
Putting Functional Gastrointestinal Disorders within the Spectrum of Inflammatory Disorders Can Improve Classification and Diagnostics of These Disorders

 , , and
, , and
Abstract
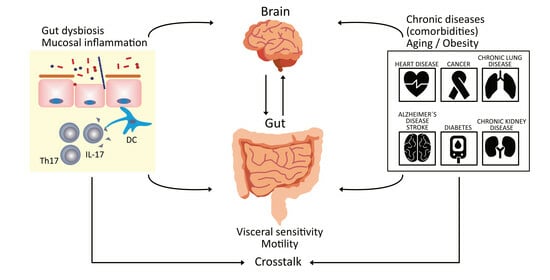
:
1. Introduction—Motivation for This Review
2. Autoinflammatory and Autoimmune Diseases
3. The Role of Inflammation in the Pathogenesis of Other Chronic Diseases
4. Inflammation in the Vital Tissue and the Whole Body’s Reaction
5. An Interplay between the Neuroendocrine, Immune, and Metabolic Pathways in Aging and Obesity as a Driver of Chronic Disease Development
6. The Role of the Gut Microbiome and the Gut Mucosal Immune System in the Development of Chronic Disease
7. Functional Gastrointestinal Disorders
8. An Association of Inflammation with FGIDs Including the Role of the Gut Microbiome
9. Discussion
10. Future Perspectives
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FGIDs | functional gastrointestinal disorders |
| IBDs | inflammatory bowel diseases |
| UC | ulcerative colitis |
| CD | Chron’s disease |
| HP | Helicobacter pylori |
| IBS | irritable bowel syndrome |
| FD | functional dyspepsia |
| DCs | dendritic cells |
| TNF-α | tumor necrosis factor-α |
| ILs | interleukins |
| IFNs | interferons |
| Treg cells | regulatory T cells |
| Teff cells | effector T cells |
| NETs | neutrophil extracellular traps |
| ROS | reactive oxygen species |
| LDGs | low-density granulocytes |
| INFIs | type I interferons |
| PRRs | pattern recognition receptors |
| PAMPs | pathogen-associated molecular patterns |
| DAMPs | danger-associated molecular patterns |
| NK cells | natural killer cells |
| Th1 cells | T helper cells type 1 |
| T2D | type 2 diabetes |
| CVD | cardiovascular diseases |
| TGF-β | transforming growth factor-β |
| naAbs | natural antibodies |
| HPA | hypothalamic-pituitary-adrenal |
| ILC1s | innate lymphoid cells type 1 |
| ILC2s | innate lymphoid type 2 cells |
| LPSs | lipopolysaccharides |
| SCFAs | short-chain fatty acids |
| BCFAs | branched-chain amino acids |
| IgA | immunolobulin A |
| FDCs | follicular dendritic cells |
| Thf cells | follicular Th cells |
| CNS | central nervous system |
| PDS | postprandial distress syndrome |
| EPS | epigastric pain syndrome |
| ENS | enteric nervous system |
| ANS | autonomic nervous system |
| ECCs | enterochromaffin cells |
| 5-HT | 5-hydroxytryptamine |
| TPH1 | tryptophan hydroxylase 1 |
| PI-IBS | post-infection irritable bowel syndrome |
| EoE | eosinophilic esophagitis |
| Th17 | T-helper 17 |
| TSLP | thymic stromal lymphoprotein |
| iNKT cells | invariant natural killer T cells |
| ILC3s | innate lymphoid type 3 cells |
| CRF | corticotropin-releasing factor |
| CRP | C-reactive protein |
| LBP | lipopolysaccharide binding protein |
| IFABP | intestinal fatty acid binding protein |
References
- Lu, Q.; Yang, M.F.; Liang, Y.J.; Xu, J.; Xu, H.M.; Nie, Y.Q.; Wang, L.S.; Yao, J.; Li, D.F. Immunology of Inflammatory Bowel Disease: Molecular Mechanisms and Therapeutics. J. Inflamm. Res. 2022, 15, 1825–1844. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Drossman, D.A. Functional gastrointestinal disorders: History, pathophysiology, clinical features and Rome IV. Gastroenterology 2016, 150, 1262–1279. [Google Scholar] [CrossRef] [PubMed]
- Drossman, D.A.; Tack, J. Rome Foundation Clinical Diagnostic Criteria for Disorders of Gut-Brain Interaction. Gastroenterology 2022, 162, 675–679. [Google Scholar] [CrossRef]
- Wei, L.; Singh, R.; Ro, S.; Ghoshal, U.C. Gut microbiota dysbiosis in functional gastrointestinal disorders: Underpinning the symptoms and pathophysiology. JGH Open 2021, 5, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Foster, J.A.; McVey Neufeld, K.A. Gut-brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef]
- Van Oudenhove, L.; Crowell, M.D.; Drossman, D.A.; Halpert, A.D.; Keefer, L.; Lackner, J.M.; Murphy, T.B.; Naliboff, B.D.; Levy, R.L. Biopsychosocial Aspects of Functional Gastrointestinal Disorders. Gastroenterology 2016, 150, 1355–1367. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, M.; Jiang, D.; Su, Q.; Shi, J. The role of inflammation in autoimmune disease: A therapeutic target. Front. Immunol. 2023, 14, 1267091. [Google Scholar] [CrossRef]
- Szekanecz, Z.; McInnes, I.B.; Schett, G.; Szamosi, S.; Benkő, S.; Szűcs, G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat. Rev. Rheumatol. 2021, 17, 585–595. [Google Scholar] [CrossRef]
