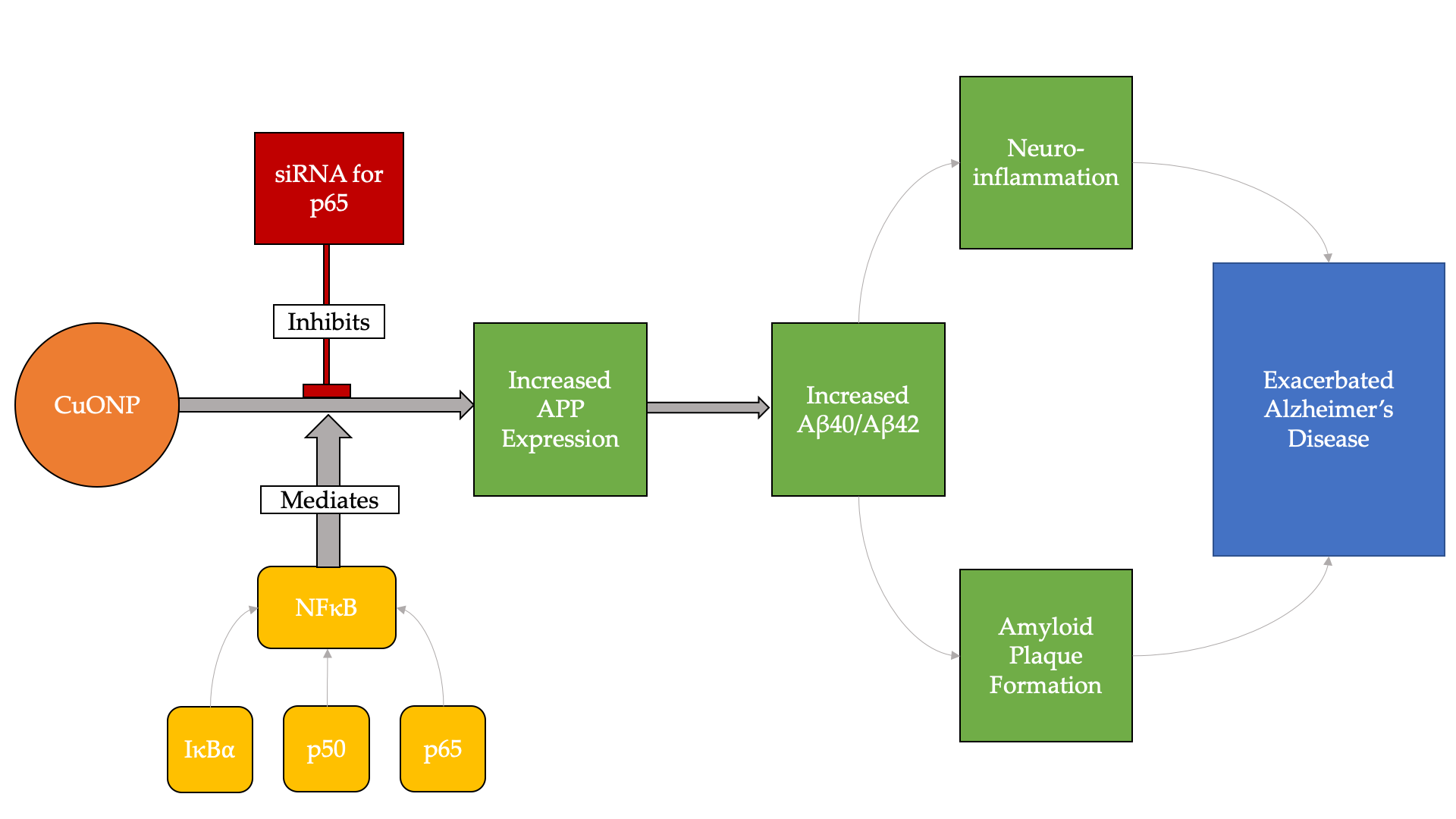
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Cell Culture
2.2. Transfection
2.3. Luciferase Reporter Assay
2.4. Western Blot
2.4.1. Nuclear and Cytoplasmic Protein Extraction
2.4.2. Nitrocellulose Membranes
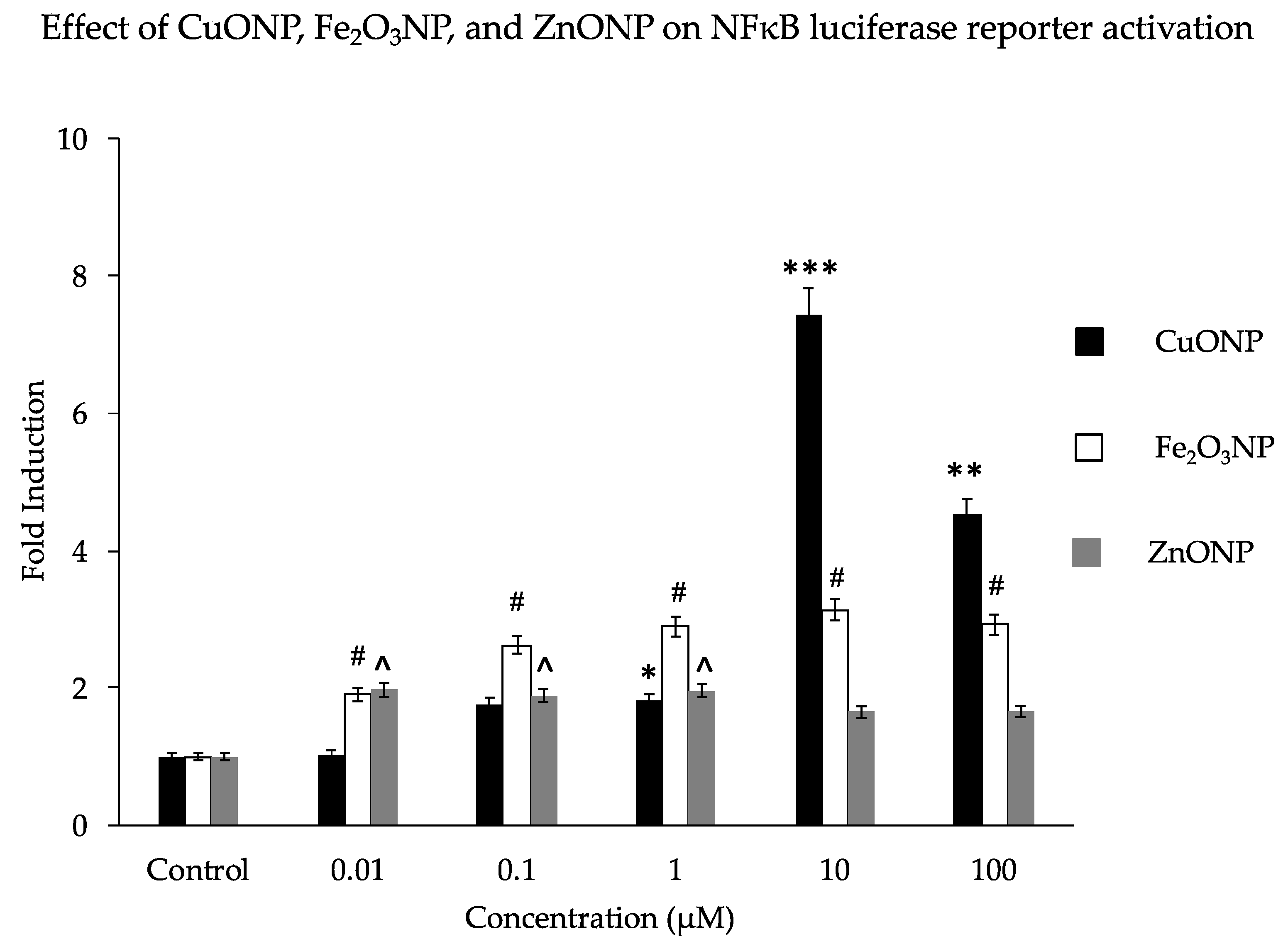
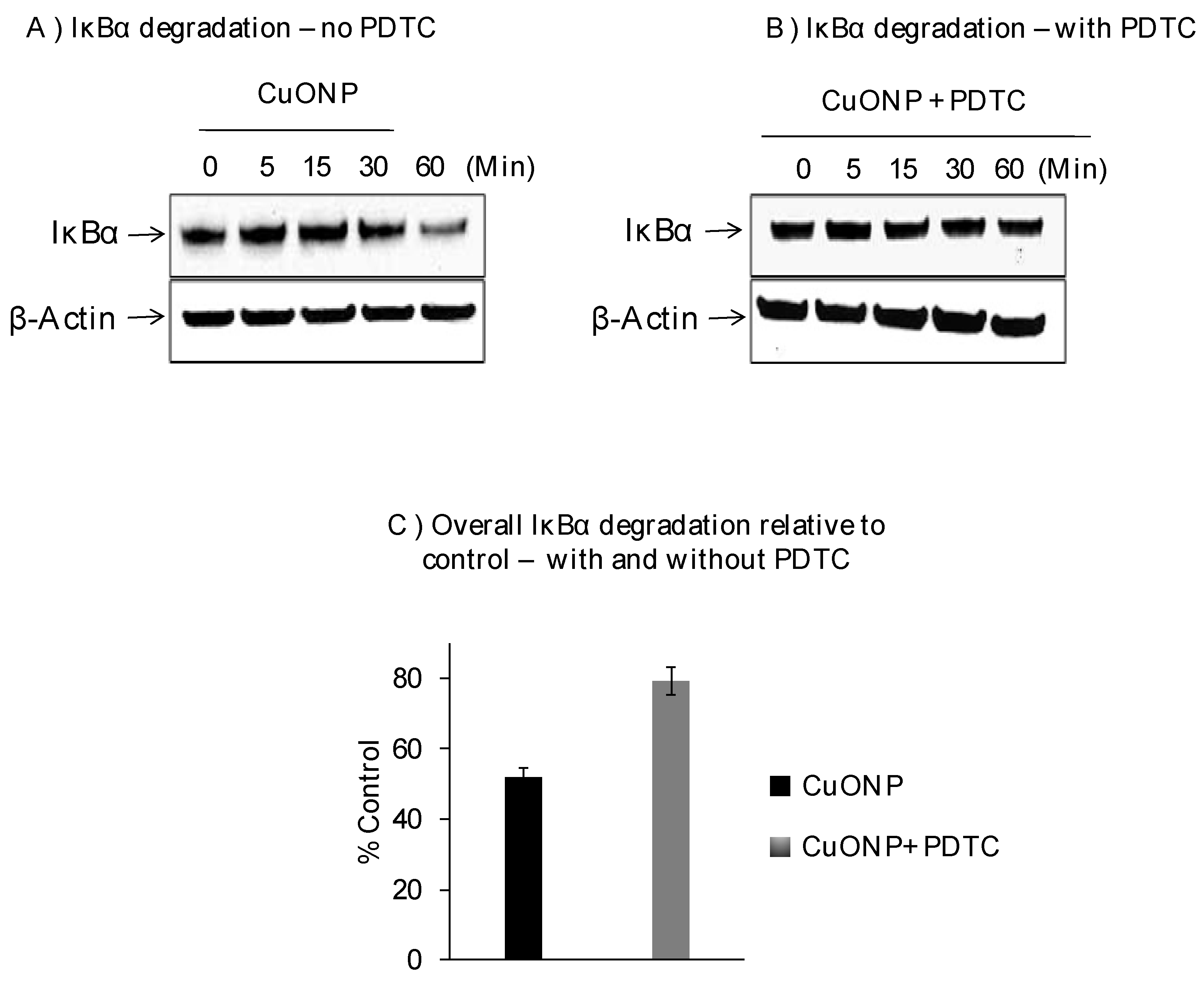
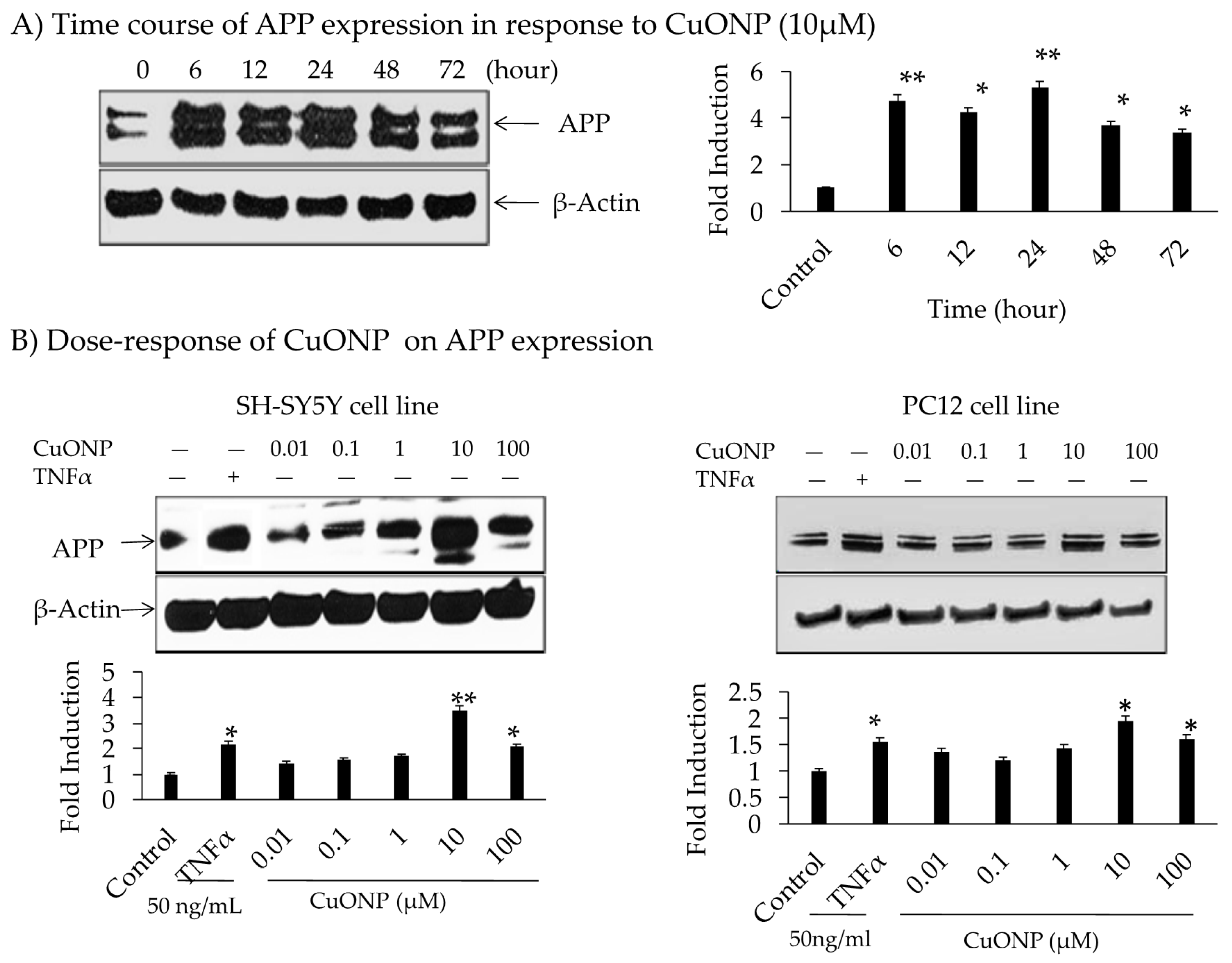
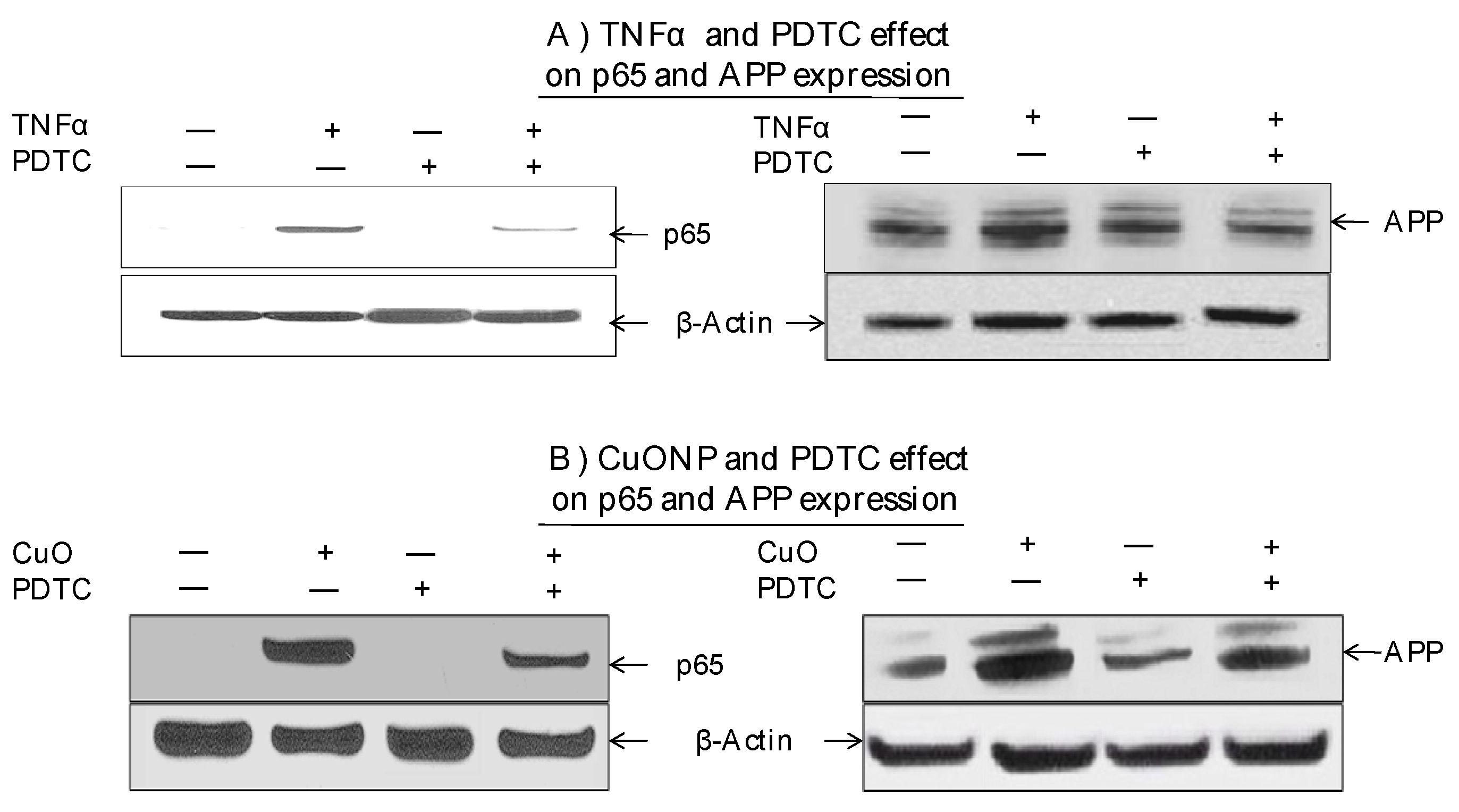
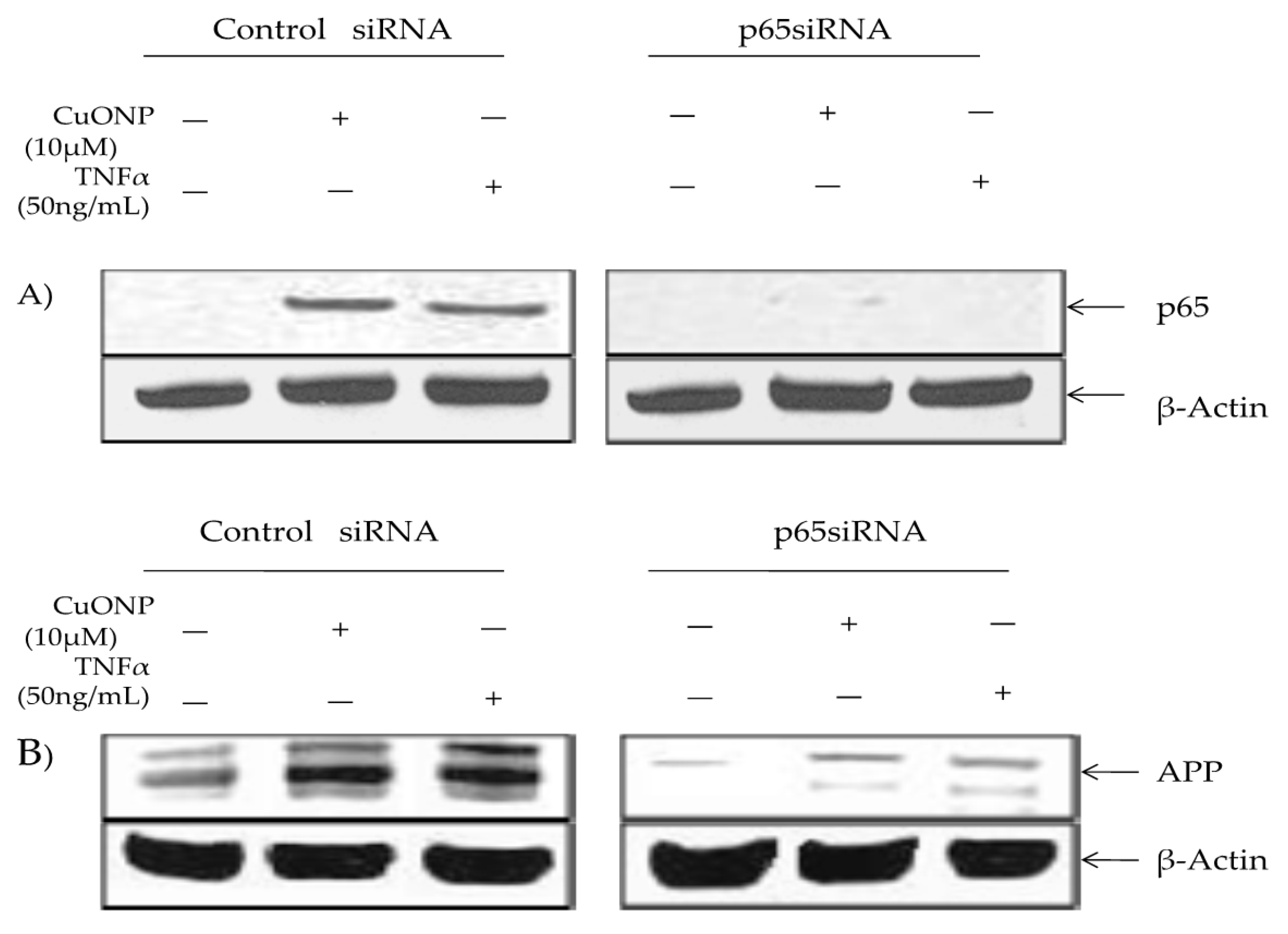
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.P.; Price, J. Human induced pluripotent stem cells as a research tool in Alzheimer’s disease. Psychol. Med. 2017, 47, 2587–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berchtold, N.C.; Cotman, C.W. Evolution in the conceptualization of dementia and Alzheimer’s disease: Greco-Roman period to the 1960s. Neurobiol. Aging 1998, 19, 173–189. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer’s-Association. 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Unzeta, M.; Esteban, G.; Bolea, I.; Fogel, W.A.; Ramsay, R.R.; Youdim, M.B.; Tipton, K.F.; Marco-Contelles, J. Multi-target directed donepezil-like ligands for Alzheimer’s disease. Front. Neurosci. 2016, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Tang, X.C. Neuroprotective effects of huperzine A: New therapeutic targets for neurodegenerative disease. Trends Pharmacol. Sci. 2006, 27, 619–625. [Google Scholar] [CrossRef]
- Sastre, M.; Walter, J.; Gentleman, S.M. Interactions between APP secretases and inflammatory mediators. J. Neuroinflamm. 2008, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.Y.; Bealgey, K.W.; Fang, Y.; Gong, Y.M.; Bao, S. Interleukin-23: Immunological roles and clinical implications. Int. J. Biochem. Cell Biol. 2009, 41, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Gahtan, E.; Overmier, J.B. Inflammatory pathogenesis in Alzheimer’s disease: Biological mechanisms and cognitive sequeli. Neurosci. Biobehav. Rev. 1999, 23, 615–633. [Google Scholar] [CrossRef]
