
Repurposing of the Cardiovascular Drug Statin for the Treatment of Cancers: Efficacy of Statin–Dipyridamole Combination Treatment in Melanoma Cell Lines
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Morphological Analysis by Optical and Fluorescence Microscopy
2.4. Cell Proliferation Assay
2.5. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.6. Western Blotting
2.7. SEM Sample Preparation and Imaging
2.8. Statistical Analysis
3. Results
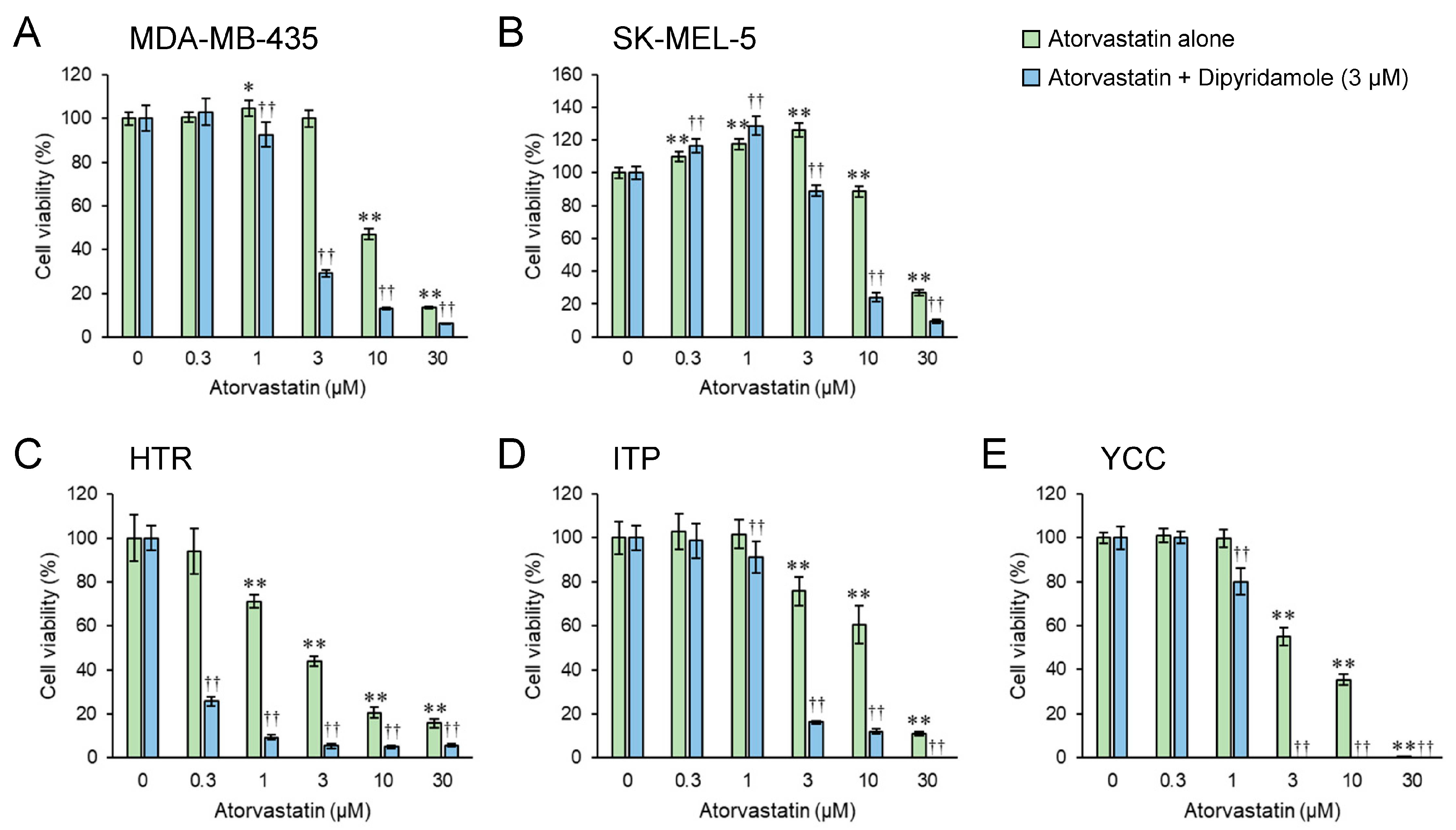
3.1. Dipyridamole Co-Treatment Augments the Cytostatic Effect of Atorvastatin in Both Human and Canine Melanoma Cell Lines
3.2. Half-Maximal Inhibitory Concentration (IC50) Value of Dipyridamole Exceeds 16.85 μM
3.3. Combined Atorvastatin and Dipyridamole Treatment Enhances the Cytostatic Effect in Both Human and Canine Melanoma Cell Lines Compared with Atorvastatin Treatment Alone
3.4. Statin and/or Dipyridamole Affect Filopodia and Lamellipodia Formation
3.5. Atorvastatin Upregulates HMGCR mRNA Expression in a Dose-Dependent Manner While Dipyridamole Tends to Downregulate It
3.6. Dipyridamole Augments Atorvastatin’s Anticancer Effect, Which Correlates with Its Attenuation of Statin-Induced Increase in HMGCR Expression
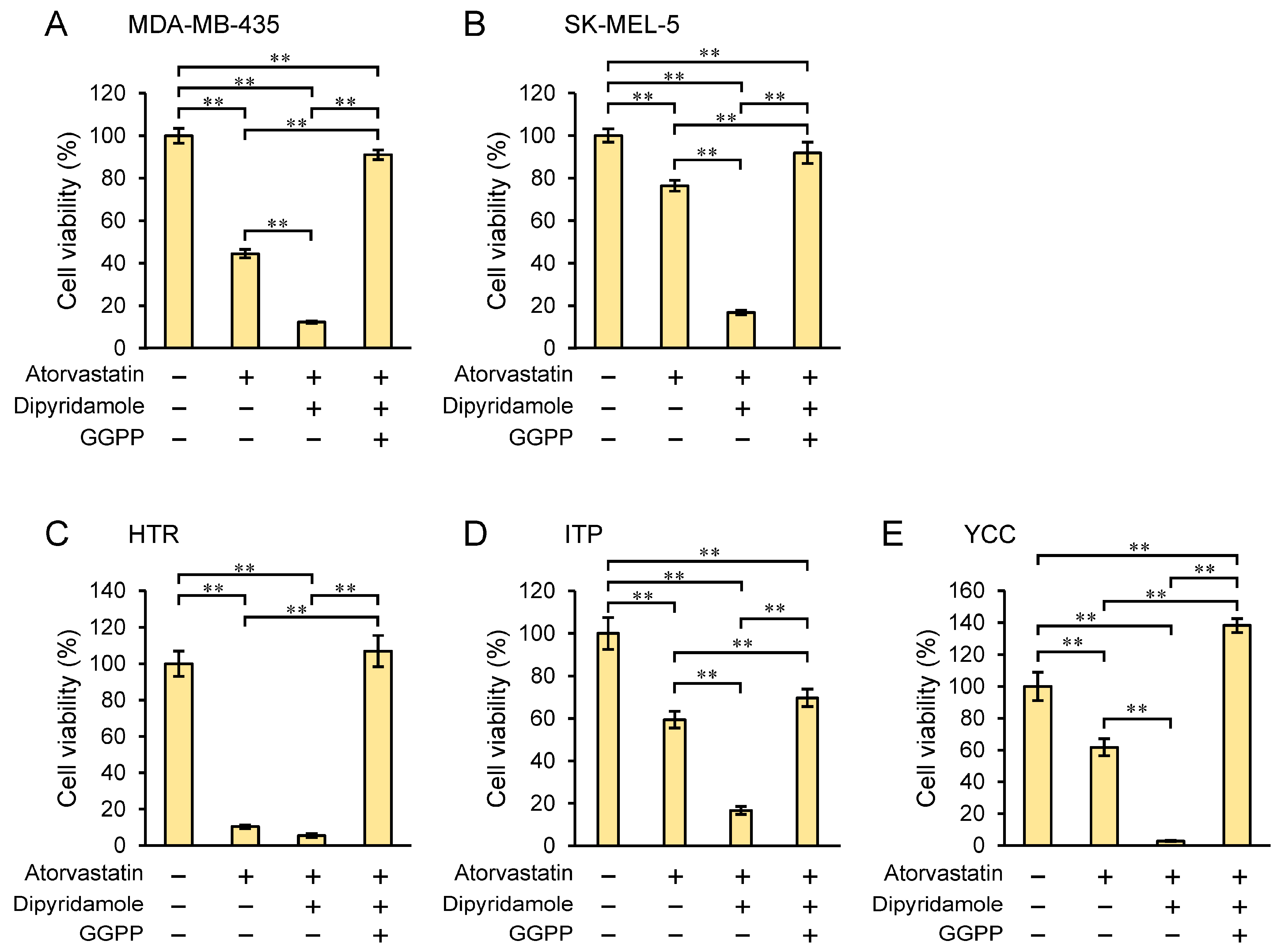
3.7. Geranylgeranyl Pyrophosphate (GGPP) Rescues Cell Proliferation Inhibited by Dipyridamole Combination with Atorvastatin
3.8. Vemurafenib Augments the Combined Inhibitory Effect of Statin and Dipyridamole
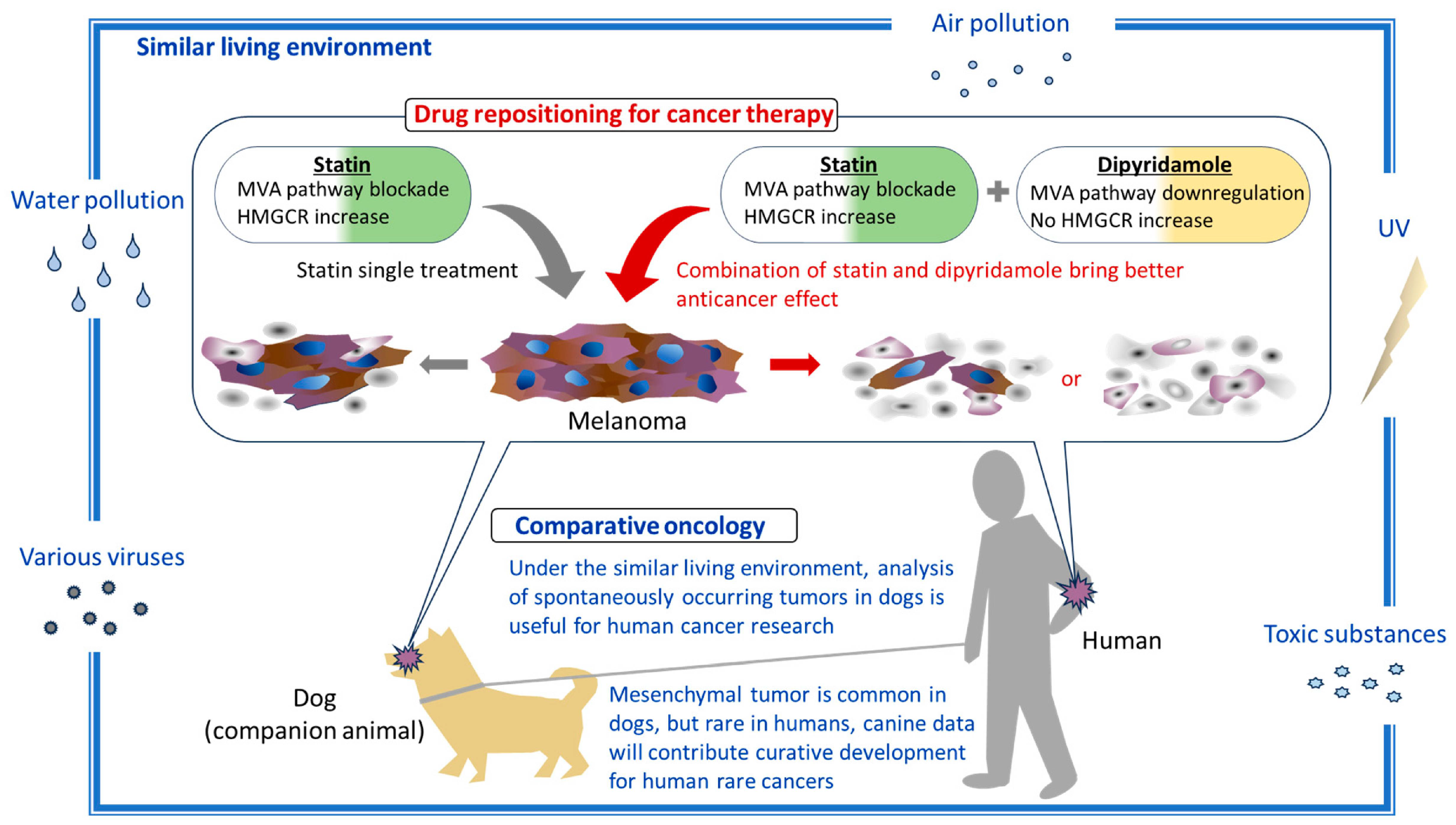
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma treatment in review. ImmunoTargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef]
- Ward-Peterson, M.; Acuña, J.M.; Alkhalifah, M.K.; Nasiri, A.M.; Al-Akeel, E.S.; Alkhaldi, T.M.; Dawari, S.A.; Aldaham, S.A. Association between race/ethnicity and survival of melanoma patients in the united states over 3 decades: A secondary analysis of SEER data. Medicine 2016, 95, e3315. [Google Scholar] [CrossRef]