- Park, H.; Bourla, A.B.; Kastner, D.L.; Colbert, R.A.; Siegel, R.M. Lighting the fires within: The cell biology of autoinflammatory diseases. Nat. Rev. Immunol. 2012, 12, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Jörg, S.; Grohme, D.A.; Erzler, M.; Binsfeld, M.; Haghikia, A.; Müller, D.N.; Linker, R.A.; Kleinewietfeld, M. Environmental factors in autoimmune diseases and their role in multiple sclerosis. Cell Mol. Life Sci. 2016, 73, 4611–4622. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018, 19, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Medina, G.; Vera-Lastra, O.; Peralta-Amaro, A.L.; Jiménez-Arellano, M.P.; Saavedra, M.A.; Cruz-Domínguez, M.P.; Jara, L.J. Metabolic syndrome, autoimmunity and rheumatic diseases. Pharmacol. Res. 2018, 133, 277–288. [Google Scholar] [CrossRef]
- Greuter, T.; Vavricka, S.R. Extraintestinal manifestations in inflammatory bowel disease—Epidemiology, genetics, and pathogenesis. Exp. Rev. Gastroenterol. Hepatol. 2019, 13, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, C.M. Shaping the spectrum—From autoinflammation to autoimmunity. Clin. Immunol. 2016, 165, 21–28. [Google Scholar] [CrossRef]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on neutrophil function in severe in-flammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allerg. Immunol. 2021, 61, 194–211. [Google Scholar] [CrossRef]
- Tamassia, N.; Bianchetto-Aguilera, F.; Arruda-Silva, F.; Gardiman, E.; Gasperini, S.; Calzetti, F.; Cassatella, M.A. Cytokine production by human neutrophils: Revisiting the “dark side of the moon”. Eur. J. Clin. Investig. 2018, 48, e12952. [Google Scholar] [CrossRef]
- Apel, F.; Zychlinsky, A.; Kenny, E.F. The role of neutrophil extracellular traps in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Wigerblad, G.; Kaplan, M.J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. 2023, 23, 274–288. [Google Scholar] [CrossRef]
- Berezin, A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, N.; Ouyang, W. Targeting the development and effector functions of TH17 cells. Semin. Immunol. 2007, 19, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef]
- Mills, K.H.G. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immuno. 2023, 23, 38–54. [Google Scholar] [CrossRef]
- Trtica Majnarić, L.; Guljaš, S.; Bosnić, Z.; Šerić, V.; Wittlinger, T. Neutrophil-to-Lymphocyte Ratio as a Cardiovascular Risk Marker May Be Less Efficient in Women Than in Men. Biomolecules 2021, 11, 528. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 2013, 25, 305–312. [Google Scholar] [CrossRef]
- Mills, C.D. Anatomy of a discovery: M1 and M2 macrophages. Front. Immunol. 2015, 6, 212. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. The spectrum of inflammatory responses. Science 2021, 374, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Rankin, L.C.; Artis, D. Beyond Host Defense: Emerging Functions of the Immune System in Regulating Complex Tissue Physiology. Cell 2018, 173, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation-Nature’s Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing “the Epidemic” of Chronic Diseases. Front. Med. 2018, 5, 316. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science 2012, 335, 936–941. [Google Scholar] [CrossRef]
- Fava, G.A.; McEwen, B.S.; Guidi, J.; Gostoli, S.; Offidani, E.; Sonino, N. Clinical characterization of allostatic overload. Psychoneuroendocrinology 2019, 108, 94–101. [Google Scholar] [CrossRef]
- Poletaev, A.; Boura, P. The immune system, natural autoantibodies and general homeostasis in health and disease. Hippokratia 2011, 15, 295–298. [Google Scholar]
- Poletaev, A.B.; Stepanyuk, V.L.; Gershwin, M.E. Integrating immunity: The immunculus and self-reactivity. J. Autoimmun. 2008, 30, 68–73. [Google Scholar] [CrossRef]
- Franceschi, C.; Valensin, S.; Bonafè, M.; Paolisso, G.; Yashin, A.I.; Monti, D.; De Benedictis, G. The network and the remodeling theories of aging: Historical background and new perspectives. Exp. Gerontol. 2000, 35, 879–896. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, Z.; Haj, T.; Kessel, A.; Toubi, E. Age-related autoimmunity. BMC Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.R. Activation of benign autoimmunity as both tumor and autoimmune disease immunotherapy: A comprehensive review. J. Autoimmun. 2014, 54, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Salvioli, S.; Garagnani, P.; de Eguileor, M.; Monti, D.; Capri, M. Immunobiography and the Heterogeneity of Immune Responses in the Elderly: A Focus on Inflammaging and Trained Immunity. Front. Immunol. 2017, 8, 982. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Cutolo, M.; Buttgereit, F.; Pongratz, G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J. Intern. Med. 2010, 267, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]