- Golde, T.E. Inflammation takes on Alzheimer disease. Nat. Med. 2002, 8, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Innate immunity, local inflammation, and degenerative disease. Sci. Aging Knowl. Environ. 2002, 2002, re3. [Google Scholar] [CrossRef] [Green Version]
- Kalaria, R.N.; Harshbarger-Kelly, M.; Cohen, D.L.; Premkumar, D.R. Molecular aspects of inflammatory and immune responses in Alzheimer’s disease. Neurobiol. Aging 1996, 17, 687–693. [Google Scholar] [CrossRef]
- Huang, X. Exposure of engineered nanomaterials and its potential contribution to Alzheimer’s pathophysiology. Biochem. Pharmacol. Open Access 2013, 2, e144. [Google Scholar]
- Carro, C.E.; Pilozzi, A.R.; Huang, X. Nanoneurotoxicity and potential nanotheranostics for Alzheimer’s disease. EC Pharmacol. Toxicol. 2019, 7, 1–7. [Google Scholar]
- Ransohoff, R.M.; Schafer, D.; Vincent, A.; Blachere, N.E.; Bar-Or, A. Neuroinflammation: Ways in which the immune system affects the brain. Neurotherapeutics 2015, 12, 896–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattan, S.I. Theories of biological aging: Genes, proteins, and free radicals. Free Radic. Res. 2006, 40, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Sinha, S.; Kawahara, T.L.; Zhang, J.Y.; Segal, E.; Chang, H.Y. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007, 21, 3244–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Schedl, P.; Song, H.J.; Garza, D.; Konsolaki, M. The Toll→NFkappaB signaling pathway mediates the neuropathological effects of the human Alzheimer’s Abeta42 polypeptide in Drosophila. PLoS ONE 2008, 3, e3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelsson, M.; Fisher, L.; Iverfeldt, K. Beta-Amyloid and interleukin-1beta induce persistent NF-kappaB activation in rat primary glial cells. Int. J. Mol. Med. 2005, 16, 449–453. [Google Scholar] [PubMed]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 2007, 178, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksen, J.L.; Sagi, S.A.; Smith, T.E.; Weggen, S.; Das, P.; McLendon, D.C.; Ozols, V.V.; Jessing, K.W.; Zavitz, K.H.; Koo, E.H.; et al. