
Unravelling the Complexity of the +33 C>G [HBB:c.-18C>G] Variant in Beta Thalassemia

 , , and
, , and
Abstract
:1. Introduction

{kind=link}
{kind=link}
| Variant Effect | Location (NG_) | Mild β | Silent β | IthaGenes ID | |
|---|---|---|---|---|---|
| Transcription | promoter | c.-151C>T | -101 C>T | IthaID: 3 | |
| c.-151C>G | -101 C>G | IthaID: 4 | |||
| c.-142C>T | -92 C>T | IthaID: 6 | |||
| c.-140C>T | -90 C>T | IthaID: 7 | |||
| c.-138C>T | -88 C>T | IthaID: 8 | |||
| c.-138C>A | -88 C>A | IthaID: 9 | |||
| c.-137C>G | -87 C>G | IthaID: 10 | |||
| c.-137C>T | -87 C>T | IthaID: 11 | |||
| c.-137C>A | -87 C>A | IthaID: 12 | |||
| c.-136C>A | -86 C>A | IthaID: 14 | |||
| c.-136C>T | -86 C>T | IthaID: 4028 | |||
| c.-136C>G | -86 C>G | IthaID: 13 | |||
| c.-123A>T | -73 A>T | IthaID: 15 | |||
| c.-81A>G | -31 A>G | IthaID: 20 | |||
| c.-80T>A | -30 T>A | IthaID: 22 | |||
| c.-79A>G | -29 A>G | IthaID: 25 | |||
| 5’UTR | c.-50A>C | CAP +1 A>C | IthaID: 34 | ||
| c.-43C>T | CAP +8 C>T | IthaID: 35 | |||
| c.-41delT | CAP +10 (-T) | CAP +10 (-T) | IthaID: 36 | ||
| c.-29G>A | CAP +22 G>A | IthaID: 38 | |||
| c.-18C>G | CAP +33 C>G | CAP +33 C>G | IthaID: 39 | ||
| RNA processing | Consensus splice site | c.92+6T>C | IVS I-6 T>C | IthaID: 111 | |
| c.316-7C>A | IVS II-844 C>A | IthaID: 219 | |||
| c.316-7C>G | IVS II-844 C>G | IthaID: 220 | |||
| Cryptic splice site | c.59A>G | CD19 AAC>AGC Hb Malay | IthaID: 79 | ||
| c.75T>A | CD 24 GGT>GGA | IthaID: 86 | |||
| c.82G>T | CD 27 GCC>TCC Hb Knossos | IthaID: 91 | |||
| Poly A site | c.*110T>C | AATAAA>AACAAA | IthaID: 272 | ||
| c.*111A>G | AATAAA>AATGAA | IthaID: 274 | |||
| c.*112A>G | AATAAA>AATAGA | IthaID: 275 | |||
| c.*113A>G | AATAAA>AATAAG | AATAAA>AATAAG | IthaID: 276 | ||
| c.*108A>C | AATAAA>CATAAA | IthaID: 270 | |||
| 3’UTR | c.*6C>G | Term CD +6 C>G (CAP +1480) | IthaID: 267 | ||
| c.*93_*105del | Term CD +90, del 13 bp (CAP +1567 to +1579) | IthaID: 269 | |||
| c.*47C>G | Term CD +47 C>G | IthaID: 268 | |||
2. Materials and Methods
2.1. Ethics Statement and Study Participants
2.2. Hematological Analyses
2.3. Molecular Analyses
2.4. Bioinformatics Analyses
3. Results
3.1. Case-Level and Segregation Data
3.2. Heterozygotes for β +33 C>G
3.3. Compound Heterozygotes for β +33 C>G with Other β-thalassemia Alleles
3.4. A Use Case for Comprehensive Genetic Analysis
3.5. Computational Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kountouris, P.; Lederer, C.W.; Fanis, P.; Feleki, X.; Old, J.; Kleanthous, M. IthaGenes: An Interactive Database for Haemoglobin Variations and Epidemiology. PLoS ONE 2014, 9, e103020. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. The Molecular Basis of β-Thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011700. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. Pathophysiology of β Thalassemia—A Guide to Molecular Therapies. Hematology 2005, 2005, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. Genetic modifiers of the beta-haemoglobinopathies. Br J Haematol. 2008, 141, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. Genetic insights into the clinical diversity of β thalassaemia. Br. J. Haematol. 2004, 124, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Cao, A. Relationship between Genotype and Phenotype: Thalassemia Intermediaa. Ann. N. Y. Acad. Sci. 1998, 850, 325–333. [Google Scholar] [CrossRef]
- Origa, R. Beta-Thalassemia. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1426/ (accessed on 23 May 2023).