- Yamazaki, N.; Kiyohara, Y.; Uhara, H.; Tsuchida, T.; Yoshida, A.; Yamada, T.; Komoto, A. Postmarketing surveillance of nivolumab plus ipilimumab combination therapy in Japanese patients with unresectable malignant melanoma. J. Dermatol. 2023, 50, 1108–1120. [Google Scholar] [CrossRef]
- Dimitriou, F.; Frauchiger, A.L.; Urosevic-Maiwald, M.; Naegeli, M.C.; Goldinger, S.M.; Barysch, M.; Franzen, D.; Kamarachev, J.; Braun, R.; Dummer, R.; et al. Sarcoid-like reactions in patients receiving modern melanoma treatment. Melanoma Res. 2018, 28, 230–236. [Google Scholar] [CrossRef]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B 2010, 86, 484–493. [Google Scholar] [CrossRef]
- Surma, S.; Mikhailidis, D.P.; Banach, M. Celebrating the 90th birthday of the scientist who discovered statins: Akira Endō. Eur. Hear. J. 2024, 45, 647–650. [Google Scholar] [CrossRef]
- Jiang, W.; Hu, J.-W.; He, X.-R.; Jin, W.-L.; He, X.-Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Longo, J.; van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as anticancer agents in the era of precision medicine. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef]
- Amin, F.; Fathi, F.; Reiner, Ž.; Banach, M.; Sahebkar, A. The role of statins in lung cancer. Arch. Med. Sci. 2022, 18, 141–152. [Google Scholar] [CrossRef]
- Lashgari, N.-A.; Roudsari, N.M.; Zadeh, S.S.T.; Momtaz, S.; Abbasifard, M.; Reiner, Ž.; Abdolghaffari, A.H.; Sahebkar, A. Statins block mammalian target of rapamycin pathway: A possible novel therapeutic strategy for inflammatory, malignant and neurodegenerative diseases. Inflammopharmacology 2023, 31, 57–75. [Google Scholar] [CrossRef]
- Pun, N.T.; Jeong, C.-H. Statin as a potential chemotherapeutic agent: Current updates as a monotherapy, combination therapy, and treatment for anti-cancer drug resistance. Pharmaceuticals 2021, 14, 470. [Google Scholar] [CrossRef]