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J. Clin. Investig. 2003, 112, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, G.P.; Yang, F.; Chu, T.; Chen, P.; Beech, W.; Teter, B.; Tran, T.; Ubeda, O.; Ashe, K.H.; Frautschy, S.A.; et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 2000, 20, 5709–5714. [Google Scholar] [CrossRef]
- Choo, X.Y.; Alukaidey, L.; White, A.R.; Grubman, A. Neuroinflammation and copper in Alzheimer’s disease. Int. J. Alzheimers Dis. 2013, 2013, 145345. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef]
- Mocchegiani, E.; Costarelli, L.; Giacconi, R.; Piacenza, F.; Basso, A.; Malavolta, M. Micronutrient (Zn, Cu, Fe)-gene interactions in ageing and inflammatory age-related diseases: Implications for treatments. Ageing Res. Rev. 2012, 11, 297–319. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ye, J.; Zhou, S.; Pi, R.; Dou, J.; Zang, L.; Chen, X.; Chao, X.; Li, W.; Liu, M.; et al. The effects of chronic copper exposure on the amyloid protein metabolisim associated genes’ expression in chronic cerebral hypoperfused rats. Neurosci. Lett. 2012, 518, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 2011, 303, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-beta homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presume, M.; Simon-Deckers, A.; Tomkiewicz-Raulet, C.; Le Grand, B.; Tran Van Nhieu, J.; Beaune, G.; Duruphty, O.; Doucet, J.; Coumoul, X.; Pairon, J.C.; et al. Exposure to metal oxide nanoparticles administered at occupationally relevant doses induces pulmonary effects in mice. Nanotoxicology 2016, 10, 1535–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persichini, T.; Percario, Z.; Mazzon, E.; Colasanti, M.; Cuzzocrea, S.; Musci, G. Copper activates the NF-kappaB pathway in vivo. Antioxid. Redox Signal. 2006, 8, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Pithadia, A.S.; Lim, M.H. Metal-associated amyloid-beta species in Alzheimer’s disease. Curr. Opin. Chem. Biol. 2012, 16, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.K.; Agrawal, M.; Marshall, F.M. Atmospheric deposition of heavy metals (Cu, Zn, Cd and Pb) in Varanasi City, India. Environ. Monit. Assess. 2008, 142, 269–278. [Google Scholar] [CrossRef]
