
Cellular Senescence in Liver Cancer: How Dying Cells Become “Zombie” Enemies
 , , ,
, , ,  ,
,  and
and 
Abstract
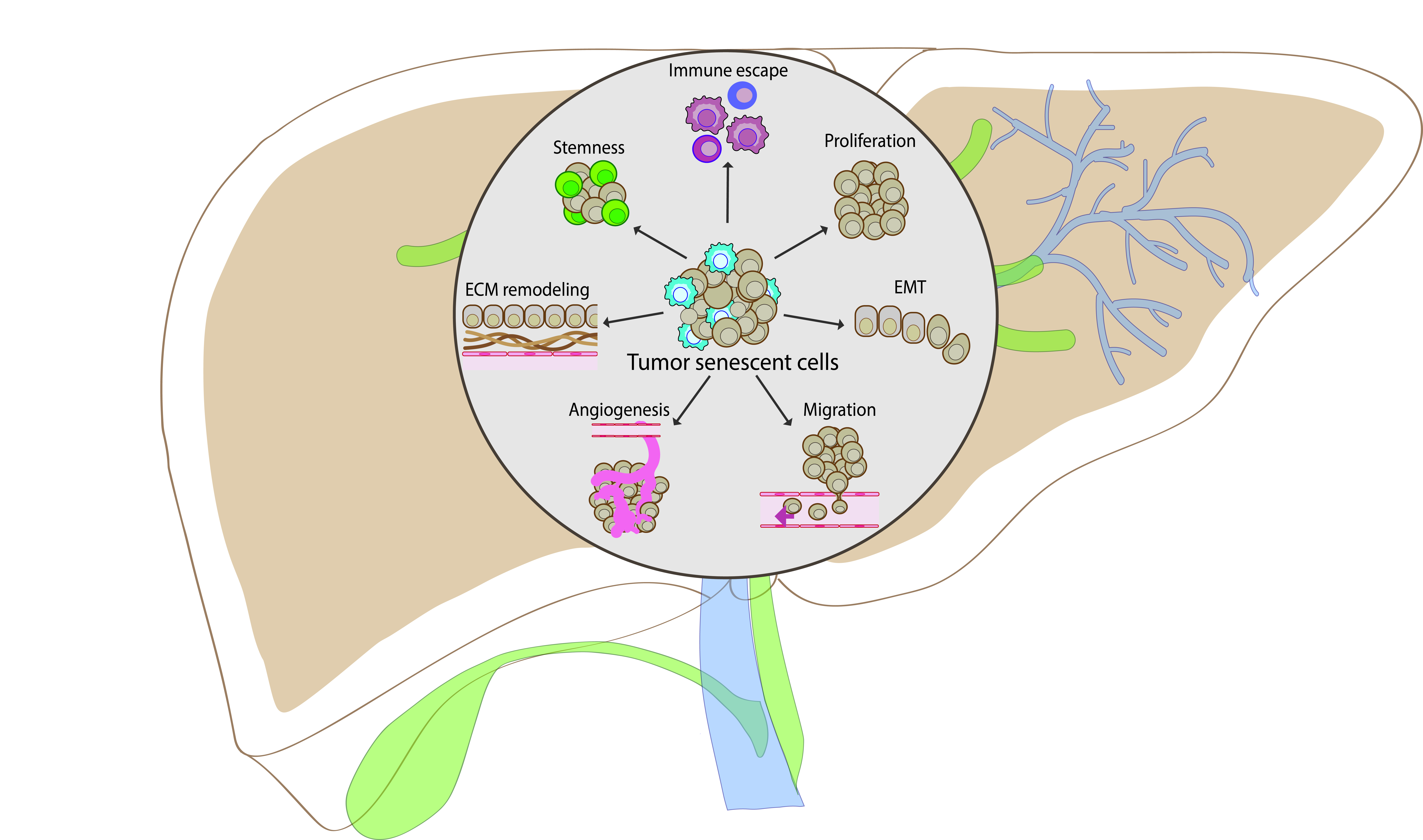
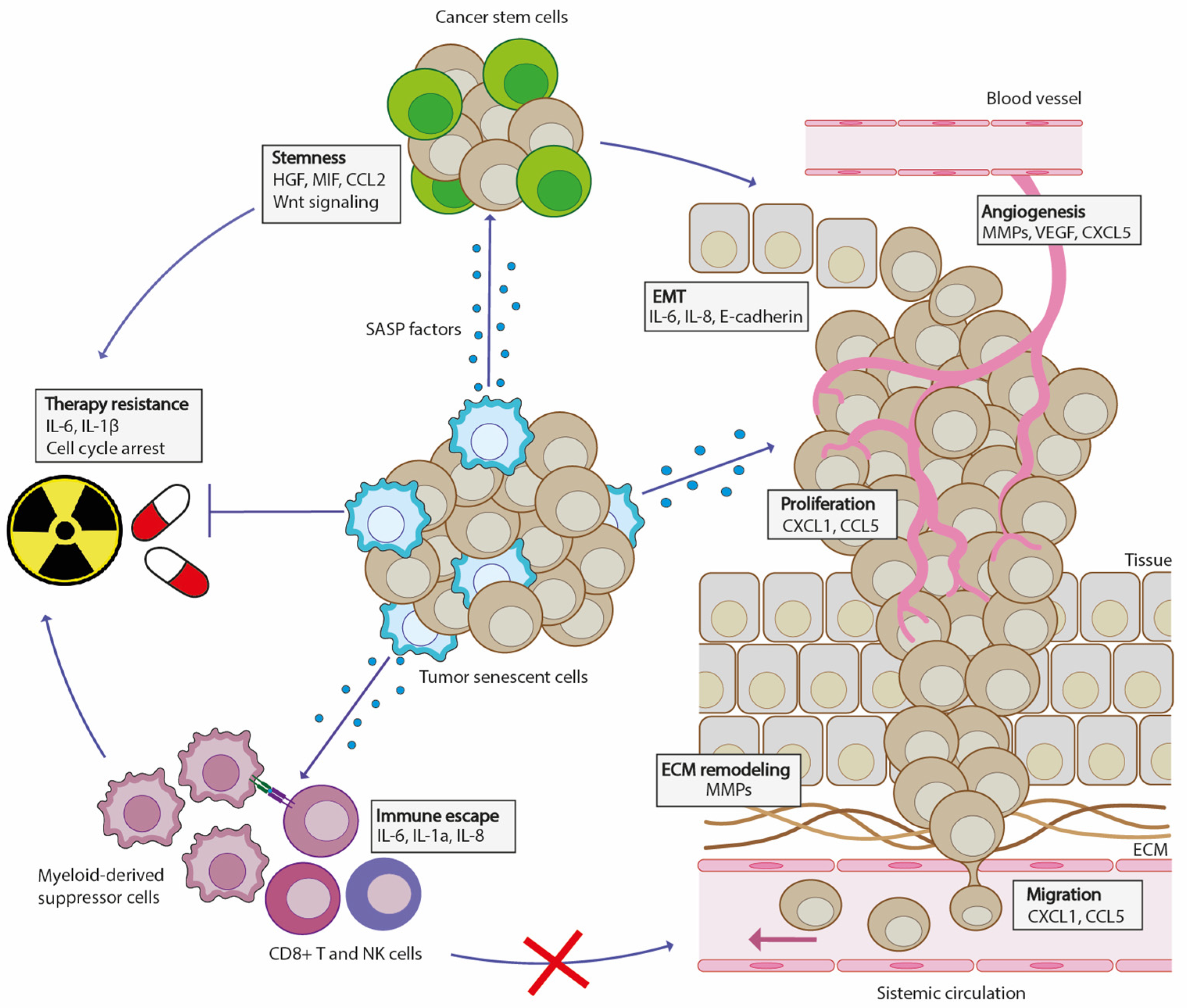
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Cellular Senescence: Origin and Distinctive Features
3. A Controversial Behavior in Cancer: When Senescent Cells Become “Zombies”
4. The Role of Cellular Senescence in Liver Cancer
4.1. Senescence in Hepatocellular Carcinoma (HCC)
4.2. Senescence in Cholangiocarcinoma (CCA)
4.3. Senescence in Colorectal Liver Metastases (CLM)
5. Conclusions and Future Challenges: How Senescence Can Be Exploited for Liver Cancer Treatments
Author Contributions
Funding
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Leonardo, A.D.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA damage triggers a prolonged p53- dependent G1 arrest ana long-term induction of Cipl in normal human fibroblasts. Genes Dev. 1994, 8, 2540–2551. [Google Scholar] [CrossRef] [PubMed]
- Gire, V.; Dulić, V. Senescence from G2 arrest, revisited. Cell Cycle 2015, 14, 297–304. [Google Scholar] [CrossRef]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for targeting senescent cells in human disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Tchkonia, T.; Niedernhofer, L.J.; Robbins, P.D.; Kirkland, J.L.; Lee, S. COVID-19 and cellular senescence. Nat. Rev. Immunol. 2023, 23, 251–263. [Google Scholar] [CrossRef]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, À.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Guo, S.; DiPietro, L.A. Factors Affecting Wound Healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef]
- Goutas, A.; Outskouni, Z.; Papathanasiou, I.; Satra, M.; Koliakos, G.; Trachana, V. Dysregulation of Caveolin-1 Phosphorylation and Nuclear Translocation Is Associated with Senescence Onset. Cells 2021, 10, 2939. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, A.; O’Loghlen, A. Cellular Senescence and Ageing: Mechanisms and Interventions. Front. Aging 2022, 3, 866718. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer—Role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Park, S.S.; Choi, Y.W.; Kim, J.H.; Kim, H.S.; Park, T.J. Senescent tumor cells: An overlooked adversary in the battle against cancer. Exp. Mol. Med. 2021, 53, 1834–1841. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Liu, P.; Tang, Q.; Chen, M.; Chen, W.; Lu, Y.; Liu, Z.; He, Z. Hepatocellular Senescence: Immunosurveillance and Future Senescence-Induced Therapy in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 589908. [Google Scholar] [CrossRef]