- Zaky, M.Y.; Fan, C.; Zhang, H.; Sun, X.-F. Unraveling the anticancer potential of statins: Mechanisms and clinical significance. Cancers 2023, 15, 4787. [Google Scholar] [CrossRef]
- Pandyra, A.; Penn, L.Z. Targeting tumor cell metabolism via the mevalonate pathway: Two hits are better than one. Mol. Cell. Oncol. 2014, 1, e969133. [Google Scholar] [CrossRef]
- Warita, K.; Warita, T.; Beckwitt, C.H.; Schurdak, M.E.; Vazquez, A.; Wells, A.; Oltvai, Z.N. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci. Rep. 2014, 4, 7593. [Google Scholar] [CrossRef]
- Ishikawa, T.; Irie, N.; Tashiro, J.; Osaki, T.; Warita, T.; Warita, K.; Naito, M. Comparison of the anticancer effects of various statins on canine oral melanoma cells. Veter-Comp. Oncol. 2023, 22, 156–161. [Google Scholar] [CrossRef]
- Ishikawa, T.; Osaki, T.; Sugiura, A.; Tashiro, J.; Warita, T.; Hosaka, Y.Z.; Warita, K. Atorvastatin preferentially inhibits the growth of high ZEB-expressing canine cancer cells. Veter-Comp. Oncol. 2022, 20, 313–323. [Google Scholar] [CrossRef]
- Collisson, E.A.; Kleer, C.; Wu, M.; De, A.; Gambhir, S.S.; Merajver, S.D.; Kolodney, M.S. Atorvastatin prevents RhoC isoprenylation, invasion, and metastasis in human melanoma cells. Mol. Cancer Ther. 2003, 2, 941–948. [Google Scholar]
- Tsubaki, M.; Takeda, T.; Kino, T.; Obata, N.; Itoh, T.; Imano, M.; Mashimo, K.; Fujiwara, D.; Sakaguchi, K.; Satou, T.; et al. Statins improve survival by inhibiting spontaneous metastasis and tumor growth in a mouse melanoma model. Am. J. Cancer Res. 2015, 5, 3186–3197. [Google Scholar] [PubMed]
- Osaki, T.; Yokoe, I.; Sunden, Y.; Ota, U.; Ichikawa, T.; Imazato, H.; Ishii, T.; Takahashi, K.; Ishizuka, M.; Tanaka, T.; et al. Efficacy of 5-Aminolevulinic acid in photodynamic detection and photodynamic therapy in veterinary medicine. Cancers 2019, 11, 495. [Google Scholar] [CrossRef]