- Sharma, S.; O’Keefe, S.J. Environmental influences on the high mortality from colorectal cancer in African Americans. Postgrad. Med. J. 2007, 83, 583–589. [Google Scholar] [CrossRef]
- Wigginton, N.S.; Haus, K.L.; Hochella, M.F., Jr. Aquatic environmental nanoparticles. J. Environ. Monit. 2007, 9, 1306–1316. [Google Scholar] [CrossRef]
- Horie, M.; Shimizu, K.; Tabei, Y. Validation of metallothionein, interleukin-8, and heme oxygenase-1 as markers for evaluation of cytotoxicity caused by metal oxide nanoparticles. Toxicol. Mech. Methods 2018, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, J.; Cho, H.-H.; Cui, K.; Li, F.; Zhou, X.; Rodgers, J.T.; Wong, S.T.C.; Huang, X. Differential cytotoxicity of metal oxide nanoparticles. J. Exp. Nanosci. 2008, 3, 321–328. [Google Scholar] [CrossRef]
- Shi, Y.; Pilozzi, A.R.; Huang, X. Exposure of CuO nanoparticles contributes to cellular apoptosis, redox stress, and Alzheimer’s Aβ amyloidosis. Int. J. Environ. Res. Public Health 2020, 17, E1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarenko, O.; Juganson, K.; Ivask, A.; Kasemets, K.; Mortimer, M.; Kahru, A. Toxicity of Ag, CuO and ZnO nanoparticles to selected environmentally relevant test organisms and mammalian cells in vitro: A critical review. Arch. Toxicol. 2013, 87, 1181–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casals, E.; Gonzalez, E.; Puntes, V.F. Reactivity of inorganic nanoparticles in biological environments: Insights into nanotoxicity mechanisms. J. Phys. D Appl. Phys. 2012, 45, 1. [Google Scholar] [CrossRef] [Green Version]
- Stark, W.J.; Stoessel, P.R.; Wohlleben, W.; Hafner, A. Industrial applications of nanoparticles. Chem. Soc. Rev. 2015, 44, 5793–5805. [Google Scholar] [CrossRef] [Green Version]
