
Activation of MAP Kinase Pathway by Polyisoprenylated Cysteinyl Amide Inhibitors Causes Apoptosis and Disrupts Breast Cancer Cell Invasion
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
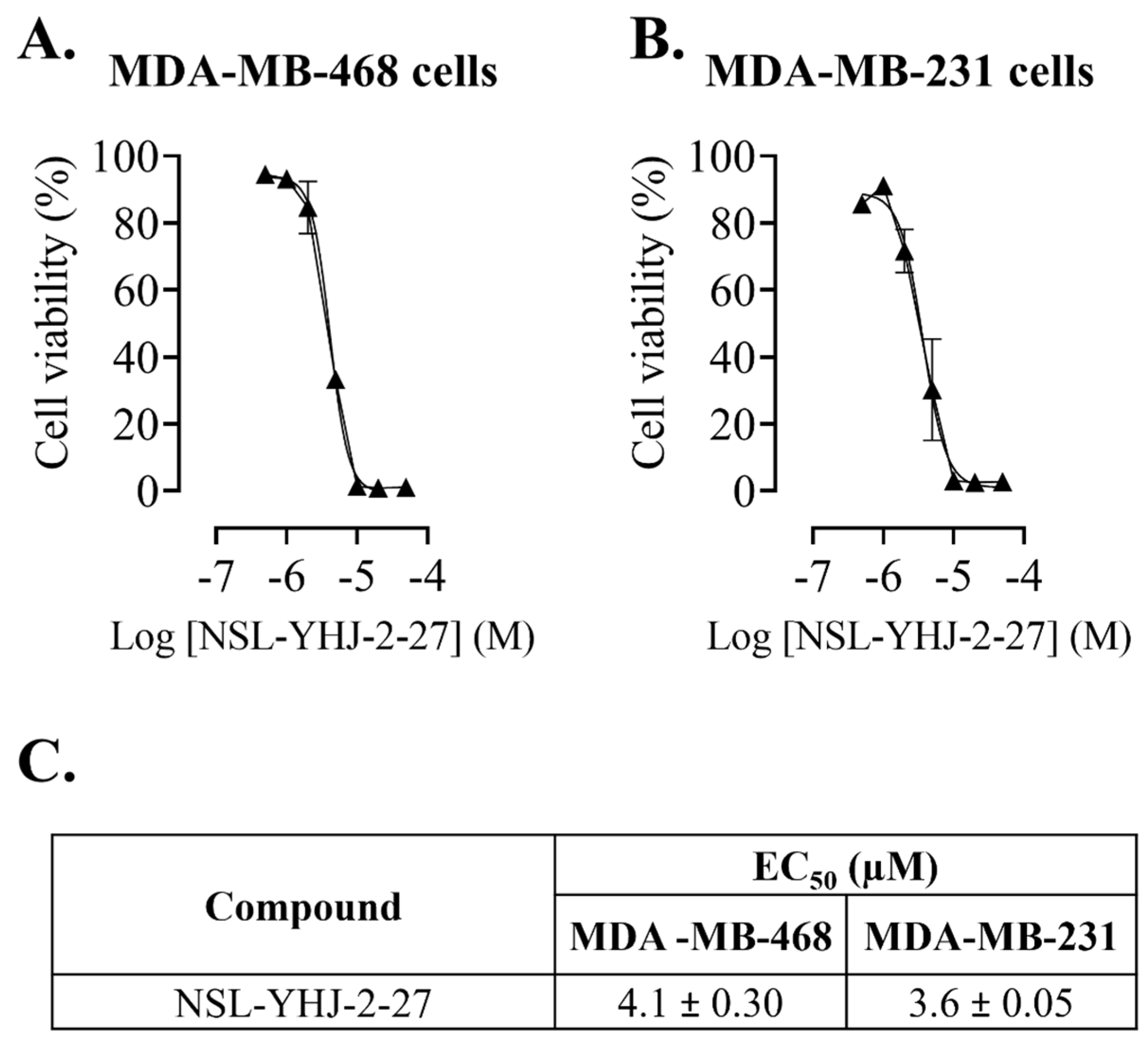
2.3. Effects of PCAIs on the Viability of Breast Cancer Cells
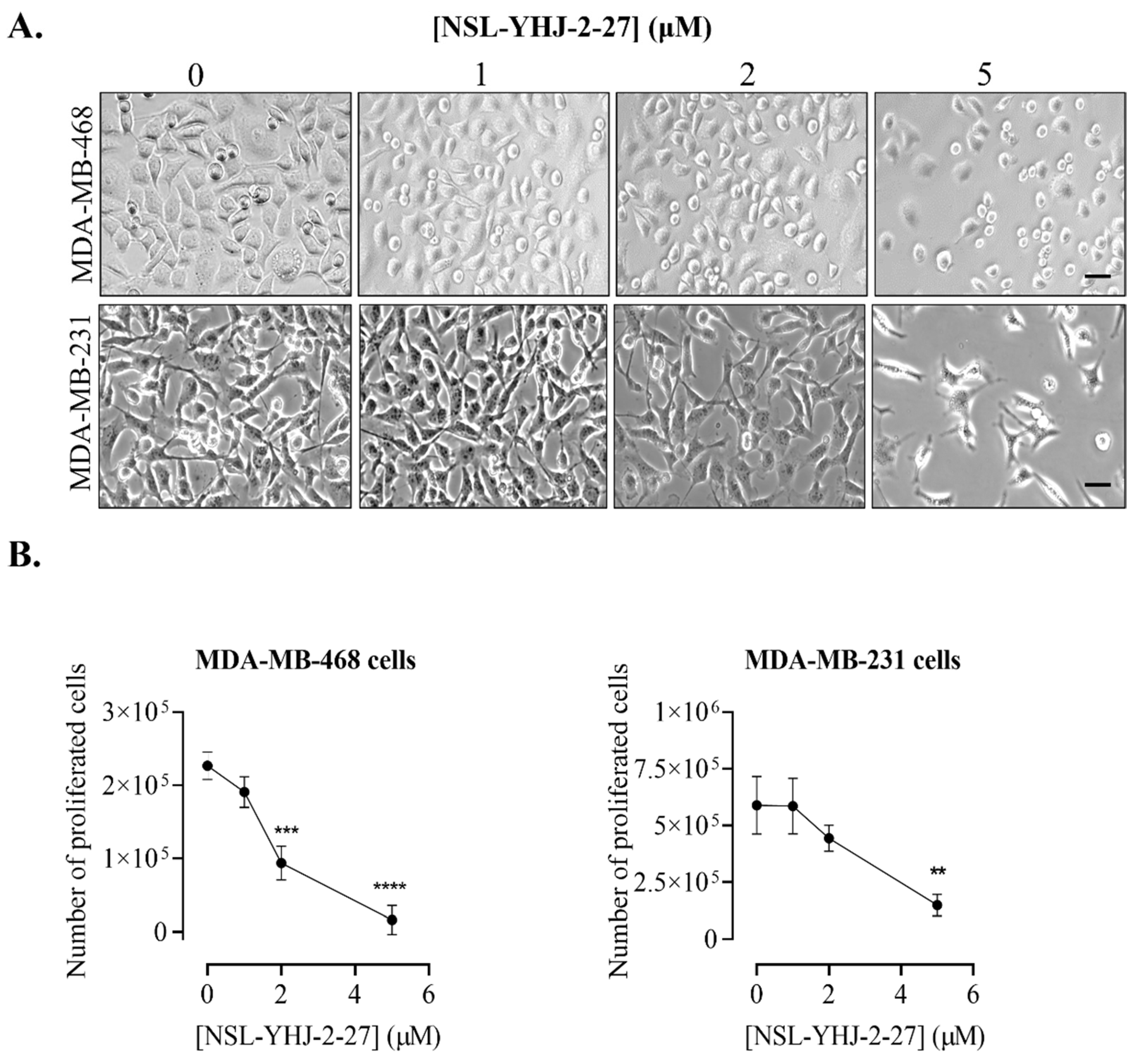
2.4. PCAIs’ Inhibition of Cell Proliferation
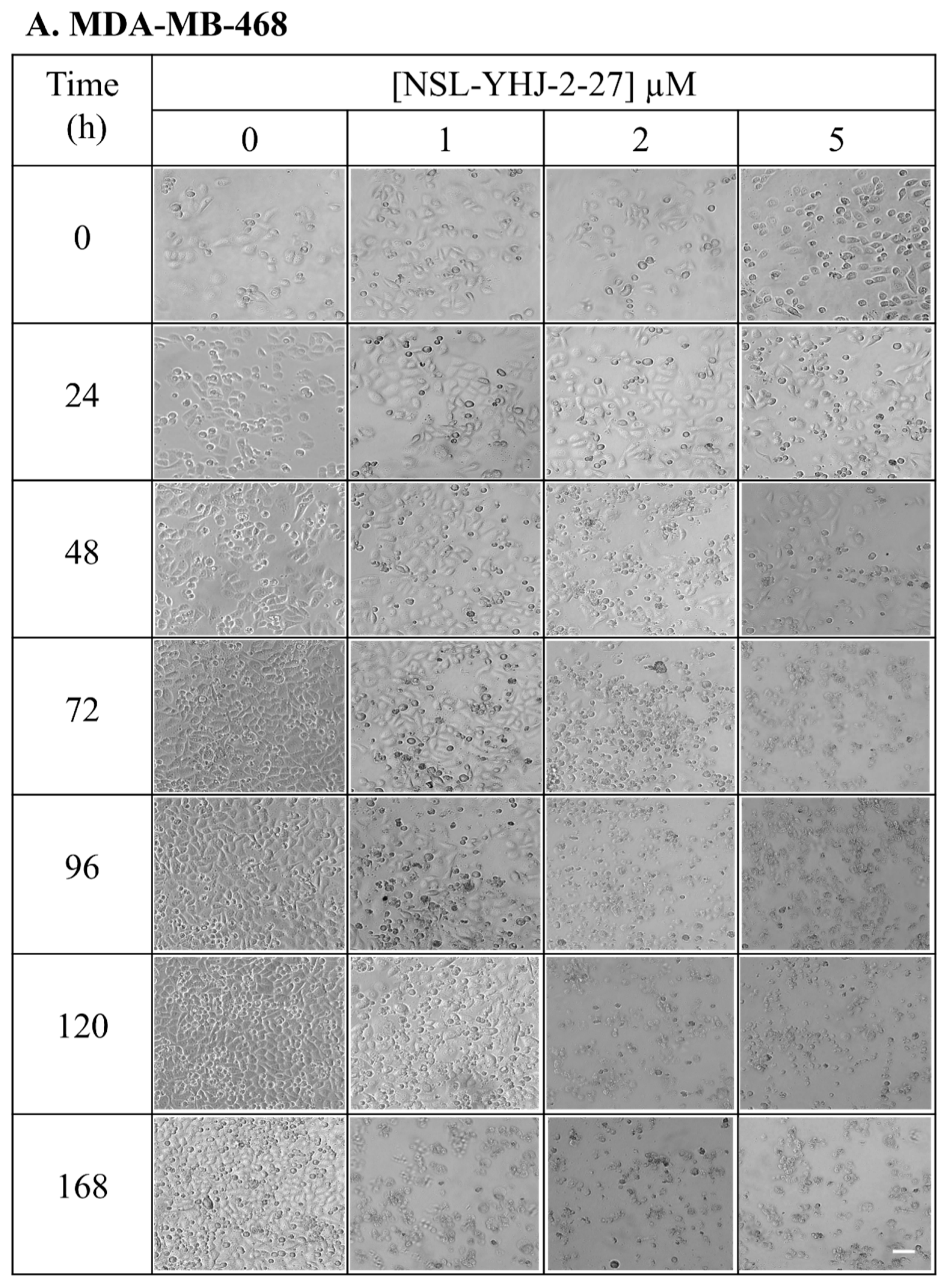
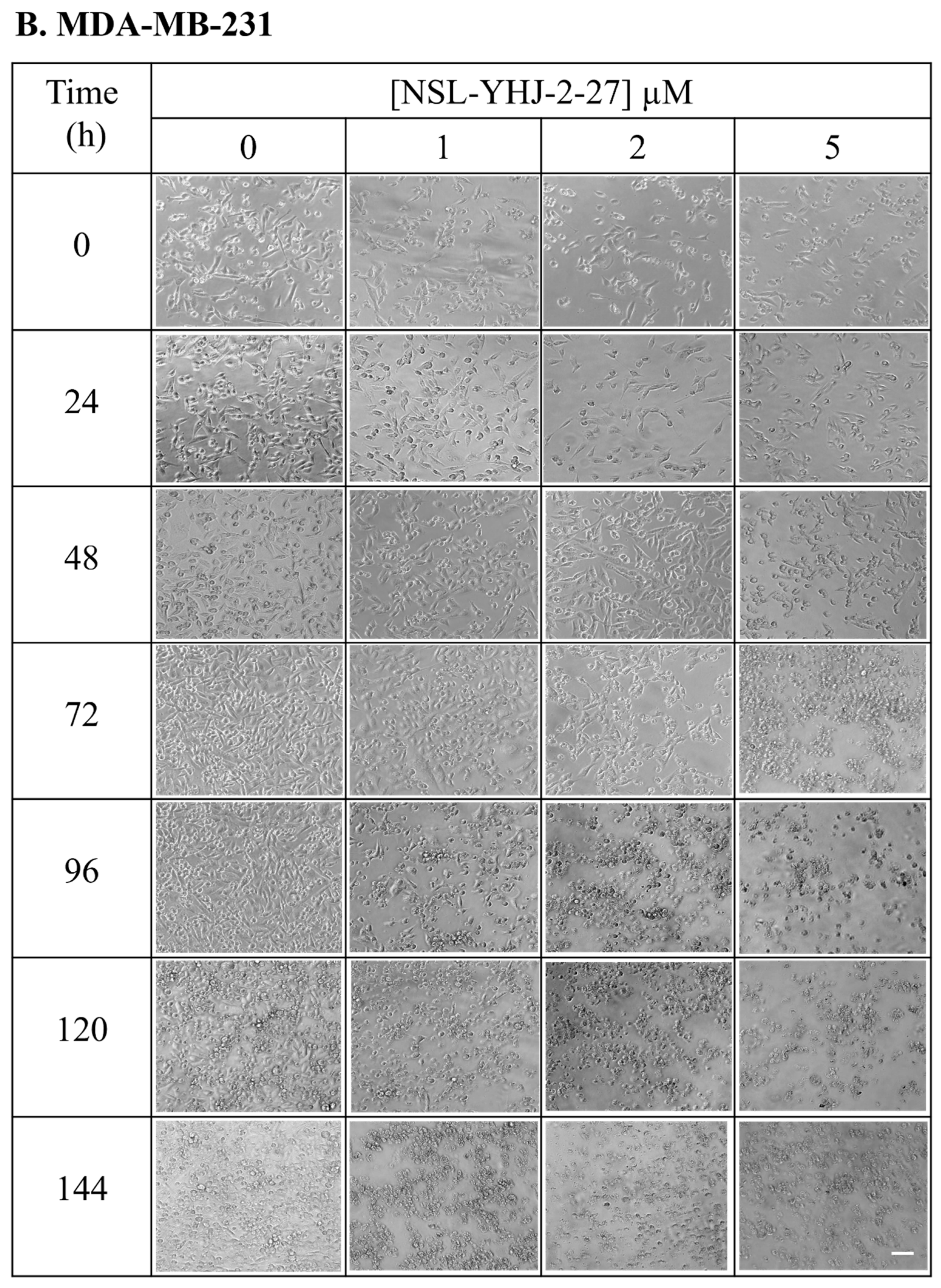
2.5. Effects of Long-Term Treatment of PCAIs on Breast Cancer Cells
2.6. Effects of PCAIs on the Phosphorylation of MAPK and PI3K/AKT Pathway Enzymes
2.7. Western Blot Assay
2.8. Apoptotic Effects of PCAIs on MDA-MB-468
2.9. Effects of PCAIs on 3D Spheroids
2.10. Effects of Inhibitors of Various RSK Isoforms on PCAsI-Induced Cell Death
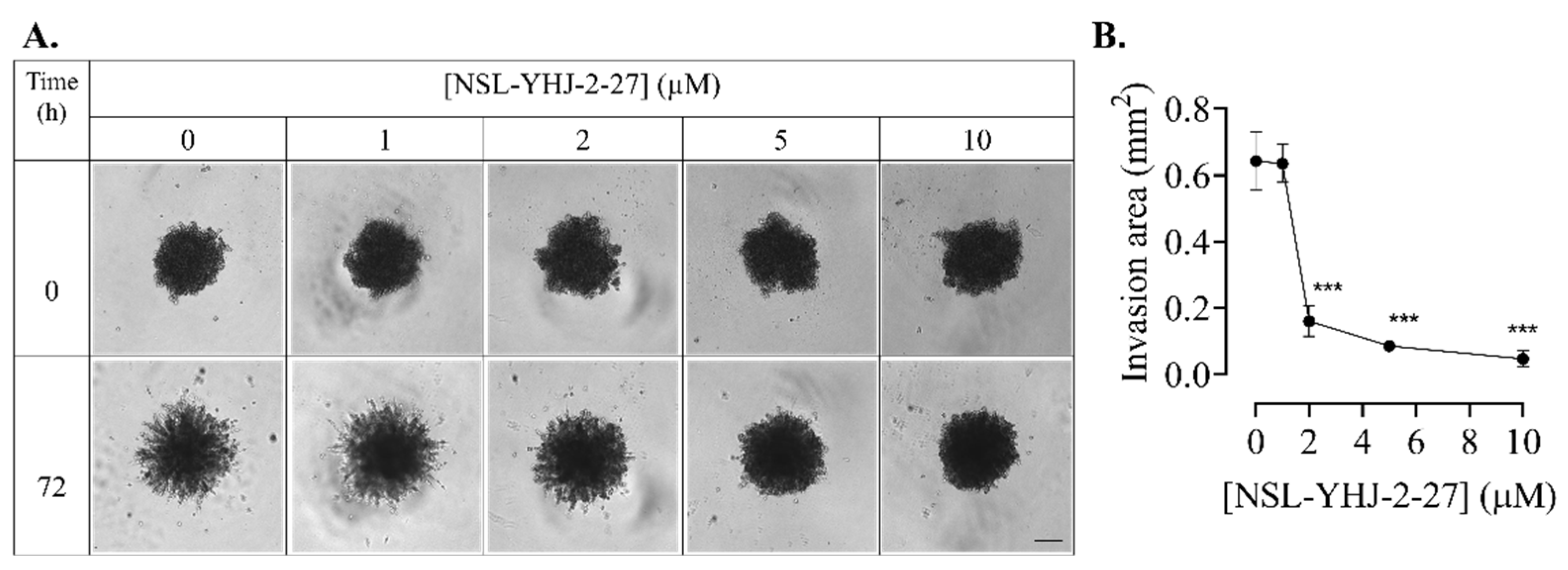
2.11. Effects of PCAIs on Breast Cancer Cell Invasion
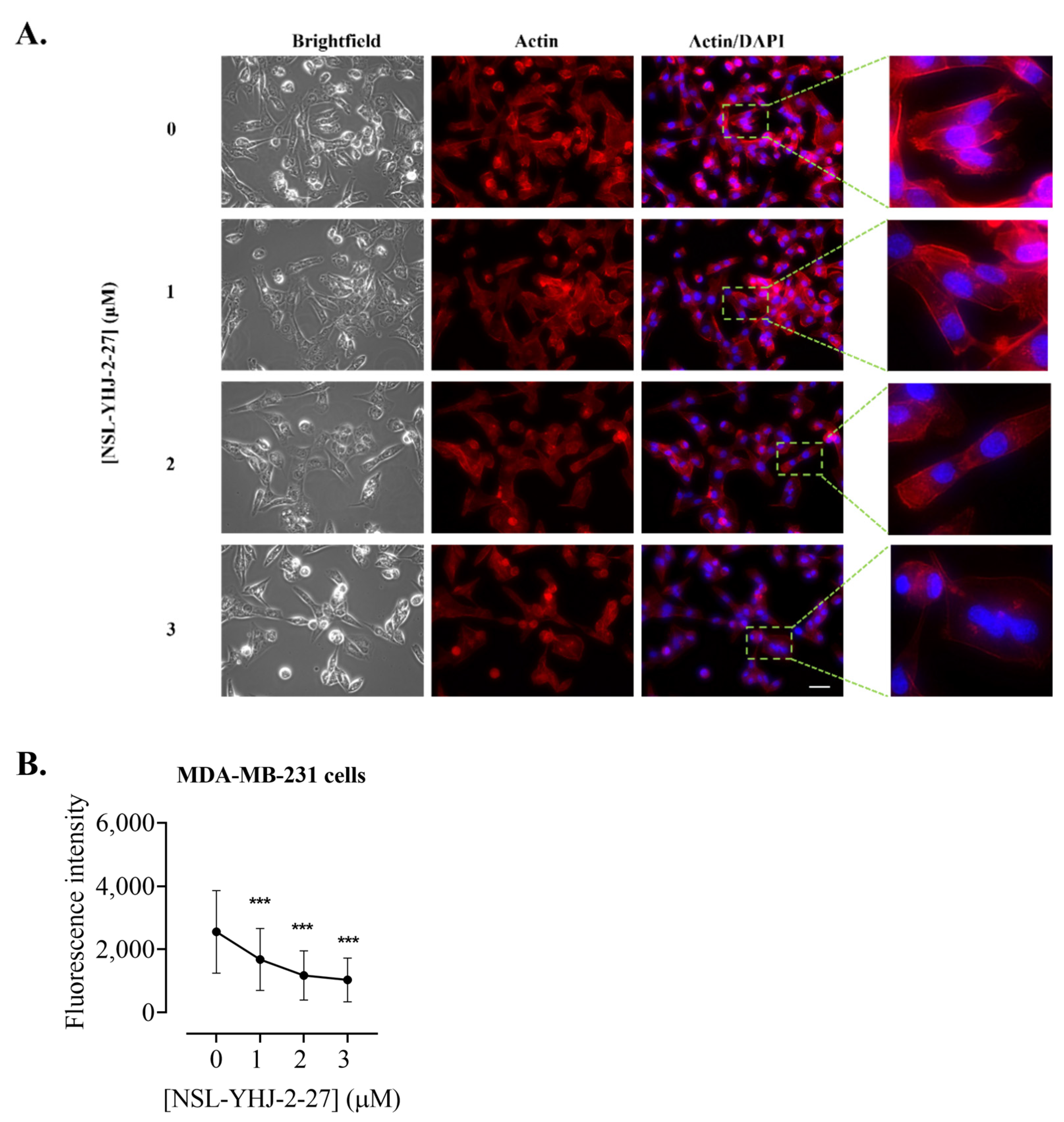
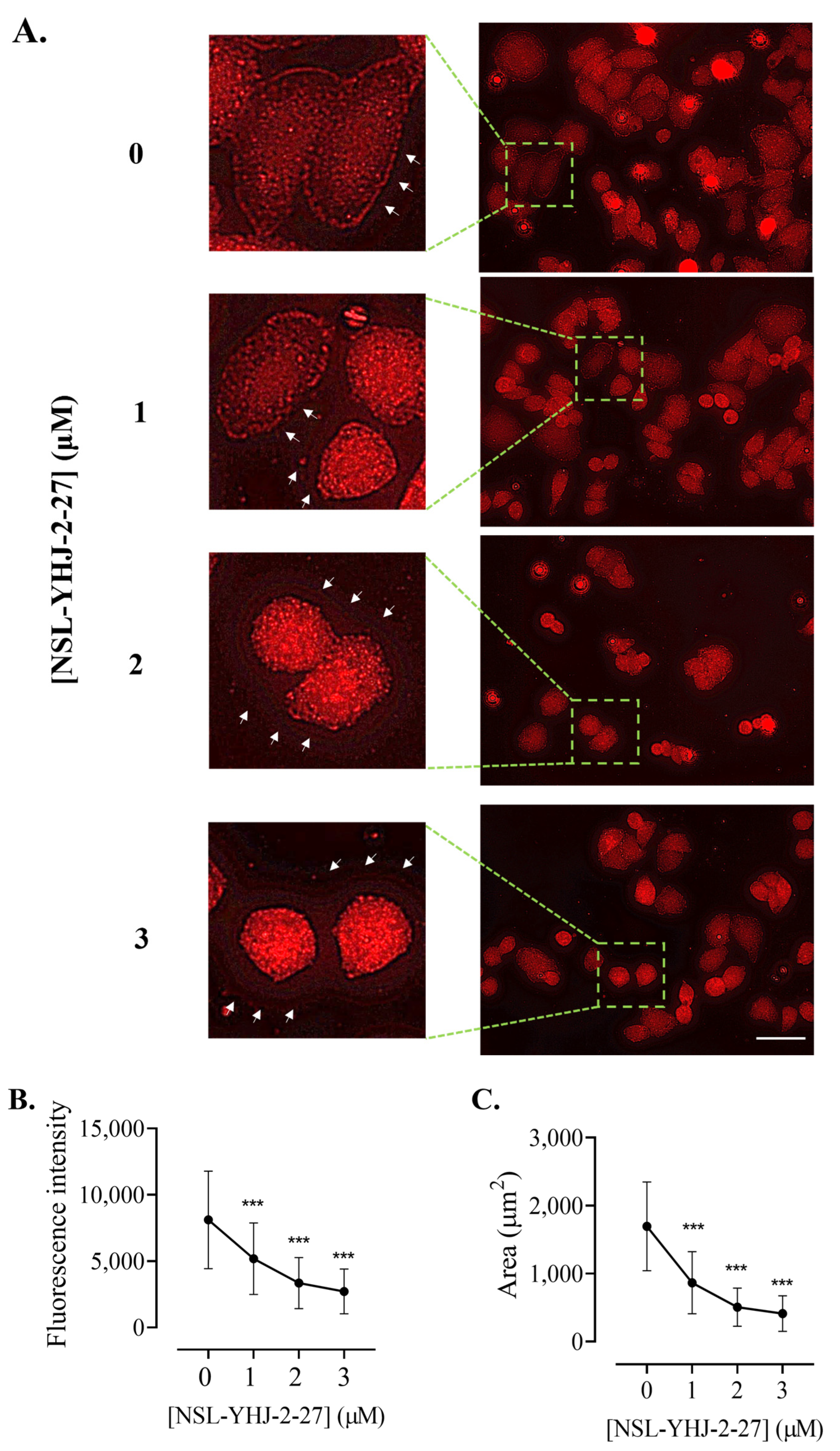
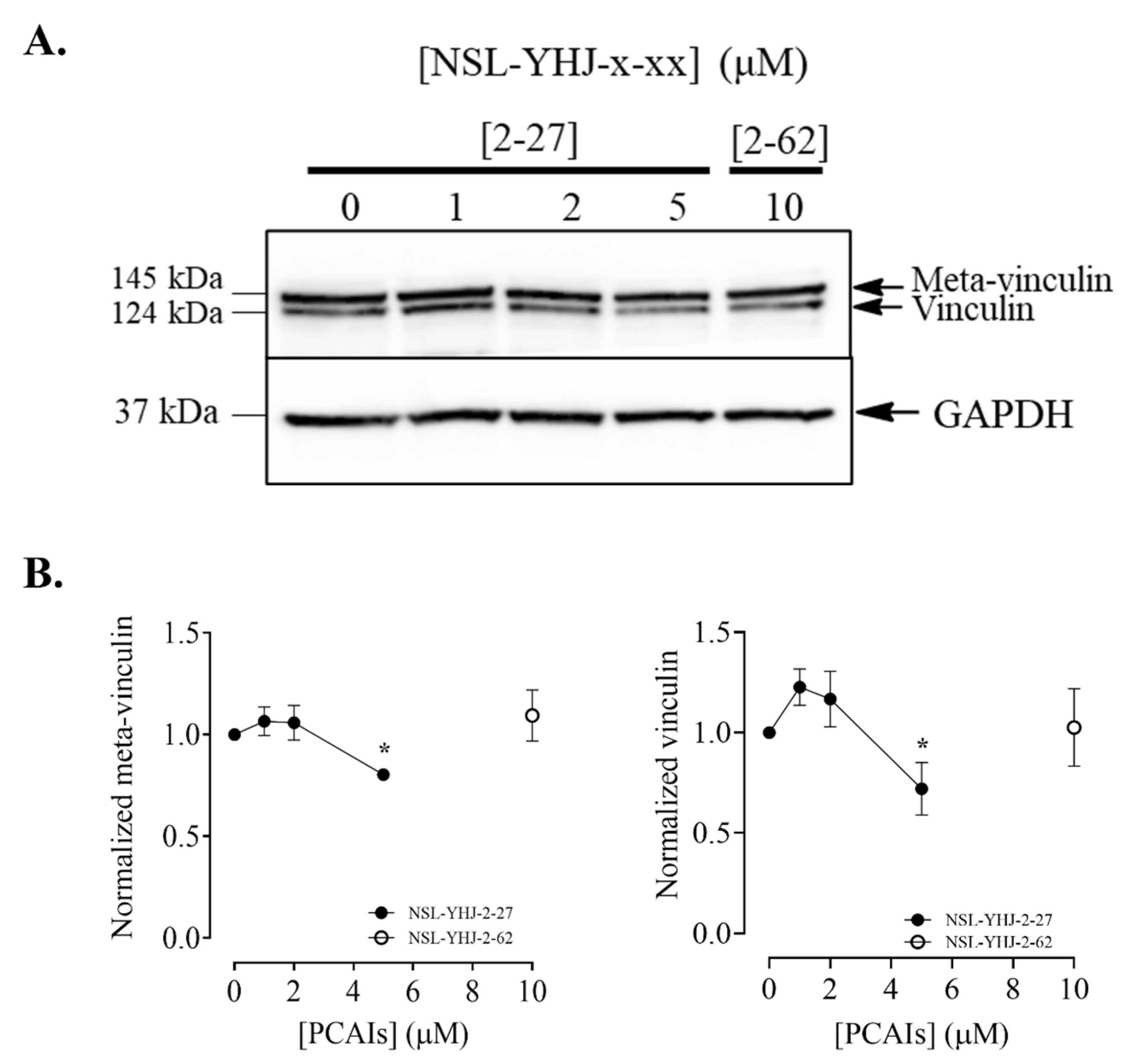
2.12. Effects of the PCAIs on Actin Filaments and Vinculin Punctates
2.13. Statistical Analysis
3. Results
3.1. PCAIs Suppress Breast Cancer Cell Viability
3.2. PCAIs Inhibit the Proliferation of Breast Cancer Cells
3.3. Long-Term Treatment of NSL-YHJ-2-27 Results in Cell Death
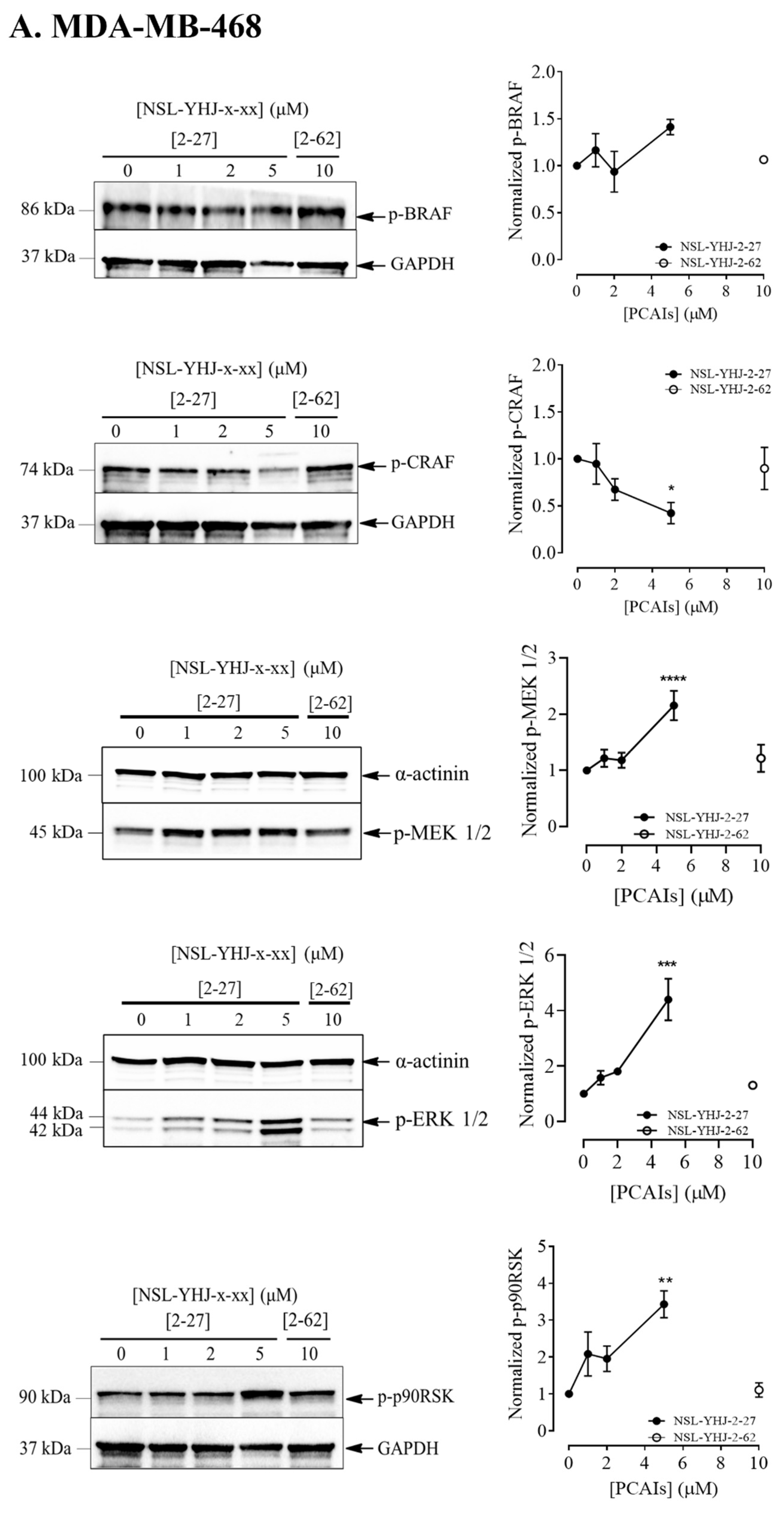
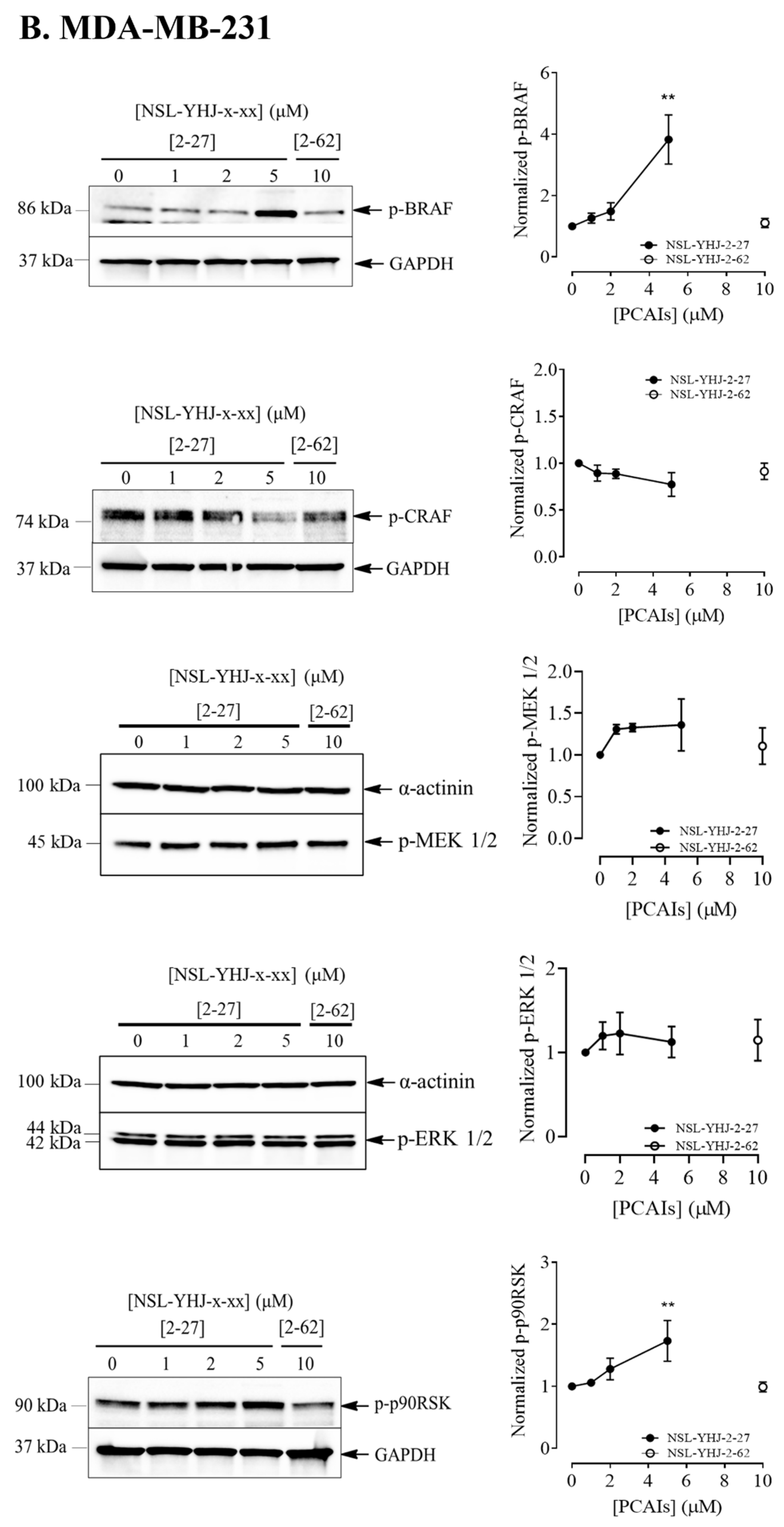
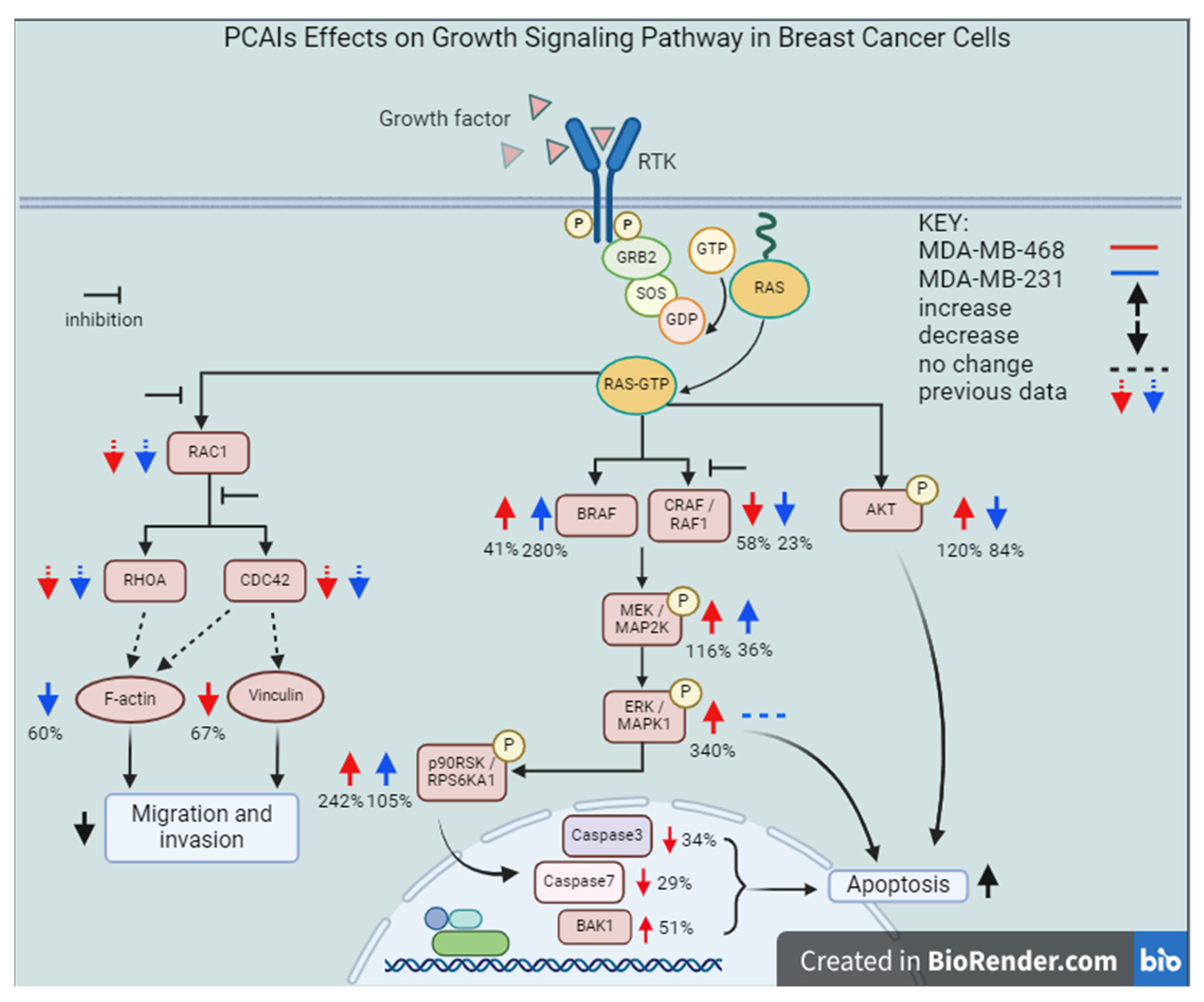
3.4. PCAIs Stimulate the Phosphorylation of MAPK Pathway Enzymes
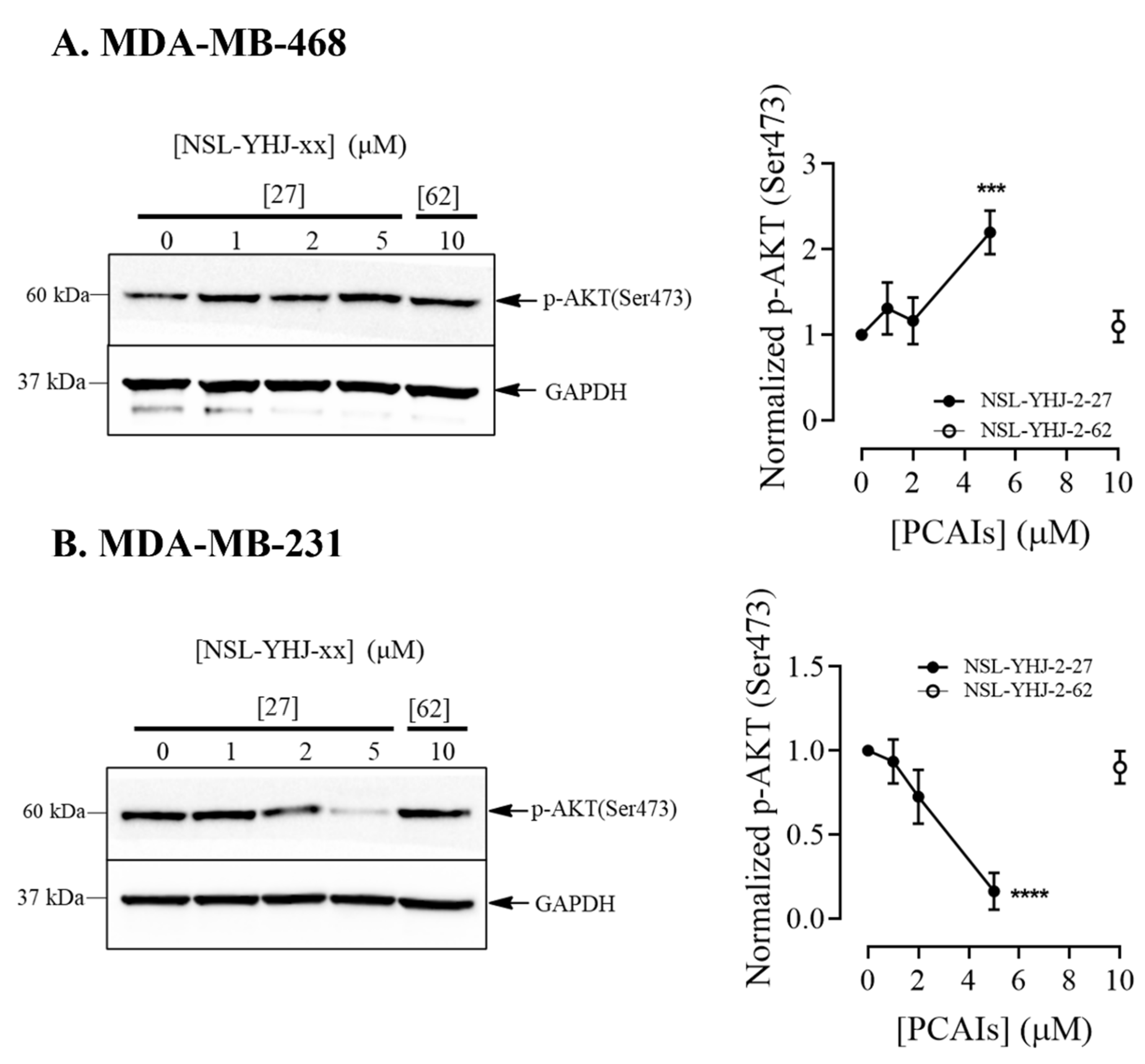
3.5. PCAIs’ Effects on AKT Pathway May Have Caused Cell Death in KRAS-Mutant Breast Cancer Cells
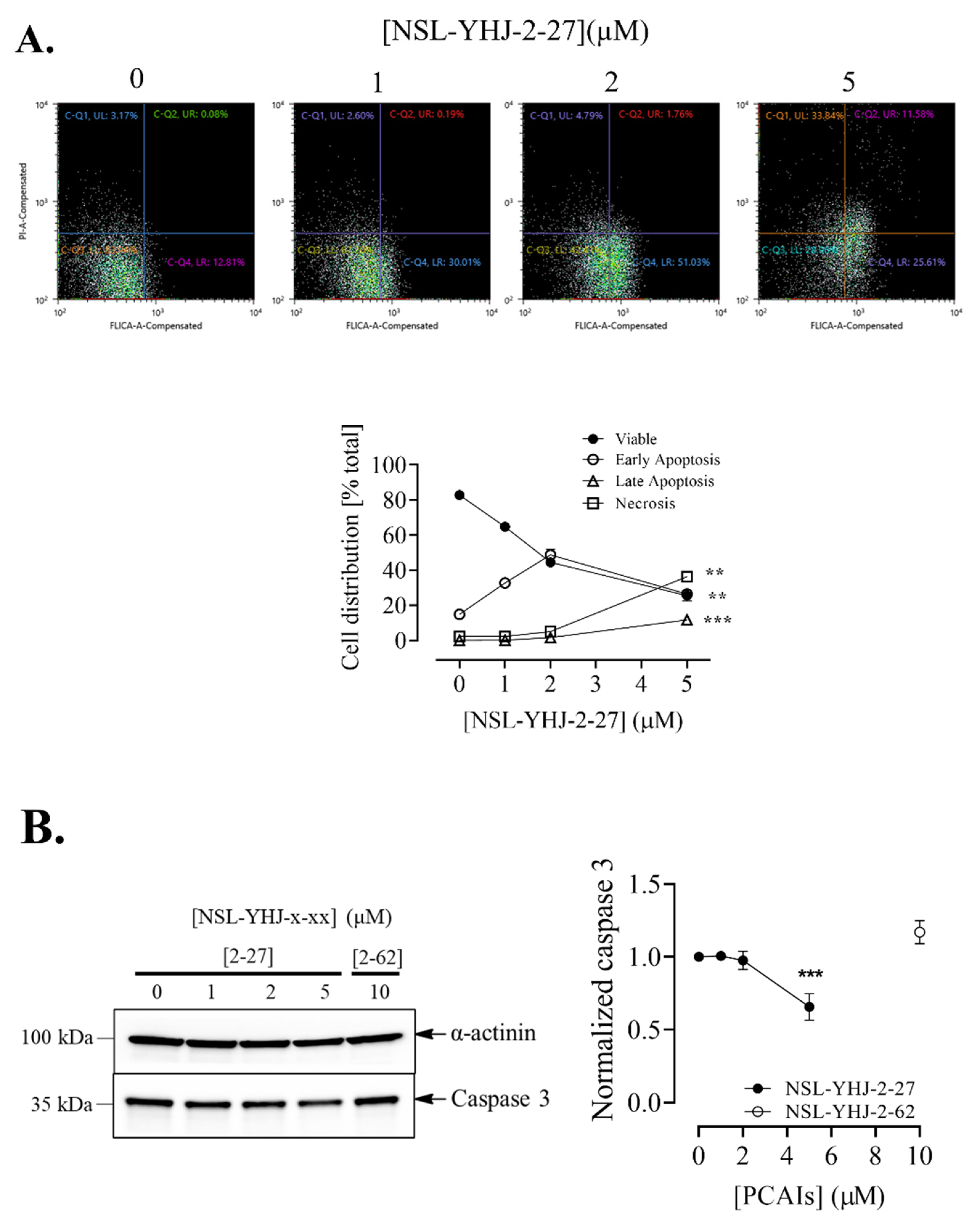
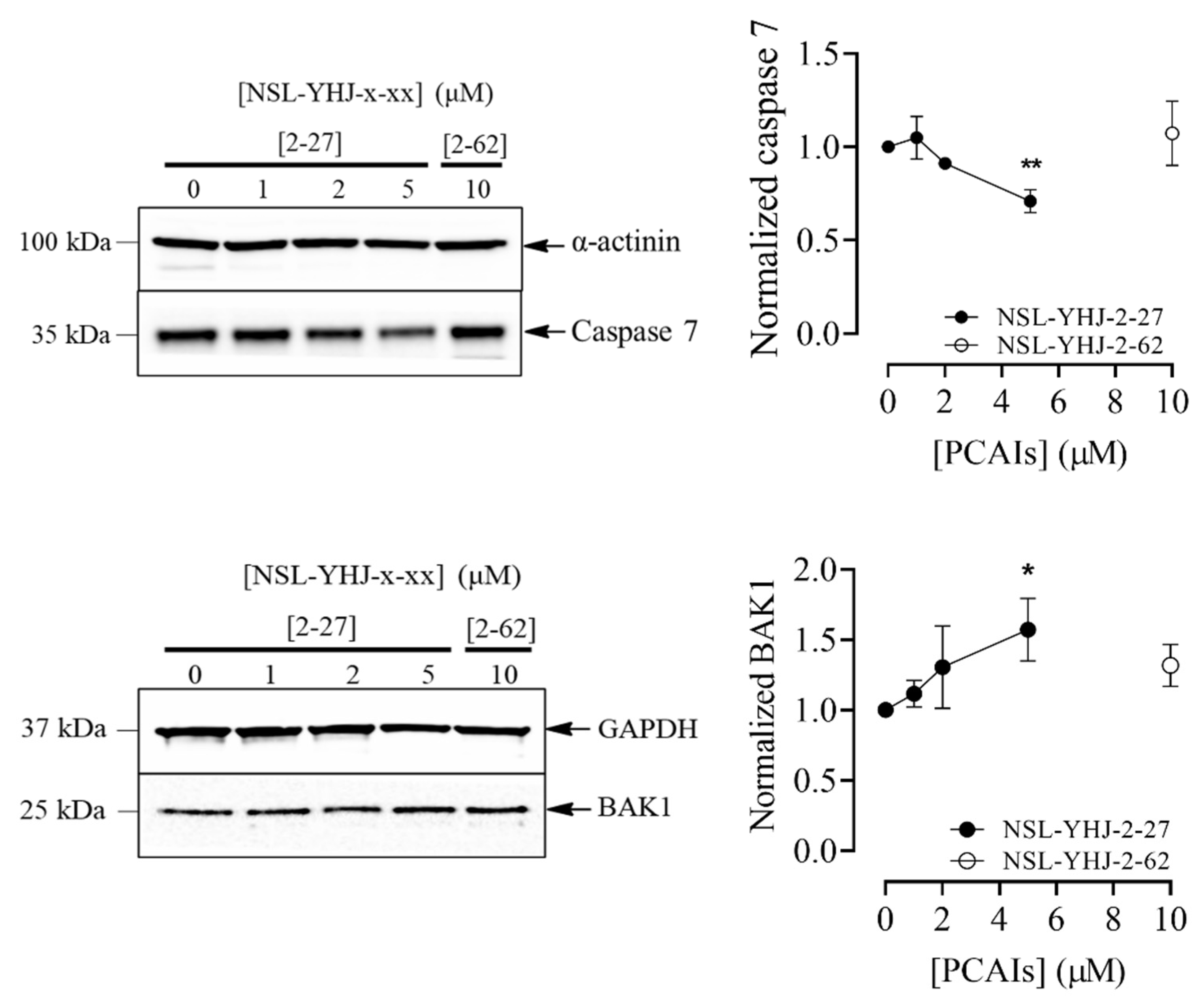
3.6. PCAIs Induce Cell Death by Apoptosis
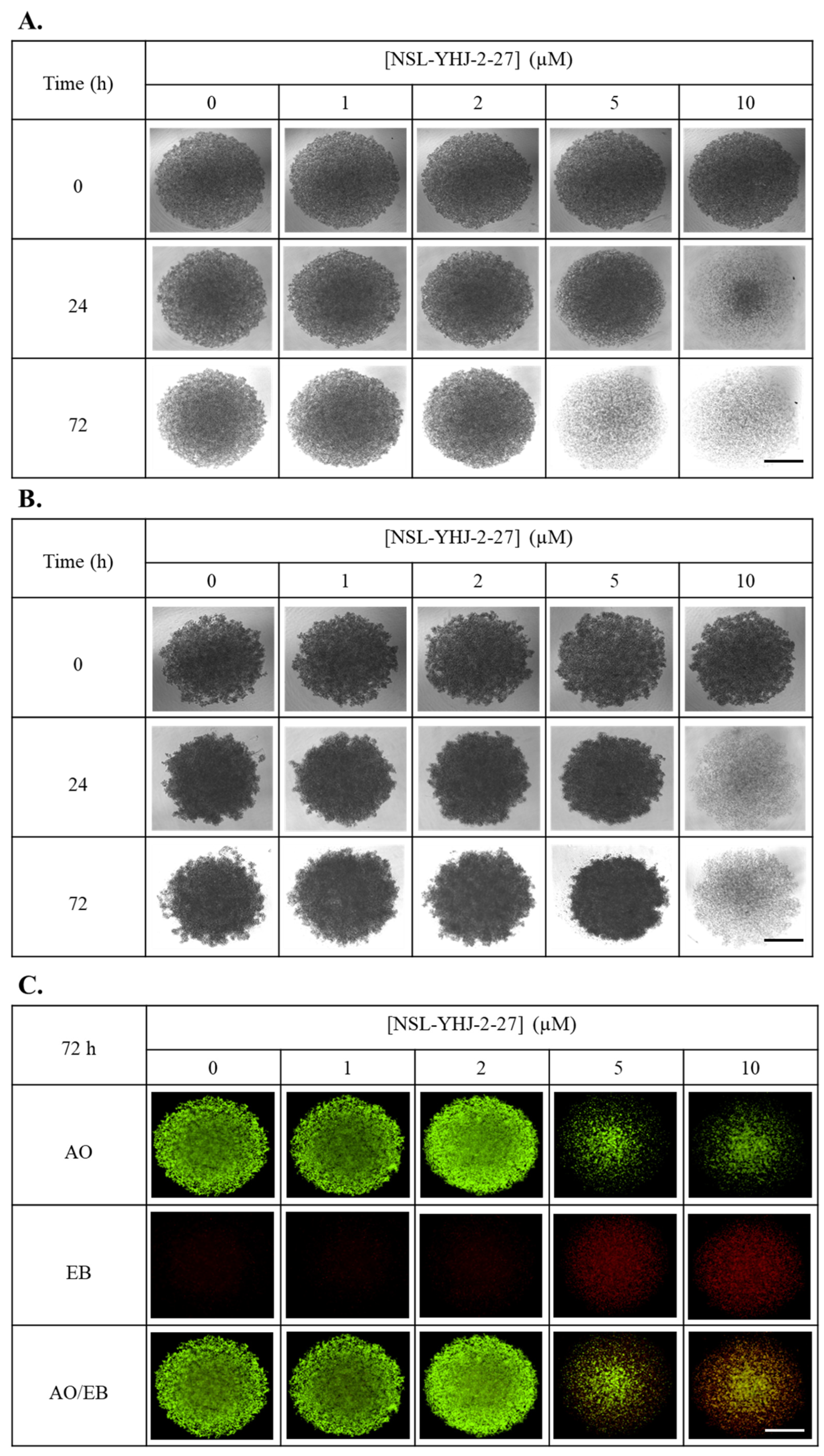
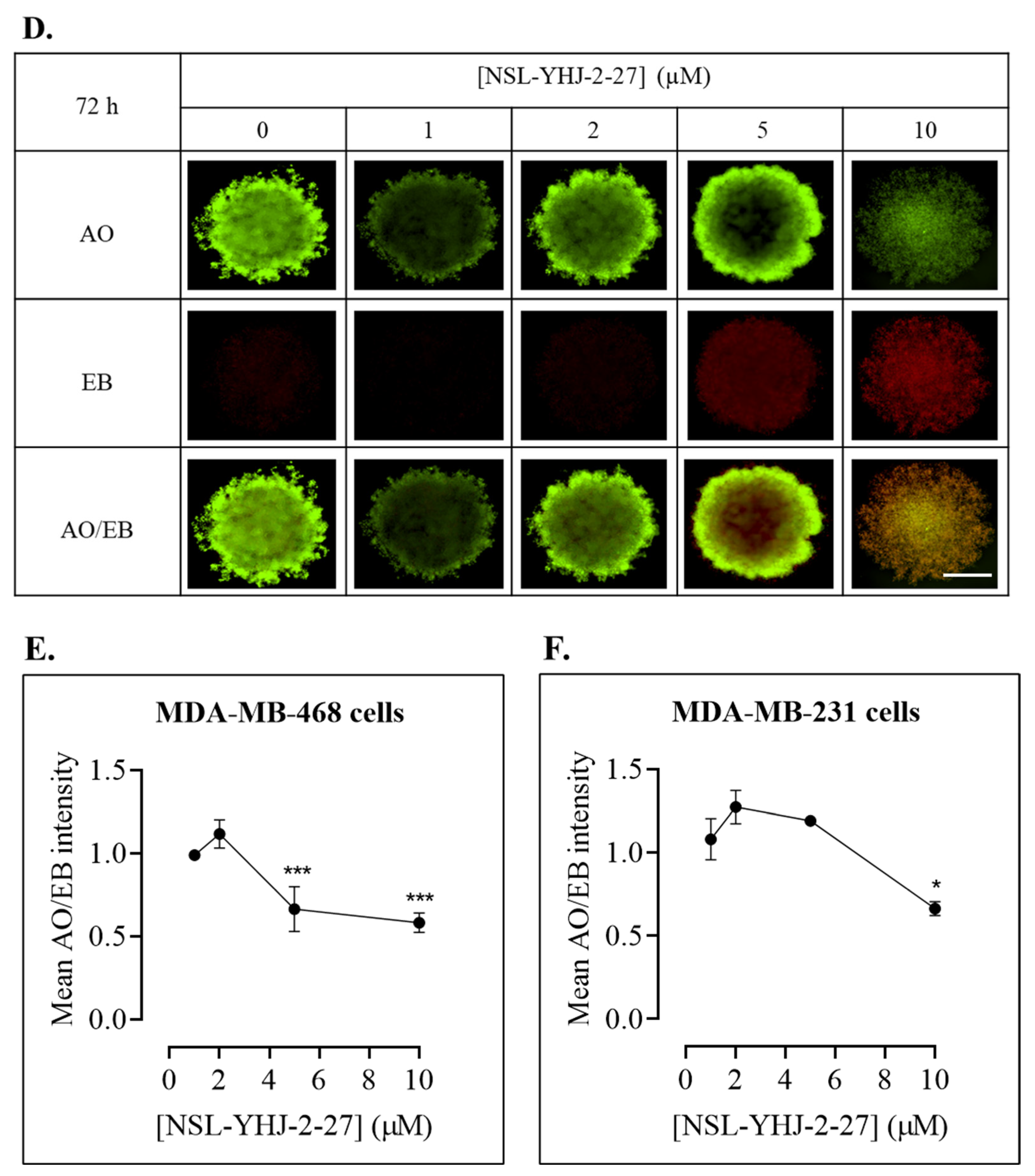
3.7. PCAIs Treatment Disaggregates Compact 3D Spheroids
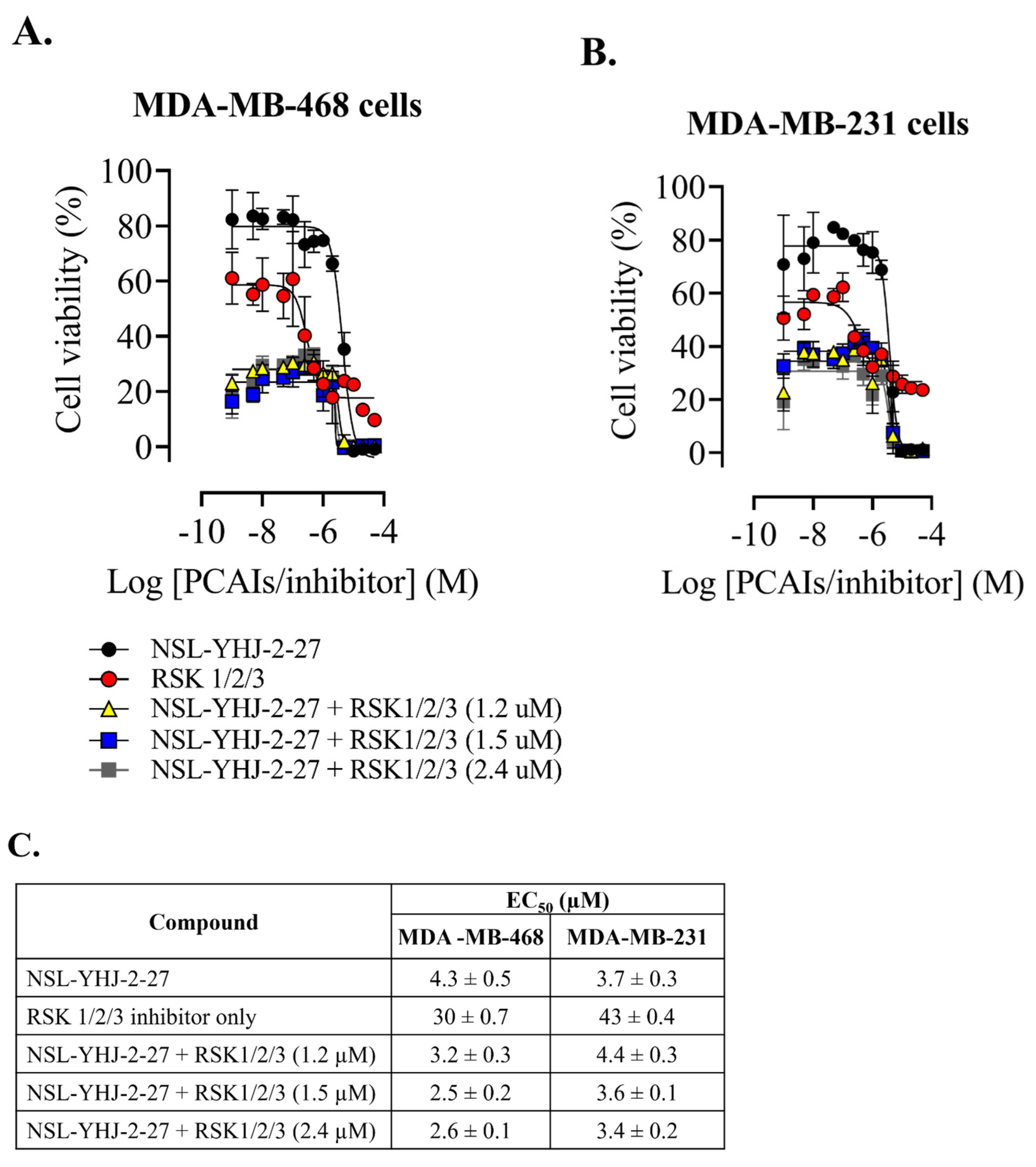
3.8. PCAIs-Induced Cell Death Is Not RSK3-Driven