- Zhou, Y.; Tashiro, J.; Kamatani, S.; Irie, N.; Suzuki, A.; Ishikawa, T.; Warita, K.; Oltvai, Z.N.; Warita, T. HMG-CoA reductase degrader, SR-12813, counteracts statin-induced upregulation of HMG-CoA reductase and augments the anticancer effect of atorvastatin. Biochem. Biophys. Res. Commun. 2023, 677, 13–19. [Google Scholar] [CrossRef]
- Nguyen, S.T.; Nguyen, H.T.-L.; Truong, K.D. Comparative cytotoxic effects of methanol, ethanol and DMSO on human cancer cell lines. Biomed. Res. Ther. 2020, 7, 3855–3859. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers–assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Rottner, K.; Stradal, T.E. Actin dynamics and turnover in cell motility. Curr. Opin. Cell Biol. 2011, 23, 569–578. [Google Scholar] [CrossRef]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Modes of invasion during tumour dissemination. Mol. Oncol. 2017, 11, 5–27. [Google Scholar] [CrossRef] [PubMed]
- Korch, C.; Hall, E.M.; Dirks, W.G.; Ewing, M.; Faries, M.; Varella-Garcia, M.; Robinson, S.; Storts, D.; Turner, J.A.; Wang, Y.; et al. Authentication of M14 melanoma cell line proves misidentification of MDA-MB-435 breast cancer cell line. Int. J. Cancer 2018, 142, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ma, M.; Yan, J.; Xu, L.; Yu, J.; Dai, J.; Xu, T.; Tang, H.; Wu, X.; Li, S.; et al. Identification of coexistence of BRAF V600E mutation and EZH2 gain specifically in melanoma as a promising target for combination therapy. J. Transl. Med. 2017, 15, 243. [Google Scholar] [CrossRef]
- Mishra, H.; Mishra, P.K.; Ekielski, A.; Jaggi, M.; Iqbal, Z.; Talegaonkar, S. Melanoma treatment: From conventional to nanotechnology. J. Cancer Res. Clin. Oncol. 2018, 144, 2283–2302. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013, 34, 508–517. [Google Scholar] [CrossRef]
- Longo, J.; Mullen, P.J.; Yu, R.; van Leeuwen, J.E.; Masoomian, M.; Woon, D.T.; Wang, Y.; Chen, E.X.; Hamilton, R.J.; Sweet, J.M.; et al. An actionable sterol-regulated feedback loop modulates statin sensitivity in prostate cancer. Mol. Metab. 2019, 25, 119–130. [Google Scholar] [CrossRef]