- Kahru, A.; Dubourguier, H.C. From ecotoxicology to nanoecotoxicology. Toxicology 2010, 269, 105–119. [Google Scholar] [CrossRef]
- Sadeghi, L.; Yousefi Babadi, V.; Espanani, H.R. Toxic effects of the Fe2O3 nanoparticles on the liver and lung tissue. Bratisl. Lek. Listy 2015, 116, 373–378. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, D.; Liu, W.; Yan, Y.; Zhou, F.; Wu, W.; Yan, Z. Reactive oxygen species trigger NF-kappaB-mediated NLRP3 inflammasome activation induced by zinc oxide nanoparticles in A549 cells. Toxicol. Ind. Health 2017, 33, 737–745. [Google Scholar] [CrossRef]
- Chien, C.C.; Yan, Y.H.; Juan, H.T.; Cheng, T.J.; Liao, J.B.; Lee, H.P.; Wang, J.S. Sustained renal inflammation following 2 weeks of inhalation of occupationally relevant levels of zinc oxide nanoparticles in Sprague Dawley rats. J. Toxic. Pathol. 2017, 30, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Andujar, P.; Simon-Deckers, A.; Galateau-Salle, F.; Fayard, B.; Beaune, G.; Clin, B.; Billon-Galland, M.A.; Durupthy, O.; Pairon, J.C.; Doucet, J.; et al. Role of metal oxide nanoparticles in histopathological changes observed in the lung of welders. Part. Fibre Toxicol. 2014, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimnia-Bajestan, E.; Niazmand, H.; Duangthongsuk, W.; Wongwises, S. Numerical investigation of effective parameters in convective heat transfer of nanofluids flowing under a laminar flow regime. Int. J. Heat Mass Transf. 2011, 54, 4376–4388. [Google Scholar] [CrossRef]
- Sharma, H.S.; Sharma, A. Nanoparticles aggravate heat stress induced cognitive deficits, blood-brain barrier disruption, edema formation and brain pathology. Prog. Brain Res. 2007, 162, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.S.; Ali, S.F.; Hussain, S.M.; Schlager, J.J.; Sharma, A. Influence of engineered nanoparticles from metals on the blood-brain barrier permeability, cerebral blood flow, brain edema and neurotoxicity. An experimental study in the rat and mice using biochemical and morphological approaches. J. Nanosci. Nanotechnol. 2009, 9, 5055–5072. [Google Scholar] [CrossRef]