3.9. PCAIs Inhibit Breast Cancer Cell Invasion into Matrigel
3.10. PCAIs Disrupt Actin Filaments and Focal Adhesion and Decrease Levels of Vinculin
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Almansour, N.M. Triple-Negative Breast Cancer: A Brief Review About Epidemiology, Risk Factors, Signaling Pathways, Treatment and Role of Artificial Intelligence. Front. Mol. Biosci. 2022, 9, 836417. [Google Scholar] [CrossRef]
- Won, K.A.; Spruck, C. Triple-negative breast cancer therapy: Current and future perspectives (Review). Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Li, Y.; Zhan, Z.; Yin, X.; Fu, S.; Deng, X. Targeted Therapeutic Strategies for Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 731535. [Google Scholar] [CrossRef]
- Sawyers, C.L. Opportunities and challenges in the development of kinase inhibitor therapy for cancer. Genes. Dev. 2003, 17, 2998–3010. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef]
- Rasti, A.R.; Guimaraes-Young, A.; Datko, F.; Borges, V.F.; Aisner, D.L.; Shagisultanova, E. PIK3CA Mutations Drive Therapeutic Resistance in Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. JCO Precis. Oncol. 2022, 6, e2100370. [Google Scholar] [CrossRef]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sanchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef]
- Krop, I.E.; Mayer, I.A.; Ganju, V.; Dickler, M.; Johnston, S.; Morales, S.N.; Yardley, D.A.; Melichar, B.; Forero-Torres, A.; Lee, S.C.; et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 811–821. [Google Scholar] [CrossRef]
- Sharma, P.; Abramson, V.G.; O’Dea, A.; Pathak, H.B.; Pessetto, Z.Y.; Wang, Y.Y.; Finke, K.; Hoffmann, M.S.; Elia, M.; Lewis, S.; et al. Clinical and biomarker results from phase I/II study of PI3K inhibitor BYL 719 (alpelisib) plus nab-paclitaxel in HER2-negative metastatic breast cancer. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Yang, S.; Liu, G. Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol. Lett. 2017, 13, 1041–1047. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Martin, P.; Pognonec, P. ERK and cell death: Cadmium toxicity, sustained ERK activation and cell death. FEBS J. 2010, 277, 39–46. [Google Scholar] [CrossRef]