- Andolfi, C.; Fisichella, P.M. Epidemiology of obesity and associated comorbidities. J. Laparoendosc. Adv. Surg. Tech. A 2018, 28, 919–924. [Google Scholar] [CrossRef]
- Tsatsoulis, A.; Paschou, S.A. Metabolically Healthy Obesity: Criteria, Epidemiology, Controversies, and Consequences. Curr. Obes. Rep. 2020, 9, 109–120. [Google Scholar] [CrossRef]
- Nedunchezhiyan, U.; Varughese, I.; Sun, A.R.; Wu, X.; Crawford, R.; Prasadam, I. Obesity, Inflammation, and Immune System in Osteoarthritis. Front. Immunol. 2022, 13, 907750. [Google Scholar] [CrossRef]
- Machado, S.A.; Pasquarelli-do-Nascimento, G.; da Silva, D.; Farias, G.R.; de Oliveira Santos, I.; Baptista, L.B.; Magalhães, K.G. Browning of the white adipose tissue regulation: New insights into nutritional and metabolic relevance in health and diseases. Nutr. Metab. 2022, 19, 61. [Google Scholar] [CrossRef]
- Razeghian-Jahromi, I.; Karimi Akhormeh, A.; Razmkhah, M.; Zibaeenezhad, M.J. Immune system and atherosclerosis: Hostile or friendly relationship. Int. J. Immunopathol. Pharmacol. 2022, 36, 3946320221092188. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Mulvagh, S.L.; Merz, C.N.; Buring, J.E.; Manson, J.E. Cardiovascular Disease in Women: Clinical Perspectives. Circ. Res. 2016, 118, 1273–1293. [Google Scholar] [CrossRef] [PubMed]
- Vijay, A.; Valdes, A.M. Role of the gut microbiome in chronic diseases: A narrative review. Eur. J. Clin. Nutr. 2022, 76, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Z.; Zhu, L.B.; Li, Z.R.; Lin, J. Bacterial colonization and intestinal mucosal barrier development. World J. Clin. Pediatr. 2013, 2, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Are we really vastly outnumbered? revisiting the ratio of bacterial to host cells in humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Conroy, M.E.; Shi, H.N.; Walker, W.A. The long-term health effects of neonatal microbial flora. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 197–201. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Hrncir, T. Gut Microbiota Dysbiosis: Triggers, Consequences, Diagnostic and Therapeutic Options. Microorganisms 2022, 10, 578. [Google Scholar] [CrossRef]
- Schlechte, J.; Skalosky, I.; Geuking, M.B.; McDonald, B. Long-distance relationships—Regulation of systemic host defense against infections by the gut microbiota. Mucosal Immunol. 2022, 15, 809–818. [Google Scholar] [CrossRef]
- Yang, W.; Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell Mol. Immunol. 2021, 18, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Kenney, M.J.; Ganta, C.K. Autonomic nervous system and immune system interactions. Compr. Physiol. 2014, 4, 1177–1200. [Google Scholar] [CrossRef] [PubMed]
- Daniel, N.; Lécuyer, E.; Chassaing, B. Host/microbiota interactions in health and diseases—Time for mucosal microbiology! Mucosal Immunol. 2021, 14, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- McGhee, J.R.; Fujihashi, K. Inside the mucosal immune system. PLoS Biol. 2012, 10, e1001397. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, D.B.; Fagarasan, S. IgA synthesis: A form of functional immune adaptation extending beyond gut. Curr. Opin. Immunol. 2012, 24, 261–268. [Google Scholar] [CrossRef]
- Stebegg, M.; Kumar, S.D.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9, 2469. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Mu, C.L.; Farzi, A.; Zhu, W.Y. Tryptophan Metabolism: A Link between the Gut Microbiota and Brain. Adv. Nutr. 2020, 11, 709–723. [Google Scholar] [CrossRef]
- Volarić, M.; Šojat, D.; Majnarić, L.T.; Vučić, D. The Association between Functional Dyspepsia and Metabolic Syndrome—The State of the Art. Int. J. Environ. Res. Public Health 2024, 21, 237. [Google Scholar] [CrossRef]
- Burns, G.; Pryor, J.; Holtmann, G.; Walker, M.M.; Talley, N.J.; Keely, S. Immune Activation in Functional Gastrointestinal Disorders. Gastroenterol. Hepatol. 2019, 15, 539–548. [Google Scholar]
- Sperber, A.D.; Bangdiwala, S.I.; Drossman, D.A.; Ghoshal, U.C.; Simren, M.; Tack, J.; Whitehead, W.E.; Dumitrascu, D.L.; Fang, X.; Fukudo, S.; et al. Worldwide Prevalence and Burden of Functional Gastrointestinal Disorders, Results of Rome Foundation Global Study. Gastroenterology 2021, 160, 99–114.e3. [Google Scholar] [CrossRef]