- Kountouris, P.; Michailidou, K.; Christou, S.; Hadjigavriel, M.; Sitarou, M.; Kolnagou, A.; Kleanthous, M.; Telfer, P. Effect of HBB genotype on survival in a cohort of transfusion-dependent thalassemia patients in Cyprus. Haematologica 2020, 106, 2458–2468. [Google Scholar] [CrossRef]
- Ho, P.J.; Rochette, J.; Fisher, C.A.; Wonke, B.; Jarvis, M.K.; Yardumian, A.; Thein, S.L. Moderate reduction of beta-globin gene transcript by a novel mutation in the 5’ untranslated region: A study of its interaction with other genotypes in two families. Blood 1996, 87, 1170–1178. [Google Scholar] [CrossRef]
- Bento, M.C.; Ribeiro, M.L.; Cunha, E.; Goncalves, P.; Martin-Nunez, G.; Tamagnini, G. b-thalassemia intermedia resulting from compound heterozigosity for an IVSI-1 (G-A) and a silent 5’ UTR +33 (C-G) mutations. Haematologica 2000, 85, 443–444. [Google Scholar] [PubMed]
- Xenophontos, M.; Minaidou, A.; Stephanou, C.; Tamana, S.; Kleanthous, M.; Kountouris, P. IthaPhen: An Interactive Database of Genotype-Phenotype Data for Hemoglobinopathies. HemaSphere 2023, 7, e922. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Stephanou, C.; Lederer, C.W.; Traeger-Synodinos, J.; Bento, C.; Harteveld, C.L.; Fylaktou, E.; Koopmann, T.T.; Halim-Fikri, H.; Michailidou, K.; et al. Adapting the ACMG/AMP variant classification framework: A perspective from the ClinGen Hemoglobinopathy Variant Curation Expert Panel. Hum. Mutat. 2022, 43, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.; Galanello, R. Beta-thalassemia. Genet. Med. 2010, 12, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Kousiappa, I.; Papasavva, T.; Christopoulos, G.; Pavlou, E.; Petrou, M.; Feleki, X.; Karitzie, E.; Phylactides, M.; Fanis, P.; et al. The molecular spectrum and distribution of haemoglobinopathies in Cyprus: A 20-year retrospective study. Sci. Rep. 2016, 6, 26371. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Ayyub, M.; Khan, S.A.; Ahmed, S.; Abbas, K.; Malik, H.S.; Tashfeen, S. Frequency of Gγ-globin promoter -158 (C>T) XmnI polymorphism in patients with homozygous/compound heterozygous beta thalassaemia. Hematol. Oncol. Stem Cell Ther. 2015, 8, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Tamana, S.; Xenophontos, M.; Minaidou, A.; Stephanou, C.; Harteveld, C.L.; Bento, C.; Traeger-Synodinos, J.; Fylaktou, I.; Yasin, N.M.; Abdul Hamid, F.S.; et al. Evaluation of in silico predictors on short nucleotide variants in HBA1, HBA2, and HBB associated with haemoglobinopathies. eLife 2022, 11, e79713. [Google Scholar] [CrossRef]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef]