- Chae, J.; Kim, J.S.; Choi, S.T.; Lee, S.G.; Ojulari, O.V.; Kang, Y.J.; Kwon, T.K.; Nam, J.O. Farrerol Induces Cancer Cell Death via ERK Activation in SKOV3 Cells and Attenuates TNF-alpha-Mediated Lipolysis. Int. J. Mol. Sci. 2021, 22, 9400. [Google Scholar] [CrossRef]
- Jung, E.J.; Kim, D.R. Apoptotic cell death in TrkA-overexpressing cells: Kinetic regulation of ERK phosphorylation and caspase-7 activation. Mol. Cells 2008, 26, 12–17. [Google Scholar] [CrossRef]
- Houles, T.; Roux, P.P. Defining the role of the RSK isoforms in cancer. Semin. Cancer Biol. 2018, 48, 53–61. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Ramos, J.W. RSK isoforms in cancer cell invasion and metastasis. Cancer Res. 2013, 73, 6099–6105. [Google Scholar] [CrossRef]
- Nakajima, H.; Ishikawa, Y.; Furuya, M.; Sano, T.; Ohno, Y.; Horiguchi, J.; Oyama, T. Protein expression, gene amplification, and mutational analysis of EGFR in triple-negative breast cancer. Breast Cancer 2014, 21, 66–74. [Google Scholar] [CrossRef]
- Suarez-Cabrera, C.; Quintana, R.M.; Bravo, A.; Casanova, M.L.; Page, A.; Alameda, J.P.; Paramio, J.M.; Maroto, A.; Salamanca, J.; Dupuy, A.J.; et al. A Transposon-based Analysis Reveals RASA1 Is Involved in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 1357–1368. [Google Scholar] [CrossRef]
- Mittal, R.; Ahmadian, M.R.; Goody, R.S.; Wittinghofer, A. Formation of a transition-state analog of the Ras GTPase reaction by Ras-GDP, tetrafluoroaluminate, and GTPase-activating proteins. Science 1996, 273, 115–117. [Google Scholar] [CrossRef]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef]
- Pegram, M.; Slamon, D. Biological rationale for HER2/neu (c-erbB2) as a target for monoclonal antibody therapy. Semin. Oncol. 2000, 27, 13–19. [Google Scholar]