- An, L.; Liu, S.; Yang, Z.; Zhang, T. Cognitive impairment in rats induced by nano-CuO and its possible mechanisms. Toxicol. Lett. 2012, 213, 220–227. [Google Scholar] [CrossRef]
- Xu, L.J.; Zhao, J.X.; Zhang, T.; Ren, G.G.; Yang, Z. In vitro study on influence of nano particles of CuO on CA1 pyramidal neurons of rat hippocampus potassium currents. Environ. Toxicol. 2009, 24, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Perreault, F.; Pedroso Melegari, S.; Henning da Costa, C.; de Oliveira Franco Rossetto, A.L.; Popovic, R.; Gerson Matias, W. Genotoxic effects of copper oxide nanoparticles in Neuro 2A cell cultures. Sci. Total Environ. 2012, 441, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Liu, G.; Huang, W.; Moir, R.D.; Vanderburg, C.R.; Lai, B.; Peng, Z.; Tanzi, R.E.; Rogers, J.T.; Huang, X. Metal exposure and Alzheimer’s pathogenesis. J. Struct. Biol. 2006, 155, 45–51. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Delgado-Chavez, R.; Mukherjee, P.S.; Kulesza, R.J.; Torres-Jardon, R.; Avila-Ramirez, J.; Villarreal-Rios, R. Hallmarks of Alzheimer disease are evolving relentlessly in Metropolitan Mexico City infants, children and young adults. APOE4 carriers have higher suicide risk and higher odds of reaching NFT stage V at ≤40 years of age. Environ. Res. 2018, 164, 475–487. [Google Scholar] [CrossRef]
- Li, F.; Zhou, X.; Zhu, J.; Ma, J.; Huang, X.; Wong, S.T. High content image analysis for human H4 neuroglioma cells exposed to CuO nanoparticles. BMC Biotechnol. 2007, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhu, B.M.; Ali, S.F.; Murdock, R.C.; Hussain, S.M.; Srivatsan, M. Copper nanoparticles exert size and concentration dependent toxicity on somatosensory neurons of rat. Nanotoxicology 2010, 4, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Altamura, S.; Muckenthaler, M.U. Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J. Alzheimers Dis. 2009, 16, 879–895. [Google Scholar] [CrossRef]