- Van den Houte, K.; Carbone, F.; Goelen, N.; Schol, J.; Masuy, I.; Arts, J.; Caenepeel, P.; Staessen, D.; Vergauwe, P.; Van Roey, G.; et al. Effects of Rome IV Definitions of Functional Dyspepsia Subgroups in Secondary Care. Clin. Gastroenterol. Hepatol. 2021, 19, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Hari, S.; Burns, G.L.; Hoedt, E.C.; Keely, S.; Talley, N.J. Eosinophils, Hypoxia-Inducible Factors, and Barrier Dysfunction in Functional Dyspepsia. Front. Allergy 2022, 3, 851482. [Google Scholar] [CrossRef] [PubMed]
- Wouters, M.M.; Vicario, M.; Santos, J. The role of mast cells in functional GI disorders. Gut 2016, 65, 155–168. [Google Scholar] [CrossRef]
- Black, C.J.; Drossman, D.A.; Talley, N.J.; Ruddy, J.; Ford, A.C. Functional gastrointestinal disorders: Advances in understanding and management. Lancet 2020, 396, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T. Functional Dyspepsia: Current Understanding and Future Perspective. Digestion 2024, 105, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Burns, G.; Carroll, G.; Mathe, A.; Horvat, J.; Foster, P.; Walker, M.M.; Talley, N.J.; Keely, S. Evidence for Local and Systemic Immune Activation in Functional Dyspepsia and the Irritable Bowel Syndrome: A Systematic Review. Am. J. Gastroenterol. 2019, 114, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Carco, C.; Young, W.; Gearry, R.B.; Talley, N.J.; McNabb, W.C.; Roy, N.C. Increasing Evidence That Irritable Bowel Syndrome and Functional Gastrointestinal Disorders Have a Microbial Pathogenesis. Front. Cell Infect. Microbiol. 2020, 10, 468. [Google Scholar] [CrossRef]
- Moloney, R.D.; Johnson, A.C.; O’Mahony, S.M.; Dinan, T.G.; Greenwood-Van Meerveld, B.; Cryan, J.F. Stress and the Microbiota-Gut-Brain Axis in Visceral Pain: Relevance to Irritable Bowel Syndrome. CNS Neurosci. Ther. 2016, 22, 102–117. [Google Scholar] [CrossRef]
- Ng, Q.X.; Soh, A.Y.S.; Loke, W.; Lim, D.Y.; Yeo, W.S. The role of inflammation in irritable bowel syndrome (IBS). J. Inflamm. Res. 2018, 11, 345–349. [Google Scholar] [CrossRef]
- Lazaridis, N.; Germanidis, G. Current insights into the innate immune system dysfunction in irritable bowel syndrome. Ann Gastroenterol. 2018, 31, 171–187. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Ivashkin, V.; Poluektov, Y.; Kogan, E.; Shifrin, O.; Sheptulin, A.; Kovaleva, A.; Kurbatova, A.; Krasnov, G.; Poluektova, E. Disruption of the pro-inflammatory, anti-inflammatory cytokines and tight junction proteins expression, associated with changes of the composition of the gut microbiota in patients with irritable bowel syndrome. PLoS ONE 2021, 16, e0252930. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Zogg, H.; Wei, L.; Bartlett, A.; Ghoshal, U.C.; Rajender, S.; Ro, S. Gut Microbial Dysbiosis in the Pathogenesis of Gastrointestinal Dysmotility and Metabolic Disorders. J. Neurogastroenterol. Motil. 2021, 27, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hou, C.; Xiao, W.; Qiu, Y. The role of mechanosensitive ion channels in the gastrointestinal tract. Front. Physiol. 2022, 13, 904203. [Google Scholar] [CrossRef] [PubMed]
- Maqoud, F.; Tricarico, D.; Mallamaci, R.; Orlando, A.; Russo, F. The Role of Ion Channels in Functional Gastrointestinal Dis-orders (FGID): Evidence of Channelopathies and Potential Avenues for Future Research and Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 11074. [Google Scholar] [CrossRef] [PubMed]
- Berumen, A.; Edwinson, A.L.; Grover, M. Post-infection Irritable Bowel Syndrome. Gastroenterol. Clin. North. Am. 2021, 50, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Klem, F.; Wadhwa, A.; Prokop, L.J.; Sundt, W.J.; Farrugia, G.; Camilleri, M.; Singh, S.; Grover, M. Prevalence, Risk Factors, and Outcomes of Irritable Bowel Syndrome After Infectious Enteritis: A Systematic Review and Meta-analysis. Gastroenterology 2017, 152, 1042–1054.e1. [Google Scholar] [CrossRef]
- Barman, M.; Unold, D.; Shifley, K.; Amir, E.; Hung, K.; Bos, N.; Salzman, N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect. Immun. 2008, 76, 907–915. [Google Scholar] [CrossRef]
- Jalanka, J.; Salonen, A.; Fuentes, S.; de Vos, W.M. Microbial signatures in post-infectious irritable bowel syndrome--toward patient stratification for improved diagnostics and treatment. Gut Microbes. 2015, 6, 364–369. [Google Scholar] [CrossRef]
- Wang, C.; Fang, X. Inflammation and Overlap of Irritable Bowel Syndrome and Functional Dyspepsia. J. Neurogastroenterol. Motil. 2021, 27, 153–164. [Google Scholar] [CrossRef]