- Naidu, R.; Yadav, M.; Nair, S.; Kutty, M.K. Expression of c-erbB3 protein in primary breast carcinomas. Br. J. Cancer 1998, 78, 1385–1390. [Google Scholar] [CrossRef]
- Quinn, C.M.; Ostrowski, J.L.; Lane, S.A.; Loney, D.P.; Teasdale, J.; Benson, F.A. c-erbB-3 protein expression in human breast cancer: Comparison with other tumour variables and survival. Histopathology 1994, 25, 247–252. [Google Scholar] [CrossRef]
- Farabaugh, S.M.; Boone, D.N.; Lee, A.V. Role of IGF1R in Breast Cancer Subtypes, Stemness, and Lineage Differentiation. Front. Endocrinol. 2015, 6, 59. [Google Scholar] [CrossRef]
- Mountzios, G.; Aivazi, D.; Kostopoulos, I.; Kourea, H.P.; Kouvatseas, G.; Timotheadou, E.; Zebekakis, P.; Efstratiou, I.; Gogas, H.; Vamvouka, C.; et al. Differential expression of the insulin-like growth factor receptor among early breast cancer subtypes. PLoS ONE 2014, 9, e91407. [Google Scholar] [CrossRef]
- Aguilar, B.J.; Nkembo, A.T.; Duverna, R.; Poku, R.A.; Amissah, F.; Ablordeppey, S.Y.; Lamango, N.S. Polyisoprenylated methylated protein methyl esterase: A putative biomarker and therapeutic target for pancreatic cancer. Eur. J. Med. Chem. 2014, 81, 323–333. [Google Scholar] [CrossRef]
- Tawfeeq, N.; Jin, Y.; Lamango, N.S. Synthetic Optimization and MAPK Pathway Activation Anticancer Mechanism of Polyisoprenylated Cysteinyl Amide Inhibitors. Cancers 2021, 13, 5757. [Google Scholar] [CrossRef]
- Amissah, F.; Duverna, R.; Aguilar, B.J.; Poku, R.A.; Kiros, G.E.; Lamango, N.S. Polyisoprenylated methylated protein methyl esterase overexpression and hyperactivity promotes lung cancer progression. Am. J. Cancer Res. 2014, 4, 116–134. [Google Scholar] [PubMed]
- Poku, R.A.; Amissah, F.; Duverna, R.; Aguilar, B.J.; Kiros, G.E.; Lamango, N.S. Polyisoprenylated methylated protein methyl esterase as a putative drug target for androgen-insensitive prostate cancer. Ecancermedicalscience 2014, 8, 459. [Google Scholar] [CrossRef]