- Kaden, D.; Bush, A.I.; Danzeisen, R.; Bayer, T.A.; Multhaup, G. Disturbed copper bioavailability in Alzheimer’s disease. Int. J. Alzheimers Dis. 2011, 2011, 345614. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zheng, J. Cholesterol promotes the interaction of Alzheimer beta-amyloid monomer with lipid bilayer. J. Mol. Biol. 2012, 421, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Condron, M.M.; Teplow, D.B.; Lyubchenko, Y.L. Nanoprobing of the effect of Cu(2+) cations on misfolding, interaction and aggregation of amyloid β peptide. J. Neuroimmune Pharmacol. 2013, 8, 262–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squitti, R.; Zito, G. Anti-copper therapies in Alzheimer’s disease: New concepts. Recent Pat. CNS Drug Discov. 2009, 4, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Tiiman, A.; Palumaa, P.; Tougu, V. The missing link in the amyloid cascade of Alzheimer’s disease—Metal ions. Neurochem. Int. 2013, 62, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Ding, M.; Xue, J.; Leng, W. The role of TLR2/JNK/NF-kappaB pathway in amyloid beta peptide-induced inflammatory response in mouse NG108-15 neural cells. Int. Immunopharmacol. 2013, 17, 880–884. [Google Scholar] [CrossRef]
- Kimura, R.; Devi, L.; Ohno, M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J. Neurochem. 2010, 113, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Chen, Y.; Zhang, H.; Ma, Q.; Zhang, Y.W.; Xu, H. Salubrinal attenuates beta-amyloid-induced neuronal death and microglial activation by inhibition of the NF-kappaB pathway. Neurobiol. Aging 2012, 33, 1007.e9–1007.e17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Hu, W. Anti-neuroinflammatory agents for the treatment of Alzheimer’s disease. Future Med. Chem. 2013, 5, 1559–1571. [Google Scholar] [CrossRef]
- Chang, K.L.; Pee, H.N.; Yang, S.; Ho, P.C. Influence of drug transporters and stereoselectivity on the brain penetration of pioglitazone as a potential medicine against Alzheimer’s disease. Sci. Rep. 2015, 5, 9000. [Google Scholar] [CrossRef] [Green Version]