- Lupu, V.V.; Ghiciuc, C.M.; Stefanescu, G.; Mihai, C.M.; Popp, A.; Sasaran, M.O.; Bozomitu, L.; Starcea, I.M.; Adam Raileanu, A.; Lupu, A. Emerging role of the gut microbiome in post-infectious irritable bowel syndrome: A literature review. World J. Gastroenterol. 2023, 29, 3241–3256. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, A.; Biglari, M.; Nasseri Moghaddam, S. Post-infectious Irritable Bowel Syndrome: A Narrative Review. Middle East. J. Dig. Dis. 2019, 11, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Y.; Deng, Z. Imbalanced shift of cytokine expression between T helper 1 and T helper 2 (Th1/Th2) in intestinal mucosa of patients with post-infectious irritable bowel syndrome. BMC Gastroenterol. 2012, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- De Winter, B.Y.; van den Wijngaard, R.M.; de Jonge, W.J. Intestinal mast cells in gut inflammation and motility disturbances. Biochim. Biophys. Acta. 2012, 1822, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.W.; Ma, Z.C.; Fu, J.; Huang, B.L.; Liu, F.J.; Sun, D.; Lan, C. Upregulated adenosine 2A receptor accelerates post-infectious irritable bowel syndrome by promoting CD4+ T cells’ T helper 17 polarization. World J. Gastroenterol. 2022, 28, 2955–2967. [Google Scholar] [CrossRef] [PubMed]
- Dellon, E.S.; Kim, H.P.; Sperry, S.L.; Rybnicek, D.A.; Woosley, J.T.; Shaheen, N.J. A phenotypic analysis shows that eosinophilic esophagitis is a progressive fibrostenotic disease. Gastrointest. Endosc. 2014, 79, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Racca, F.; Pellegatta, G.; Cataldo, G.; Vespa, E.; Carlani, E.; Pelaia, C.; Paoletti, G.; Messina, M.R.; Nappi, E.; Canonica, G.W.; et al. Type 2 Inflammation in Eosinophilic Esophagitis: From Pathophysiology to Therapeutic Targets. Front. Physiol. 2022, 12, 815842. [Google Scholar] [CrossRef]
- Khokhar, D.; Marella, S.; Idelman, G.; Chang, J.W.; Chehade, M.; Hogan, S.P. Eosinophilic esophagitis: Immune mechanisms and therapeutic targets. Clin. Exp. Allergy 2022, 52, 1142–1156. [Google Scholar] [CrossRef]
- Arias, Á.; Lucendo, A.J. Molecular basis and cellular mechanisms of eosinophilic esophagitis for the clinical practice. Expert. Rev. Gastroenterol. Hepatol. 2019, 13, 99–117. [Google Scholar] [CrossRef]
- Wąsik, J.; Małecka-Wojciesko, E. Eosinophilic Esophagitis—What Do We Know So Far? J. Clin. Med. 2023, 12, 2259. [Google Scholar] [CrossRef]
- O’Shea, K.M.; Aceves, S.S.; Dellon, E.S.; Gupta, S.K.; Spergel, J.M.; Furuta, G.T.; Rothenberg, M.E. Pathophysiology of Eosinophilic Esophagitis. Gastroenterol. 2018, 154, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.A.M.; Akhter, S.; Khan, M.R.; Saha, M.; Roy, P.K. Association of duodenal eosinophilia with Helicobacter pylori-negative functional dyspepsia. Arab. J. Gastroenterol. 2020, 21, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.P.; Holck, S.; Janulaityte-Günther, D.; Kupcinskas, L.; Kiudelis, G.; Jonaitis, L.; Janciauskas, D.; Holck, P.; Bennedsen, M.; Permin, H.; et al. Gastric inflammatory markers and interleukins in patients with functional dyspepsia, with and without Helicobacter pylori infection. FEMS Immunol. Med. Microbiol. 2005, 44, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Fairlie, T.; Brown, G.; Jones, M.P.; Eslick, G.D.; Duncanson, K.; Thapar, N.; Keely, S.; Koloski, N.; Shahi, M.; et al. Duodenal Eosinophils and Mast Cells in Functional Dyspepsia: A Systematic Review and Meta-Analysis of Case-Control Studies. Clin. Gastroenterol. Hepatol. 2022, 20, 2229–2242.e29. [Google Scholar] [CrossRef] [PubMed]
- Eladham, M.W.; Selvakumar, B.; Saheb Sharif-Askari, N.; Saheb Sharif-Askari, F.; Ibrahim, S.M.; Halwani, R. Unraveling the gut-Lung axis: Exploring complex mechanisms in disease interplay. Heliyon 2024, 10, e24032. [Google Scholar] [CrossRef]
- Kuruvilla, M.E.; Lee, F.E.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin Rev Allergy Immunol. 2019, 56, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, M.; Jacobs, I.; Wauters, L.; Vanuytsel, T. Immune Activation in Functional Dyspepsia: Bystander Becoming the Suspect. Front. Neurosci. 2022, 16, 831761. [Google Scholar] [CrossRef]
- Bennet, S.M.; Polster, A.; Törnblom, H.; Isaksson, S.; Capronnier, S.; Tessier, A.; Le Nevé, B.; Simrén, M.; Öhman, L. Global Cytokine Profiles and Association With Clinical Characteristics in Patients with Irritable Bowel Syndrome. Am. J. Gastroenterol. 2016, 111, 1165–1176. [Google Scholar] [CrossRef]