- Ntantie, E.; Allen, M.J.; Fletcher, J.; Nkembo, A.T.; Lamango, N.S.; Ikpatt, O.F. Suppression of focal adhesion formation may account for the suppression of cell migration, invasion and growth of non-small cell lung cancer cells following treatment with polyisoprenylated cysteinyl amide inhibitors. Oncotarget 2018, 9, 25781–25795. [Google Scholar] [CrossRef] [PubMed]
- Nkembo, A.T.; Amissah, F.; Ntantie, E.; Poku, R.A.; Salako, O.O.; Ikpatt, O.F.; Lamango, N.S. Polyisoprenylated Cysteinyl Amide Inhibitors Deplete K-Ras and Induce Caspase-dependent Apoptosis in Lung Cancer Cells. Curr. Cancer Drug Targets 2019, 19, 838–851. [Google Scholar] [CrossRef]
- Fomina-Yadlin, D.; Kubicek, S.; Walpita, D.; Dancik, V.; Hecksher-Sorensen, J.; Bittker, J.A.; Sharifnia, T.; Shamji, A.; Clemons, P.A.; Wagner, B.K.; et al. Small-molecule inducers of insulin expression in pancreatic alpha-cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15099–15104. [Google Scholar] [CrossRef]
- Testa, J.R.; Tsichlis, P.N. AKT signaling in normal and malignant cells. Oncogene 2005, 24, 7391–7393. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef]
- Moelling, K.; Schad, K.; Bosse, M.; Zimmermann, S.; Schweneker, M. Regulation of Raf-Akt Cross-talk. J. Biol. Chem. 2002, 277, 31099–31106. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, T.; Sun, Q.; Niu, M.; Jiang, Y.; Pang, D. Downregulation of Ras GTPase-activating protein 1 is associated with poor survival of breast invasive ductal carcinoma patients. Oncol. Rep. 2015, 33, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.J.; Der, C.J. Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res. Treat. 1995, 35, 133–144. [Google Scholar] [CrossRef]
- Nkembo, A.T.; Salako, O.; Poku, R.A.; Amissah, F.; Ntantie, E.; Flores-Rozas, H.; Lamango, N.S. Disruption of actin filaments and suppression of pancreatic cancer cell viability and migration following treatment with polyisoprenylated cysteinyl amides. Am. J. Cancer Res. 2016, 6, 2532–2546. [Google Scholar]