- Chami, L.; Buggia-Prevot, V.; Duplan, E.; Del Prete, D.; Chami, M.; Peyron, J.F.; Checler, F. Nuclear factor-kappaB regulates betaAPP and beta- and gamma-secretases differently at physiological and supraphysiological Abeta concentrations. J. Biol. Chem. 2015, 290, 29758. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wang, Z.Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017, 35, 265–290. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease: How far have we come in the clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Kounatidis, I.; Chtarbanova, S.; Cao, Y.; Hayne, M.; Jayanth, D.; Ganetzky, B.; Ligoxygakis, P. NF-kappaB immunity in the brain determines fly lifespan in healthy aging and age-related neurodegeneration. Cell Rep. 2017, 19, 836–848. [Google Scholar] [CrossRef]
- Cacciottolo, M.; Wang, X.; Driscoll, I.; Woodward, N.; Saffari, A.; Reyes, J.; Serre, M.L.; Vizuete, W.; Sioutas, C.; Morgan, T.E.; et al. Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl. Psychiatry 2017, 7, e1022. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mou, X.; Pilozzi, A.; Tailor, B.; Yi, J.; Cahill, C.; Rogers, J.; Huang, X. Exposure to CuO Nanoparticles Mediates NFκB Activation and Enhances Amyloid Precursor Protein Expression. Biomedicines 2020, 8, 45. https://doi.org/10.3390/biomedicines8030045
Mou X, Pilozzi A, Tailor B, Yi J, Cahill C, Rogers J, Huang X. Exposure to CuO Nanoparticles Mediates NFκB Activation and Enhances Amyloid Precursor Protein Expression. Biomedicines. 2020; 8(3):45. https://doi.org/10.3390/biomedicines8030045
Chicago/Turabian StyleMou, Xiaoyang, Alexander Pilozzi, Breeya Tailor, Jing Yi, Catherine Cahill, Jack Rogers, and Xudong Huang. 2020. "Exposure to CuO Nanoparticles Mediates NFκB Activation and Enhances Amyloid Precursor Protein Expression" Biomedicines 8, no. 3: 45. https://doi.org/10.3390/biomedicines8030045