- Goral, V.; Kucukoner, M.; Buyukbayram, H. Mast cells count and serum cytokine levels in patients with irritable bowel syndrome. Hepato-Gastroenterol. 2010, 57, 751–754. [Google Scholar]
- Seyedmirzaee, S.; Hayatbakhsh, M.M.; Ahmadi, B.; Baniasadi, N.; Bagheri Rafsanjani, A.M.; Nikpoor, A.R.; Mohammadi, M. Serum immune biomarkers in irritable bowel syndrome. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 631–637. [Google Scholar] [CrossRef]
- Knowles, S.R.; Skvarc, D.; Ford, A.C.; Palsson, O.S.; Bangdiwala, S.I.; Sperber, A.D.; Mikocka-Walus, A. Negative Impact of Disorders of Gut-Brain Interaction on Health-Related Quality of Life: Results From the Rome Foundation Global Epidemiology Survey. Gastroenterology 2023, 164, 655–658.e10. [Google Scholar] [CrossRef] [PubMed]
- Tak, L.M.; Rosmalen, J.G. Dysfunction of stress responsive systems as a risk factor for functional somatic syndromes. J. Psychosom. Res. 2010, 68, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Skoluda, N.; Ali, N.; Nater, U.M.; Mewes, R. Hair cortisol levels in women with medically unexplained symptoms. J. Psychiatr. Res. 2022, 146, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Sartor, M.L.; Scott, F.; Cleare, A.J. Inflammatory proteins are altered in chronic fatigue syndrome-A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2019, 107, 69–83. [Google Scholar] [CrossRef]
- Bashashati, M.; Moradi, M.; Sarosiek, I. Interleukin-6 in irritable bowel syndrome: A systematic review and meta-analysis of IL-6 (-G174C) and circulating IL-6 levels. Cytokine 2017, 99, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Kamp, K.J.; Han, C.; Shulman, R.J.; Cain, K.C.; Barney, P.; Opp, M.R.; Chang, L.; Burr, R.L.; Heitkemper, M.M. Cytokine Levels and Symptoms Among Women with Irritable Bowel Syndrome: Considering the Role of Hormonal Contraceptive Use. Biol. Res. Nurs. 2021, 23, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Finlay, S.; Rudd, D.; McDermott, B.; Sarnyai, Z. Allostatic load and systemic comorbidities in psychiatric disorders. Psychoneuroendocrinology 2022, 140, 105726. [Google Scholar] [CrossRef] [PubMed]
- Mostafaei, S.; Kabir, K.; Kazemnejad, A.; Feizi, A.; Mansourian, M.; Hassanzadeh Keshteli, A.; Afshar, H.; Arzaghi, S.M.; Rasekhi Dehkordi, S.; Adibi, P.; et al. Explanation of somatic symptoms by mental health and personality traits: Application of Bayesian regularized quantile regression in a large population study. BMC Psychiatry 2019, 19, 207. [Google Scholar] [CrossRef]
- Brouillet, J.Z.; Boltri, M.; Lengvenyte, A.; Lajnef, M.; Richard, J.R.; Barrau, C.; Strumila, R.; Coyac, M.; Wu, C.L.; Boukouaci, W.; et al. Association of markers of inflammation and intestinal permeability in suicidal patients with major mood disorders. J. Affect. Disord. Rep. 2023, 14, 100624. [Google Scholar] [CrossRef]
- Murni, A.W.; Darwin, E.; Zubir, N.; Nurdin, A.E. Analyzing Determinant Factors for Pathophysiology of Functional Dyspepsia Based on Plasma Cortisol Levels, IL-6 and IL-8 Expressions and H. pylori Activity. Acta Medica Indones 2018, 50, 38–45. [Google Scholar]
- Konturek, P.C.; Brzozowski, T.; Konturek, S.J. Stress and the gut: Pathophysiology, clinical consequences, diagnostic approach, and treatment options. J. Physiol. Pharmacol. 2011, 62, 591–599. [Google Scholar] [PubMed]
- Jacobs, I.; Ceulemans, M.; Wauters, L.; Breynaert, C.; Vermeire, S.; Verstockt, B.; Vanuytsel, T. Role of Eosinophils in Intestinal Inflammation and Fibrosis in Inflammatory Bowel Disease: An Overlooked Villain? Front. Immunol. 2021, 12, 754413. [Google Scholar] [CrossRef] [PubMed]
- Wauters, L.; Burns, G.; Ceulemans, M.; Walker, M.M.; Vanuytsel, T.; Keely, S.; Talley, N.J. Duodenal inflammation: An emerging target for functional dyspepsia? Expert. Opin. Ther. Targets 2020, 24, 511–523. [Google Scholar] [CrossRef]
- Robida, P.A.; Puzzovio, P.G.; Pahima, H.; Levi-Schaffer, F.; Bochner, B.S. Human eosinophils and mast cells: Birds of a feather flock together. Immunol. Rev. 2018, 282, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Nishida, A. Alteration of the Gut Microbiome in Inflammatory Bowel Disease. Digestion 2023, 104, 16–23. [Google Scholar] [CrossRef]
- Pittayanon, R.; Lau, J.T.; Yuan, Y.; Leontiadis, G.I.; Tse, F.; Surette, M.; Moayyedi, P. Gut Microbiota in Patients With Irritable Bowel Syndrome-A Systematic Review. Gastroenterology 2019, 157, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Hermes, G.D.A.; Reijnders, D.; Kootte, R.S.; Goossens, G.H.; Smidt, H.; Nieuwdorp, M.; Blaak, E.E.; Zoetendal, E.G. Individual and cohort-specific gut microbiota patterns associated with tissue-specific insulin sensitivity in overweight and obese males. Sci. Rep. 2020, 10, 7523. [Google Scholar] [CrossRef]