- Ozturk, M.; Arslan-Ergul, A.; Bagislar, S.; Senturk, S.; Yuzugullu, H. Senescence and immortality in hepatocellular carcinoma. Cancer Lett. 2009, 286, 103–113. [Google Scholar] [CrossRef]
- Spinelli, R.; Baboota, R.K.; Gogg, S.; Beguinot, F.; Blüher, M.; Nerstedt, A.; Smith, U. Increased cell senescence in human metabolic disorders. J. Clin. Investig. 2023, 133, e169922. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.L.; Gasek, N.S.; Kuchel, G.A.; Xu, M. The heterogeneity of cellular senescence: Insights at the single-cell level. Trends Cell Biol. 2023, 33, 9–17. [Google Scholar] [CrossRef]
- Rossiello, F.; Herbig, U.; Longhese, M.P.; Fumagalli, M.; d’Adda Di Fagagna, F. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 2014, 26, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Sadasivam, S.; DeCaprio, J.A. The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Masgras, I.; Carrera, S.; de Verdier, P.J.; Brennan, P.; Majid, A.; Makhtar, W.; Tulchinsky, E.; Jones, G.D.D.; Roninson, I.B.; Macip, S. Reactive Oxygen Species and Mitochondrial Sensitivity to Oxidative Stress Determine Induction of Cancer Cell Death by p21. J. Biol. Chem. 2012, 287, 9845–9854. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.-D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99.e6. [Google Scholar] [CrossRef]
- Omori, S.; Wang, T.-W.; Johmura, Y.; Kanai, T.; Nakano, Y.; Kido, T.; Susaki, E.A.; Nakajima, T.; Shichino, S.; Ueha, S.; et al. Generation of a p16 Reporter Mouse and Its Use to Characterize and Target p16high Cells In Vivo. Cell Metab. 2020, 32, 814–828.e6. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.I.; et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 2017, 9, 1867–1884. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef] [PubMed]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef] [PubMed]
- Dikovskaya, D.; Cole, J.J.; Mason, S.M.; Nixon, C.; Karim, S.A.; McGarry, L.; Clark, W.; Hewitt, R.N.; Sammons, M.A.; Zhu, J.; et al. Mitotic Stress Is an Integral Part of the Oncogene-Induced Senescence Program that Promotes Multinucleation and Cell Cycle Arrest. Cell Rep. 2015, 12, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; Chang, C.; Lin, B.; Wu, Y.; Wu, M.; Lin, L.; Huang, W.; Holz, J.D.; Sheu, T.; Lee, J.; et al. Up-regulation of cofilin-1 in cell senescence associates with morphological change and p27kip1-mediated growth delay. Aging Cell 2021, 20, e13288. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Kashyap, S.; Chini, E.; Von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Liu, J.; Cai, S.-Z.; Zhou, Y.; Zhang, X.-P.; Liu, D.-F.; Jiang, R.; Wang, Y.-P. Senescence as A Consequence of Ginsenoside Rg1 Response on K562 Human Leukemia Cell Line. Asian Pac. J. Cancer Prev. 2012, 13, 6191–6196. [Google Scholar] [CrossRef]
- Druelle, C.; Drullion, C.; Deslé, J.; Martin, N.; Saas, L.; Cormenier, J.; Malaquin, N.; Huot, L.; Slomianny, C.; Bouali, F.; et al. ATF6α regulates morphological changes associated with senescence in human fibroblasts. Oncotarget 2016, 7, 67699–67715. [Google Scholar] [CrossRef]
- Lloyd, A.C. The Regulation of Cell Size. Cell 2013, 154, 1194–1205. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Tsuji, S.; Minami, S.; Hashimoto, R.; Konishi, Y.; Suzuki, T.; Kondo, T.; Sasai, M.; Torii, S.; Ono, C.; Shichinohe, S.; et al. SARS-CoV-2 infection triggers paracrine senescence and leads to a sustained senescence-associated inflammatory response. Nat. Aging 2022, 2, 115–124. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Tripathi, U.; Misra, A.; Tchkonia, T.; Kirkland, J.L. Impact of Senescent Cell Subtypes on Tissue Dysfunction and Repair: Importance and Research Questions. Mech. Ageing Dev. 2021, 198, 111548. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; De Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef]
- Ryu, S.J.; Oh, Y.S.; Park, S.C. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 2007, 14, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone Modifications in Senescence-Associated Resistance to Apoptosis by Oxidative Stress. Redox Biol. 2013, 1, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Scudellari, M. To stay young, kill zombie cells. Nature 2017, 550, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Park, T.J. Cellular senescence in cancer. BMB Rep. 2019, 52, 42–46. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Wang, H.J.; Tan, Y.Z. Wnt/β-Catenin Signaling Induces the Aging of Mesenchymal Stem Cells through the DNA Damage Response and the p53/p21 Pathway. PLoS ONE 2011, 6, e21397. [Google Scholar] [CrossRef]