- Pandyra, A.; Mullen, P.J.; Kalkat, M.; Yu, R.; Pong, J.T.; Li, Z.; Trudel, S.; Lang, K.S.; Minden, M.D.; Schimmer, A.D.; et al. Immediate utility of two approved agents to target both the metabolic mevalonate pathway and its restorative feedback loop. Cancer Res 2014, 74, 4772–4782. [Google Scholar] [CrossRef]
- Raghu, V.K.; Beckwitt, C.H.; Warita, K.; Wells, A.; Benos, P.V.; Oltvai, Z.N. Biomarker identification for statin sensitivity of cancer cell lines. Biochem. Biophys. Res. Commun. 2018, 495, 659–665. [Google Scholar] [CrossRef]
- Baptista, D.; Ferreira, P.G.; Rocha, M. A systematic evaluation of deep learning methods for the prediction of drug synergy in cancer. PLoS Comput. Biol. 2023, 19, e1010200. [Google Scholar] [CrossRef]
- Held, M.A.; Langdon, C.G.; Platt, J.T.; Graham-Steed, T.; Liu, Z.; Chakraborty, A.; Bacchiocchi, A.; Koo, A.; Haskins, J.W.; Bosenberg, M.W.; et al. Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. Cancer Discov. 2013, 3, 52–67. [Google Scholar] [CrossRef]
- Levine, B.D.; Cagan, R.L. Drosophila lung cancer models identify trametinib plus statin as candidate therapeutic. Cell Rep. 2016, 14, 1477–1487. [Google Scholar] [CrossRef]
- Beckwitt, C.H.; Shiraha, K.; Wells, A. Lipophilic statins limit cancer cell growth and survival, via involvement of Akt signaling. PLoS ONE 2018, 13, e0197422. [Google Scholar] [CrossRef]
- van Leeuwen, J.E.; Ba-Alawi, W.; Branchard, E.; Cruickshank, J.; Schormann, W.; Longo, J.; Silvester, J.; Gross, P.L.; Andrews, D.W.; Cescon, D.W.; et al. Computational pharmacogenomic screen identifies drugs that potentiate the anti-breast cancer activity of statins. Nat. Commun. 2022, 13, 6323. [Google Scholar] [CrossRef]
- Guo, S.; Stins, M.; Ning, M.; Lo, E.H. Amelioration of inflammation and cytotoxicity by dipyridamole in brain endothelial cells. Cerebrovasc. Dis. 2010, 30, 290–296. [Google Scholar] [CrossRef]