- Brancaleoni, V.; Di Pierro, E.; Motta, I.; Cappellini, M.D. Laboratory diagnosis of thalassemia. Int. J. Lab. Hematol. 2016, 38, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Vadolas, J.; Nefedov, M.; Wardan, H.; Mansooriderakshan, S.; Voullaire, L.; Jamsai, D.; Williamson, R.; Ioannou, P.A. Humanized beta-thalassemia mouse model containing the common IVSI-110 splicing mutation. J Biol Chem. 2006, 281, 7399–7405. [Google Scholar] [CrossRef] [PubMed]
- Uludağ, A.; Uysal, A.; Uludağ, A.; Ertekin, Y.H.; Tekin, M.; Kütük, B.; Silan, F.; Özdemir, Ö. Prevalence and mutations of β-thalassemia trait and abnormal hemoglobins in premarital screening in Çanakkale province, Turkey. Balk. J. Med. Genet. BJMG 2016, 19, 29. [Google Scholar] [CrossRef] [PubMed]
- Lo Giudice, C.; Zambelli, F.; Chiara, M.; Pavesi, G.; Tangaro, M.A.; Picardi, E.; Pesole, G. UTRdb 2.0: A comprehensive, expert curated catalog of eukaryotic mRNAs untranslated regions. Nucleic Acids Res. 2023, 51, D337–D344. [Google Scholar] [CrossRef] [PubMed]
- Sgourou, A.; Routledge, S.; Antoniou, M.; Papachatzopoulou, A.; Psiouri, L.; Athanassiadou, A. Thalassaemia mutations within the 5′UTR of the human β-globin gene disrupt transcription. Br. J. Haematol. 2004, 124, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.J.; Sloane-Stanley, J.; Athanassiadou, A.; Wood, W.G.; Thein, S.L. An in vitro system for expression analysis of mutations of the β-globin gene: Validation and application to two mutations in the 5’ UTR. Br. J. Haematol. 1999, 106, 938–947. [Google Scholar] [CrossRef]
- Lewis, B.A.; Kim, T.-K.; Orkin, S.H. A downstream element in the human β-globin promoter: Evidence of extended sequence-specific transcription factor IID contacts. Proc. Natl. Acad. Sci. USA 2000, 97, 7172–7177. [Google Scholar] [CrossRef]
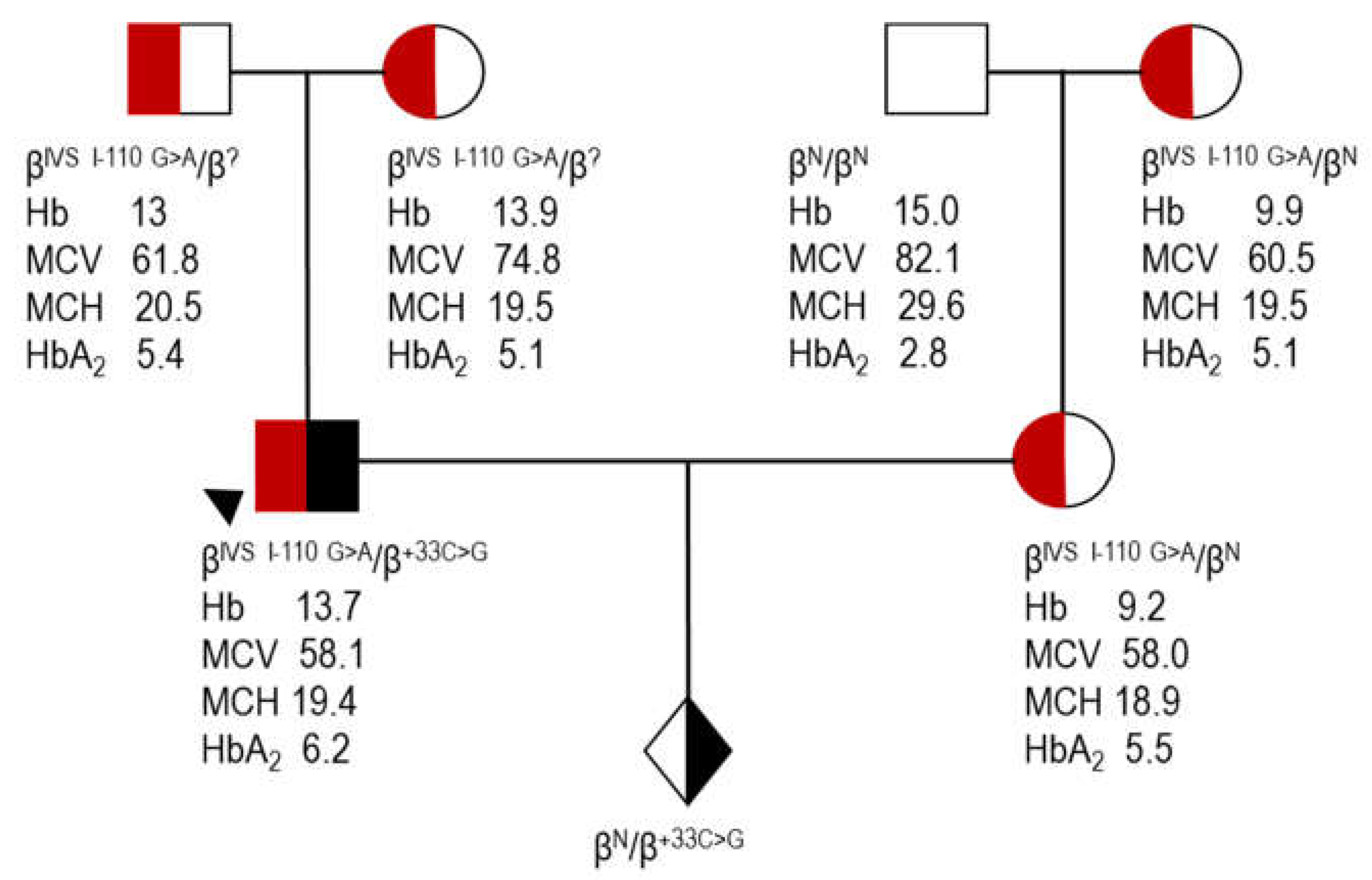
 proband (case 7, Table 4); Hb, hemoglobin; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; HbA2, A2 hemoglobin; ?, unknown β allele as a result of direct detection methods utilized for the identification of the most common β variants.