- Poku, R.A.; Salako, O.O.; Amissah, F.; Nkembo, A.T.; Ntantie, E.; Lamango, N.S. Polyisoprenylated cysteinyl amide inhibitors induce caspase 3/7- and 8-mediated apoptosis and inhibit migration and invasion of metastatic prostate cancer cells. Am. J. Cancer Res. 2017, 7, 1515–1527. [Google Scholar] [PubMed]
- Tawfeeq, N.; Lazarte, J.M.S.; Jin, Y.; Gregory, M.D.; Lamango, N.S. Polyisoprenylated cysteinyl amide inhibitors deplete singly polyisoprenylated monomeric G-proteins in lung and breast cancer cell lines. Oncotarget 2023, 14, 243–257. [Google Scholar] [CrossRef]
- Ntantie, E.; Fletcher, J.; Amissah, F.; Salako, O.O.; Nkembo, A.T.; Poku, R.A.; Ikpatt, F.O.; Lamango, N.S. Polyisoprenylated cysteinyl amide inhibitors disrupt actin cytoskeleton organization, induce cell rounding and block migration of non-small cell lung cancer. Oncotarget 2017, 8, 31726–31744. [Google Scholar] [CrossRef]
- Villalobo, A.; Ishida, H.; Vogel, H.J.; Berchtold, M.W. Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochimica et Biophysica Acta (BBA)—Mol. Cell Res. 2018, 1865, 507–521. [Google Scholar] [CrossRef]
- Shimura, T.; Takenaka, Y.; Tsutsumi, S.; Hogan, V.; Kikuchi, A.; Raz, A. Galectin-3, a Novel Binding Partner of β-Catenin. Cancer Res. 2004, 64, 6363–6367. [Google Scholar] [CrossRef]
- Weise, K.; Kapoor, S.; Werkmüller, A.; Möbitz, S.; Zimmermann, G.; Triola, G.; Waldmann, H.; Winter, R. Dissociation of the K-Ras4B/PDEδ Complex upon Contact with Lipid Membranes: Membrane Delivery Instead of Extraction. J. Am. Chem. Soc. 2012, 134, 11503–11510. [Google Scholar] [CrossRef]
- Agamasu, C.; Ghirlando, R.; Taylor, T.; Messing, S.; Tran, T.H.; Bindu, L.; Tonelli, M.; Nissley, D.V.; McCormick, F.; Stephen, A.G. KRAS Prenylation Is Required for Bivalent Binding with Calmodulin in a Nucleotide-Independent Manner. Biophys. J. 2019, 116, 1049–1063. [Google Scholar] [CrossRef]