- Liu, Y.; Cone, J.; Le, S.N.; Fong, M.; Tao, L.; Shoaf, S.E.; Bricmont, P.; Czerwiec, F.S.; Kambayashi, J.; Yoshitake, M.; et al. Cilostazol and dipyridamole synergistically inhibit human platelet aggregation. J. Cardiovasc. Pharmacol. 2004, 44, 266–273. [Google Scholar] [CrossRef]
- Bennaceur, K.; Atwill, M.; Al Zhrany, N.; Hoffmann, J.; Keavney, B.; Breault, D.; Richardson, G.; von Zglinicki, T.; Saretzki, G.; Spyridopoulos, I. Atorvastatin induces T cell proliferation by a telomerase reverse transcriptase (TERT) mediated mechanism. Atherosclerosis 2014, 236, 312–320. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Eynde, K.V.D.; et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018, 9, 2500. [Google Scholar] [CrossRef]
- Liang, J.; Yu, D.; Luo, C.; Bennett, C.; Jedrychowski, M.; Gygi, S.P.; Widlund, H.R.; Puigserver, P. Epigenetic suppression of PGC1α (PPARGC1A) causes collateral sensitivity to HMGCR-inhibitors within BRAF-treatment resistant melanomas. Nat. Commun. 2023, 14, 3251. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Yu, P.; Chen, X.-S.; Wei, H.; Cao, S.-J.; Zhang, M.; Zhang, Y.; Tao, Y.-G.; Cao, D.-S.; Qiu, F.; et al. Identification of HMGCR as the anticancer target of physapubenolide against melanoma cells by in silico target prediction. Acta Pharmacol. Sin. 2022, 43, 1594–1604. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Obata, N.; Kawashima, K.; Tabata, M.; Imano, M.; Satou, T.; Nishida, S. Combination therapy with dacarbazine and statins improved the survival rate in mice with metastatic melanoma. J. Cell. Physiol. 2019, 234, 17975–17989. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Xu, H.; Tang, Q.; Xia, H.; Bi, F. Dipyridamole enhances the cytotoxicities of trametinib against colon cancer cells through combined targeting of HMGCS1 and MEK pathway. Mol. Cancer Ther. 2020, 19, 135–146. [Google Scholar] [CrossRef] [PubMed]
- van der Weyden, L.; Patton, E.E.; A Wood, G.; Foote, A.K.; Brenn, T.; Arends, M.J.; Adams, D.J. Cross-species models of human melanoma. J. Pathol. 2016, 238, 152–165. [Google Scholar] [CrossRef]