- Osanai, M.; Murata, M.; Nishikiori, N.; Chiba, H.; Kojima, T.; Sawada, N. Occludin-mediated premature senescence is a fail-safe mechanism against tumorigenesis in breast carcinoma cells. Cancer Sci. 2007, 98, 1027–1034. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A Senescence Program Controlled by p53 and p16INK4a Contributes to the Outcome of Cancer Therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; van Deursen, J.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. JNCI J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, J.S. Exploiting tumor cell senescence in anticancer therapy. BMB Rep. 2014, 47, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B. Tumor Cell Senescence in Cancer Treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Sabin, R.J.; Anderson, R.M. Cellular Senescence—Its role in cancer and the response to ionizing radiation. Genome Integr. 2011, 2, 7. [Google Scholar] [CrossRef]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef]
- Coppé, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor Suppressor and Aging Biomarker p16INK4a Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef]
- Zhu, Y.; Xu, L.; Zhang, J.; Hu, X.; Liu, Y.; Yin, H.; Lv, T.; Zhang, H.; Liu, L.; An, H.; et al. Sunitinib induces cellular senescence via p53/ D ec1 activation in renal cell carcinoma cells. Cancer Sci. 2013, 104, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Däbritz, J.H.M.; Yu, Y.; Milanovic, M.; Schönlein, M.; Rosenfeldt, M.T.; Dörr, J.R.; Kaufmann, A.M.; Dörken, B.; Schmitt, C.A. CD20-Targeting Immunotherapy Promotes Cellular Senescence in B-Cell Lymphoma. Mol. Cancer Ther. 2016, 15, 1074–1081. [Google Scholar] [CrossRef]
- Zhu, H.; Blake, S.; Kusuma, F.K.; Pearson, R.B.; Kang, J.; Chan, K.T. Oncogene-induced senescence: From biology to therapy. Mech. Ageing Dev. 2020, 187, 111229. [Google Scholar] [CrossRef] [PubMed]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 7031–7036. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Reimann, M.; Lee, S.; Loddenkemper, C.; Dörr, J.R.; Tabor, V.; Aichele, P.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Tumor Stroma-Derived TGF-β Limits Myc-Driven Lymphomagenesis via Suv39h1-Dependent Senescence. Cancer Cell 2010, 17, 262–272. [Google Scholar] [CrossRef]
- Yang, J.; Liu, M.; Hong, D.; Zeng, M.; Zhang, X. The Paradoxical Role of Cellular Senescence in Cancer. Front. Cell Dev. Biol. 2021, 9, 722205. [Google Scholar] [CrossRef]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (SASP) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Yang, G.; Rosen, D.G.; Zhang, Z.; Bast, R.C.; Mills, G.B.; Colacino, J.A.; Mercado-Uribe, I.; Liu, J. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 16472–16477. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Colombatti, A. The Inflammatory Chemokine CCL5 and Cancer Progression. Mediat. Inflamm. 2014, 2014, 292376. [Google Scholar] [CrossRef] [PubMed]