proband (case 7, Table 4); Hb, hemoglobin; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; HbA2, A2 hemoglobin; ?, unknown β allele as a result of direct detection methods utilized for the identification of the most common β variants.
proband (case 7, Table 4); Hb, hemoglobin; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; HbA2, A2 hemoglobin; ?, unknown β allele as a result of direct detection methods utilized for the identification of the most common β variants.
proband (case 7, Table 4); Hb, hemoglobin; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; HbA2, A2 hemoglobin; ?, unknown β allele as a result of direct detection methods utilized for the identification of the most common β variants.

| Heterozygotes | Compound Heterozygotes | |||||
|---|---|---|---|---|---|---|
| Variable | n | Mean ± SD | Median (min–max) | n | Mean ± SD | Median (min–max) |
| Sex at birth | ||||||
| Female | 16 | 7 | ||||
| Male | 10 | 4 | ||||
| Age (years) | ||||||
| Female | 16 | 37.4 ± 17.38 | 30 (16–66) | 7 | 37.43 ± 17.9 | 34 (15–61) |
| Male | 10 | 32.2 ± 14.7 | 29 (17–70) | 4 | 25.5 ± 19.33 | 23.5 (4–51) |
| Hb (g/dL) | ||||||
| Female | 12 | 13.14 ± 0.96 | 13.4 (11.6–14.3) | 7 | 8.86 ± 0.56 | 8.7 (8–9.6) |
| Male | 6 | 15.47 ± 1.17 | 15.6 (13.7–17.2) | 3 | 10.13 ± 3.11 | 8.7 (8–13.7) |
| RBC (×106) | ||||||
| Female | 12 | 4.72 ± 0.3 | 4.73 (4.23–5.35) | 7 | 4.62 ± 0.48 | 4.56 (3.99–5.24) |
| Male | 6 | 5.38 ± 0.43 | 5.28 (4.98–6.22) | 3 | 5.31 ± 1.54 | 4.64 (4.21–7.07) |
| PCV (%) | ||||||
| Female | 12 | 39.9 ± 1.99 | 40.05 (36.4–42.4) | 7 | 28.01 ± 2.49 | 26.8 (25.2–31.9) |
| Male | 6 | 46.02 ± 2.95 | 46.55 (41.7–49.9) | 3 | 31.23 ± 8.6 | 27.3 (25.3–41.1) |
| MCV (fL) | 26 | 85.19 ± 5.39 | 85.4 (69.9–93.5) | 10 | 60.28 ± 3.59 | 61.05 (54.5–65.2) |
| MCH (pg) | 26 | 28.12 ± 2.04 | 28.3 (22.7–31.3) | 10 | 19.22 ± 1.33 | 19.4 (17.2–21.3) |
| MCHC (g/dL) | 18 | 33.05 ± 0.99 | 33.05 (30.5–34.7) | 10 | 31.87 ± 1.0 | 31.85 (29.8–33.3) |
| HbA2 (%) | 26 | 3.02 ± 0.27 | 3 (2.4–3.6) | 9 | 5.84 ± 0.37 | 5.8 (5.2–6.4) |
| RDW (%) | 10 | 13.89 ± 1.8 | 13.55 (11.9–18.7) | |||
| HbF (%) | 7 | 3.97 ± 2.71 | 3.1 (1.5–8.2) | |||
| Transfusion phenotype | ||||||
| NTDT | 6 | |||||
| TDT | 3 | |||||
| Bone deformities | ||||||
| Present | 1 | |||||