- Stevenson, V.B.; Klahn, S.; LeRoith, T.; Huckle, W.R. Canine melanoma: A review of diagnostics and comparative mechanisms of disease and immunotolerance in the era of the immunotherapies. Front. Veter-Sci. 2023, 9, 1046636. [Google Scholar] [CrossRef]
- Torres, C.G.; Olivares, A.; Stoore, C. Simvastatin exhibits antiproliferative effects on spheres derived from canine mammary carcinoma cells. Oncol. Rep. 2015, 33, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Baba, K.; Kambayashi, S.; Okuda, M. Effect of simvastatin on cell proliferation and Ras activation in canine tumour cells. Veter-Comp. Oncol. 2021, 19, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Vigneau, A.; Rico, C.; Boerboom, D.; Paquet, M. Statins downregulateYAPandTAZand exert anti-cancer effects in canine mammary tumour cells. Veter-Comp. Oncol. 2022, 20, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, S.; Rush, J.; Freeman, L. Short-term effects of atorvastatin in normal dogs and dogs with congestive heart failure due to myxomatous mitral valve disease. J. Veter-Intern. Med. 2013, 27, 985–989. [Google Scholar] [CrossRef]
- Afrin, S.; Rahman, Y.; Sarwar, T.; Ansari, M.O.; Ahmed, S.; Alam Rizvi, M.; Shadab, G.H.A.; Tabish, M. Cytotoxic and genotoxic evaluation of dipyridamole and its alternative therapeutic potential in cancer therapy: An in vitro and in vivo approach. J. Mol. Struct. 2021, 1249, 131626. [Google Scholar] [CrossRef]
- Kubota, T.; Fujisaki, K.; Itoh, Y.; Yano, T.; Sendo, T.; Oishi, R. Apoptotic injury in cultured human hepatocytes induced by HMG-CoA reductase inhibitors. Biochem. Pharmacol. 2004, 67, 2175–2186. [Google Scholar] [CrossRef]
- Shu, N.; Hu, M.; Ling, Z.; Liu, P.; Wang, F.; Xu, P.; Zhong, Z.; Sun, B.; Zhang, M.; Li, F.; et al. The enhanced atorvastatin hepatotoxicity in diabetic rats was partly attributed to the upregulated hepatic Cyp3a and SLCO1B1. Sci. Rep. 2016, 6, 33072. [Google Scholar] [CrossRef]