- Wang, T.; Rijnaarts, I.; Hermes, G.D.A.; de Roos, N.M.; Witteman, B.J.M.; de Wit, N.J.W.; Govers, C.; Smidt, H.; Zoetendal, E.G. Fecal Microbiota Signatures Are Not Consistently Related to Symptom Severity in Irritable Bowel Syndrome. Dig. Dis. Sci. 2022, 67, 5137–5148. [Google Scholar] [CrossRef]
- Matenchuk, B.A.; Mandhane, P.J.; Kozyrskyj, A.L. Sleep, circadian rhythm, and gut microbiota. Sleep Med. Rev. 2020, 53, 101340. [Google Scholar] [CrossRef]
- Powell, N.; Walker, M.M.; Talley, N.J. Gastrointestinal eosinophils in health, disease and functional disorders. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 146–156. [Google Scholar] [CrossRef]
- Cinicola, B.L.; Pulvirenti, F.; Capponi, M.; Bonetti, M.; Brindisi, G.; Gori, A.; De Castro, G.; Anania, C.; Duse, M.; Zicari, A.M. Selective IgA Deficiency and Allergy: A Fresh Look to an Old Story. Medicina 2022, 58, 129. [Google Scholar] [CrossRef]
- Odineal, D.D.; Gershwin, M.E. The epidemiology and clinical manifestations of autoimmunity in selective IgA deficiency. Clin. Rev. Allergy Immunol. 2020, 58, 107–133. [Google Scholar] [CrossRef]
- Liefferinckx, C.; De Grève, Z.; Toubeau, J.F.; Perée, H.; Quertinmont, E.; Tafciu, V.; Minsart, C.; Rahmouni, S.; Georges, M.; Vallée, F.; et al. New approach to determine the healthy immune variations by combining clustering methods. Sci. Rep. 2021, 11, 8917. [Google Scholar] [CrossRef]
- Vanuytsel, T.; Bercik, P.; Boeckxstaens, G. Understanding neuroimmune interactions in disorders of gut-brain interaction: From functional to immune-mediated disorders. Gut 2023, 72, 787–798. [Google Scholar] [CrossRef]
- Sabatino, A.; Regolisti, G.; Brusasco, I.; Cabassi, A.; Morabito, S.; Fiaccadori, E. Alterations of intestinal barrier and microbiota in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 924–933. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
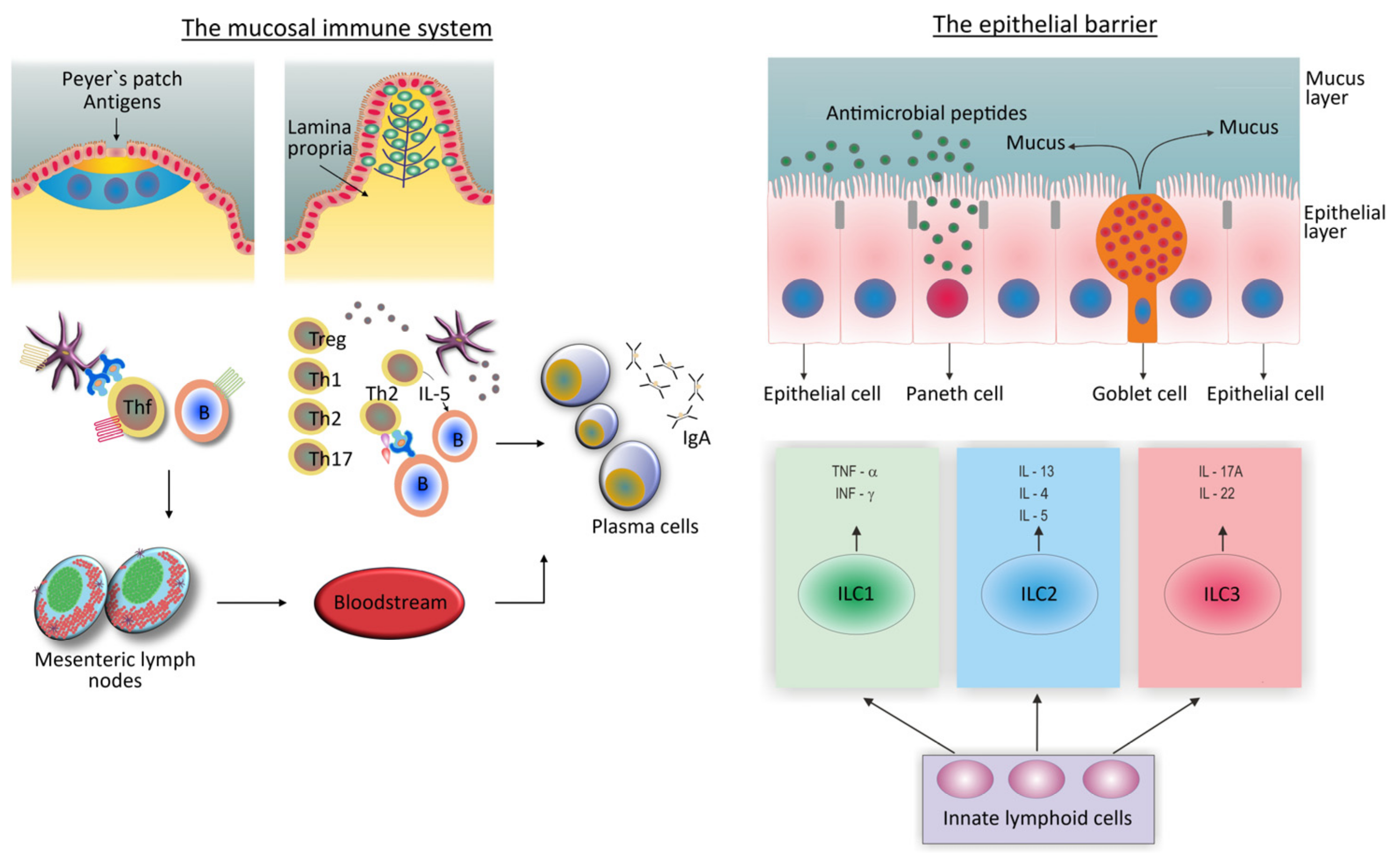{kind=link}
{kind=link}
| Post-Infection Bowel Syndrome |
|---|
| Mast cells, eosinophils, T lymphocytes and macrophages release inflammatory factors (TNF-α, IFN-γ, IL-1, 6, and 8), as well as histamine, leukotriene, and 5-HT, leading to increased intestinal permeability via the reorganization of proteins associated with tight junctions and visceral hypersensitivity. The molecule 5-HT is a crucial neurotransmitter that has a major impact on the functioning of the brain–gut axis, and it is released by ECs. Pain perception brought on by colon distention may be amplified by a long-term high concentration of 5-HT binding to the 5-HT3 receptors on the nociceptive neurons of the vagus in the colorectal mucosa [90,91,92]. |
| Th1 and Th2 cytokine expression varies in the intestinal mucosa, suggesting that the PI-IBS was caused by an immune dysregulation mechanism. Th1-derived cytokine expression (IFN-γ) is increased while Th2-derived cytokine expression (IL-10) is decreased, indicating a shift towards Th1 immunodominance which may lead to chronic low-grade inflammation [93,94]. |
| T helper 17 (Th17) polarizations are observed in IBS, and adenosine and its receptors are involved in inflammation by promoting the Th17 polarization of CD4+ T cells [95]. |
| Eosinophilic Esophagitis |
| EoE is characterized by extensive eosinophilic inflammation causing esophageal-function abnormalities [96]. |
| Epithelial cells and DCs release cytokines (IL-25 and IL-33), and thymic stromal lymphoprotein (TSLP) leading to the activation of invariant natural killer T (iNKT) cells, adaptive CD4+ effector memory Th2 cells, and ILC2. The predominant reaction is the Th2 immune response, secreting IL-4, IL-5, IL-13, IL-15, eotaxin-3, and periostin [97,98]. |
| IL-5 causes eosinophils to multiply and extend from the bone marrow to all layers of the esophagus. They then degranulate, triggering the release of various molecules linked to the remodeling of tissue [99]. |
| Eotaxin-3 is overexpressed causing esophageal mucosal inflammation as a result of environmental antigens.Mast cells can potentially regulate the disease—number of mast cells has been shown to be large even in clinical remission [100]. |
| TGF-β causes mucosal remodeling and smooth muscle dysfunction [101]. |
| Inflammation in FD |
|---|