- Brtva, T.R.; Drugan, J.K.; Ghosh, S.; Terrell, R.S.; Campbell-Burk, S.; Bell, R.M.; Der, C.J. Two Distinct Raf Domains Mediate Interaction with Ras. J. Biol. Chem. 1995, 270, 9809–9812. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef]
- Jilaveanu, L.B.; Zito, C.R.; Aziz, S.A.; Conrad, P.J.; Schmitz, J.C.; Sznol, M.; Camp, R.L.; Rimm, D.L.; Kluger, H.M. C-Raf is associated with disease progression and cell proliferation in a subset of melanomas. Clin. Cancer Res. 2009, 15, 5704–5713. [Google Scholar] [CrossRef]
- Hagemann, C.; Rapp, U.R. Isotype-specific functions of Raf kinases. Exp. Cell Res. 1999, 253, 34–46. [Google Scholar] [CrossRef]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef]
- Murphy, L.O.; Blenis, J. MAPK signal specificity: The right place at the right time. Trends Biochem. Sci. 2006, 31, 268–275. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Woods, D.; Parry, D.; Cherwinski, H.; Bosch, E.; Lees, E.; McMahon, M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol. Cell Biol. 1997, 17, 5598–5611. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.S.; Hahn, W.C.; Gupta, P.; Weinberg, R.A. Genes involved in senescence and immortalization. Curr. Opin. Cell Biol. 2000, 12, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Guise, S.; Braguer, D.; Carles, G.; Delacourte, A.; Briand, C. Hyperphosphorylation of tau is mediated by ERK activation during anticancer drug-induced apoptosis in neuroblastoma cells. J. Neurosci. Res. 2001, 63, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Romeo, Y.; Roux, P.P. Paving the way for targeting RSK in cancer. Expert. Opin. Ther. Targets 2011, 15, 5–9. [Google Scholar] [CrossRef]
- Bignone, P.A.; Lee, K.Y.; Liu, Y.; Emilion, G.; Finch, J.; Soosay, A.E.R.; Charnock, F.M.L.; Beck, S.; Dunham, I.; Mungall, A.J.; et al. RPS6KA2, a putative tumour suppressor gene at 6q27 in sporadic epithelial ovarian cancer. Oncogene 2007, 26, 683–700. [Google Scholar] [CrossRef]