- Zhang, Q.; Qu, H.; Chen, Y.; Luo, X.; Chen, C.; Xiao, B.; Ding, X.; Zhao, P.; Lu, Y.; Chen, A.F.; et al. Atorvastatin induces mitochondria-dependent ferroptosis via the modulation of Nrf2-xCT/GPx4 axis. Front. Cell Dev. Biol. 2022, 10, 806081. [Google Scholar] [CrossRef]
- Kaufmann, P.; Török, M.; Zahno, A.; Waldhauser, K.M.; Brecht, K.; Krähenbühl, S. Toxicity of statins on rat skeletal muscle mitochondria. Cell. Mol. Life Sci. 2006, 63, 2415–2425. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Melanoma | IC50 Atorvastatin (μM) | IC50 Combination ¶ (μM) | Reduction (%) |
|---|---|---|---|
| MDA-MB-435 | 9.583 | 2.180 | 77 |
| SK-MEL-5 | 21.769 | 5.618 | 74 |
| HTR | 2.286 | 0.184 | 92 |
| ITP | 14.466 | 1.715 | 88 |
| YCC | 3.507 | 1.117 | 68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irie, N.; Mizoguchi, K.; Warita, T.; Nakano, M.; Sasaki, K.; Tashiro, J.; Osaki, T.; Ishikawa, T.; Oltvai, Z.N.; Warita, K. Repurposing of the Cardiovascular Drug Statin for the Treatment of Cancers: Efficacy of Statin–Dipyridamole Combination Treatment in Melanoma Cell Lines. Biomedicines 2024, 12, 698. https://doi.org/10.3390/biomedicines12030698
Irie N, Mizoguchi K, Warita T, Nakano M, Sasaki K, Tashiro J, Osaki T, Ishikawa T, Oltvai ZN, Warita K. Repurposing of the Cardiovascular Drug Statin for the Treatment of Cancers: Efficacy of Statin–Dipyridamole Combination Treatment in Melanoma Cell Lines. Biomedicines. 2024; 12(3):698. https://doi.org/10.3390/biomedicines12030698
Chicago/Turabian StyleIrie, Nanami, Kana Mizoguchi, Tomoko Warita, Mirai Nakano, Kasuga Sasaki, Jiro Tashiro, Tomohiro Osaki, Takuro Ishikawa, Zoltán N. Oltvai, and Katsuhiko Warita. 2024. "Repurposing of the Cardiovascular Drug Statin for the Treatment of Cancers: Efficacy of Statin–Dipyridamole Combination Treatment in Melanoma Cell Lines" Biomedicines 12, no. 3: 698. https://doi.org/10.3390/biomedicines12030698