| Levels of systemic cytokines and eosinophil infiltration in patients with FD, especially those with PDS, are increased. The eosinophil–mast cell axis secretes chemical mediators influencing visceral hypersensitivity and gastrointestinal motility and leading to neuromuscular and epithelial dysfunction [102,103]. |
| Eosinophil infiltration causes low-grade inflammation in up to 40% of FD patients. When cells degranulate, symptoms occur, along with impaired mucosal integrity and structural and neuronal abnormalities [104]. |
| Unregulated or disrupted activation of mast cells can interfere with gut homeostasis, causing tissue dysfunction, and increasing inflammation [73]. |
| Studies have shown increased levels of TNF-α, IL-1β, and IL-6, all of which are associated with the Th17 pathway. The proliferation and inflammatory activity of Th17 and ILC2 populations may reduce Treg and innate lyphoide type 3 cell (ILC3) populations if homeostasis is disrupted in FGID patients [69]. |
| The observational studies confirm a close association between FD and asthma. This association is especially reasonable when asthma is viewed as an umbrella diagnosis for several diseases with distinct and interrelating inflammatory pathways [87,88]. |
| Contents flow through the mucosa due to enhanced duodenal mucosal permeability. The immune cells causing low-grade mucosal inflammation are the ones that identify them. The enhanced permeability of the duodenal mucosa and weakened epithelial barrier are the results of inflammatory cells’ release of histamine, tryptase, and cytokines, which modify submucosal afferent neurons [75,105,106,107]. |
| Inflammation in IBS |
|---|
| Higher serum levels of TNF-α and IL-17 were found to be negatively connected with quality-of-life scores and to be correlated with IBS patients’ discomfort and severity of symptoms [80]. |
| Some studies found no relationship between the severity of the overall symptoms and the expression of the cytokines. IBS patients had higher serum levels of the pro-inflammatory cytokines IL-6 and IL-8 and lower serum levels of the anti-inflammatory IL-10. The correlation between certain clinical symptoms and inflammatory cytokines implies that immunological activation might be significant for diarrhea-predominant IBS patients [108,109,110]. |
| Studies reveal higher numbers and volume of mast cells in IBS patients compared with healthy controls (mast cells counts and density vary among studies and within different segments of the intestine). Mast cells’ mediators modify enteric nerve and motor function, and they have a role in the pathophysiology of IBS. DCs (elevated in some patients with IBS) also contribute to the pathophysiology by inducing visceral hypersensitivity, activating the microcirculation, and prolonging intestinal activity. They can release corticotropin-releasing factor (CRF), which causes changes in visceral hypersensitivity and intestinal motility [80]. |
| Neuroinflammation plays a role in the pathogenesis of IBS through the “gut–brain” axis, altering neuroendocrine pathways and glucocorticoid receptor genes, resulting in a generalized pro-inflammatory phenotype [79]. |
| Pro-inflammatory bacterial species such Enterobacteriaceae are more prevalent than Bifidobacterium and Lactobacillus. The Firmicutes/Bacteroidetes ratio has been changed. These changes can cause a dissolving of mucosal glycoproteins in the intestinal barrier, causing leakage and creating favorable conditions for prolonging the inflammatory state [82]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šojat, D.; Volarić, M.; Keškić, T.; Volarić, N.; Cerovečki, V.; Trtica Majnarić, L. Putting Functional Gastrointestinal Disorders within the Spectrum of Inflammatory Disorders Can Improve Classification and Diagnostics of These Disorders. Biomedicines 2024, 12, 702. https://doi.org/10.3390/biomedicines12030702
Šojat D, Volarić M, Keškić T, Volarić N, Cerovečki V, Trtica Majnarić L. Putting Functional Gastrointestinal Disorders within the Spectrum of Inflammatory Disorders Can Improve Classification and Diagnostics of These Disorders. Biomedicines. 2024; 12(3):702. https://doi.org/10.3390/biomedicines12030702
Chicago/Turabian StyleŠojat, Dunja, Mile Volarić, Tanja Keškić, Nikola Volarić, Venija Cerovečki, and Ljiljana Trtica Majnarić. 2024. "Putting Functional Gastrointestinal Disorders within the Spectrum of Inflammatory Disorders Can Improve Classification and Diagnostics of These Disorders" Biomedicines 12, no. 3: 702. https://doi.org/10.3390/biomedicines12030702