| Absent | 8 | |||||
| Splenomegaly | ||||||
| Present | 3 | |||||
| Absent | 6 | |||||
| Hepatomegaly | ||||||
| Present | 0 | |||||
| Absent | 8 | |||||
| Splenectomy | ||||||
| Yes | 0 | |||||
| No | 9 | |||||
| Case | Sex-Age | Hb g/dL | RBC ×106 | RDW % | PCV % | MCV fL | MCH pg | MCHC g/dL | HbA2 % | α Genotype |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F-66 | 13.7 | 4.86 | 41.5 | 85.4 | 28.2 | 33 | 2.8 | ||
| 2 | F-30 | 14 | 4.79 | 40.7 | 85 | 29.2 | 32.6 | 3.2 | ||
| 3 | F-23 | 11.6 | 4.53 | 38 | 83.9 | 25.6 | 30.5 | 2.8 | αα/-α3.7 | |
| 4 | F-30 | 11.9 | 4.23 | 12.9 | 36.9 | 87.1 | 28 | 32.2 | 3 | αα/αα |
| 5 | F-65 | 14.3 | 4.93 | 14.1 | 42.1 | 85.4 | 29 | 34 | 2.9 | |
| 6 | F-56 | 13.9 | 5.35 | 42.4 | 79.3 | 26 | 32.8 | 2.4 | αα/-α3.7 | |
| 7 | F-23 | 11.7 | 4.97 | 18.7 | 36.4 | 73.2 | 23.5 | 32.1 | 2.7 | αα/αα |
| 8 | F-44 | 14 | 4.48 | 13.3 | 41.9 | 93.5 | 31.3 | 33.4 | 2.7 | |
| 9 | F-51 | 12.7 | 4.9 | 13.9 | 40 | 81.6 | 25.9 | 31.8 | 2.6 | |
| 10 | F-58 | 13.6 | 4.43 | 13.6 | 40.1 | 90.5 | 30.7 | 33.9 | 3.3 | |
| 11 | F-27 | 13.1 | 4.56 | 13.2 | 39.6 | 86.8 | 28.7 | 33.1 | 3.3 | |
| 12 | F-27 | 69.9 | 22.7 | 3.5 | -α3.7/-α3.7 | |||||
| 13 | F-22 | 90.3 | 30.2 | 3 | ||||||
| 14 | F-25 | 91 | 29.6 | 3.2 | ||||||
| 15 | F-16 | 90.1 | 28.5 | 3 | ||||||
| 16 | F-21 | 13.2 | 4.66 | 39.3 | 84.3 | 28.3 | 33.6 | 2.9 | ||
| 17 | M-70 | 13.7 | 4.98 | 41.7 | 83.7 | 27.5 | 32.9 | 2.9 | ||
| 18 | M-33 | 15.7 | 5.29 | 11.9 | 47.8 | 90.3 | 29.7 | 32.9 | 3 | αα/αα |
| 19 | M-34 | 17.2 | 6.22 | 49.9 | 80.2 | 27.7 | 34.7 | 3.1 | ||
| 20 | M-32 | 15.5 | 5.19 | 13.5 | 46.3 | 89.2 | 29.9 | 33.5 | 3.3 | |
| 21 | M-26 | 88.2 | 28.3 | 3 | ||||||
| 22 | M-38 | 14.8 | 5.35 | 13.8 | 43.6 | 81.5 | 27.7 | 33.9 | 3 | |
| 23 | M-25 | 86.9 | 29 | 3 | ||||||
| 24 | M-17 | 85.3 | 27.7 | 3.1 | ||||||
| 25 | M-22 | 83.6 | 27.9 | 3.2 | ||||||
| 26 | M-25 | 15.9 | 5.27 | 46.8 | 88.8 | 30.2 | 34 | 3.6 |
| Case | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex/Age | F/46 | F/61 | F/58 | F/23 | M/4 | M/23 | M/24 | F/25 | F/34 | M/51 | F/15 |
| Hb g/dL | 9 | 9.5 | 8 | 8.7 | 8 | 13.7 | 8.7 | 8.5 | 8.7 | 9.6 | |
| RBC × 106 | 5.24 | 5.13 | 4.14 | 4.38 | 4.64 | 7.07 | 4.56 | 3.99 | 4.21 | 4.92 | |
| MCV fL | 55.5 | 62.2 | 60.9 | 61.2 | 54.5 | 58.1 | 58.6 | 65.2 | 64.8 | 61.8 | |