- Thakur, A.; Sun, Y.; Bollig, A.; Wu, J.; Biliran, H.; Banerjee, S.; Sarkar, F.H.; Liao, D.J. Anti-invasive and antimetastatic activities of ribosomal protein S6 kinase 4 in breast cancer cells. Clin. Cancer Res. 2008, 14, 4427–4436. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.G.; Yi, J.K.; Kim, H.S.; Lee, H.Y.; Lee, K.M.; Yi, M.; Ahn, S.; Shin, H.C.; Ju, J.H.; Shin, I.; et al. Phosphorylation of p90RSK is associated with increased response to neoadjuvant chemotherapy in ER-positive breast cancer. BMC Cancer 2012, 12, 585. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Park, Y.; Chen, C.C.; Xu, P.Z.; Chen, M.L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef]
- Izdebska, M.; Zielinska, W.; Grzanka, D.; Gagat, M. The Role of Actin Dynamics and Actin-Binding Proteins Expression in Epithelial-to-Mesenchymal Transition and Its Association with Cancer Progression and Evaluation of Possible Therapeutic Targets. Biomed. Res. Int. 2018, 2018, 4578373. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.L.; Herraiz, C.; Sanz-Moreno, V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014, 5, e29019. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure |
|---|---|
| NSL-YHJ-2-27 |  |
| NSL-YHJ-2-62 |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazarte, J.M.S.; Lamango, N.S. Activation of MAP Kinase Pathway by Polyisoprenylated Cysteinyl Amide Inhibitors Causes Apoptosis and Disrupts Breast Cancer Cell Invasion. Biomedicines 2024, 12, 470. https://doi.org/10.3390/biomedicines12030470
Lazarte JMS, Lamango NS. Activation of MAP Kinase Pathway by Polyisoprenylated Cysteinyl Amide Inhibitors Causes Apoptosis and Disrupts Breast Cancer Cell Invasion. Biomedicines. 2024; 12(3):470. https://doi.org/10.3390/biomedicines12030470
Chicago/Turabian StyleLazarte, Jassy Mary S., and Nazarius S. Lamango. 2024. "Activation of MAP Kinase Pathway by Polyisoprenylated Cysteinyl Amide Inhibitors Causes Apoptosis and Disrupts Breast Cancer Cell Invasion" Biomedicines 12, no. 3: 470. https://doi.org/10.3390/biomedicines12030470