| MCH pg | 17.2 | 18.5 | 19.4 | 19.9 | 17.2 | 19.4 | 19.1 | 21.3 | 20.7 | 19.5 | |
| MCHC g/dL | 30.9 | 29.8 | 31.8 | 32.5 | 31.6 | 33.3 | 32.6 | 32.7 | 31.9 | 31.6 | |
| PCV % | 29.1 | 31.9 | 25.2 | 26.8 | 25.3 | 41.1 | 26.7 | 26 | 27.3 | 30.4 | |
| HbA2 % | 5.4 | NA | 6 | 5.8 | 6 | 6.2 | 6.4 | 5.8 | 5.2 | 5.8 | |
| HbF % | 3.1 | 1.6 | 1.5 | 4 | NA | 8.2 | 2.2 | 7.2 | |||
| β/α ratio | 0.31 | 0.25 | |||||||||
| Film | ++ | ++++ | + | +++ | ++++ | ++ | +++ | ||||
| β genotype | CD39/+33 | CD39/+33 | CD39/+33 | CD39/+33 | IVSI-110/+33 | IVSI-110/+33 | IVSI-110/+33 | IVSI-110/+33 | IVSI-110/+33 | IVSI-110/+33 | IVSI-1/+33 |
| α genotype | αα/αα | αα/αα | αα/αα | αα/αα | αα/αα | αα/αα | αα/-α3.7 | αα/αα | αα/αα | αα/αα | αα/αα |
| XmnI | −/− | −/− | −/− | −/− | −/− | −/− | +/− | −/− | +/− | −/− | −/− |
| Thalassemiaphenotype | NTDT | NTDT | TDT | TDT | NTDT | TDT | NTDT | NTDT | NTDT | ||
| Transfusion frequency | Rare (once post birth, once post surgery) | Never | Regular (began at 31 yr) | Regular (began at 35 yr) | Never | Regular (began at 36 mo) | Never | Rare (post both pregnancies) | Never | Never | Never |
| Bonedeformities | None | None | Facial | None | None | None | None | None | None | ||
| Splenomegaly | No | No | Yes | No | Yes | No | No | No | Barely palpable | ||
| Hepatomegaly | No | No | No | No | No | No | No | No | |||
| Splenectomy | No | No | No | No | No | No | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stephanou, C.; Petrou, M.; Kountouris, P.; Makariou, C.; Christou, S.; Hadjigavriel, M.; Kleanthous, M.; Papasavva, T. Unravelling the Complexity of the +33 C>G [HBB:c.-18C>G] Variant in Beta Thalassemia. Biomedicines 2024, 12, 296. https://doi.org/10.3390/biomedicines12020296
Stephanou C, Petrou M, Kountouris P, Makariou C, Christou S, Hadjigavriel M, Kleanthous M, Papasavva T. Unravelling the Complexity of the +33 C>G [HBB:c.-18C>G] Variant in Beta Thalassemia. Biomedicines. 2024; 12(2):296. https://doi.org/10.3390/biomedicines12020296
Chicago/Turabian StyleStephanou, Coralea, Miranda Petrou, Petros Kountouris, Christiana Makariou, Soteroula Christou, Michael Hadjigavriel, Marina Kleanthous, and Thessalia Papasavva. 2024. "Unravelling the Complexity of the +33 C>G [HBB:c.-18C>G] Variant in Beta Thalassemia" Biomedicines 12, no. 2: 296. https://doi.org/10.3390/biomedicines12020296