- Alba, J.; Barcia, R.; Gutiérrez-Berzal, J.; Ramos-Martínez, J.I. Could inhibition of metalloproteinases be used to block the process of metastasis? Cell Biochem. Funct. 2022, 40, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Vilotić, A.; Nacka-Aleksić, M.; Pirković, A.; Bojić-Trbojević, Ž.; Dekanski, D.; Jovanović Krivokuća, M. IL-6 and IL-8: An Overview of Their Roles in Healthy and Pathological Pregnancies. Int. J. Mol. Sci. 2022, 23, 14574. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Komoda, K.; Mikawa, R.; Asai, A.; Sugimoto, M. Cellular senescence promotes cancer metastasis by enhancing soluble E-cadherin production. iScience 2021, 24, 103022. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xu, Z.-Q.; Zong, Y.-P.; Ou, B.-C.; Shen, X.-H.; Feng, H.; Zheng, M.-H.; Zhao, J.-K.; Lu, A.-G. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 2019, 10, 178. [Google Scholar] [CrossRef]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.-H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Dou, Z.; Berger, S.L. Senescence Elicits Stemness: A Surprising Mechanism for Cancer Relapse. Cell Metab. 2018, 27, 710–711. [Google Scholar] [CrossRef]
- Muñoz-Galván, S.; Lucena-Cacace, A.; Perez, M.; Otero-Albiol, D.; Gomez-Cambronero, J.; Carnero, A. Tumor cell-secreted PLD increases tumor stemness by senescence-mediated communication with microenvironment. Oncogene 2019, 38, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.R.; Ritchie, S.; Pereira, B.A.; Timpson, P. Overcoming the senescence-associated secretory phenotype (SASP): A complex mechanism of resistance in the treatment of cancer. Mol. Oncol. 2021, 15, 3242–3255. [Google Scholar] [CrossRef] [PubMed]
- Battram, A.M.; Bachiller, M.; Martín-Antonio, B. Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword. Int. J. Mol. Sci. 2020, 21, 4346. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Pretzsch, E.; Nieß, H.; Bösch, F.; Westphalen, C.; Jacob, S.; Neumann, J.; Werner, J.; Heinemann, V.; Angele, M. Age and metastasis—How age influences metastatic spread in cancer. Colorectal cancer as a model. Cancer Epidemiol. 2022, 77, 102112. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, A.; Chakraborty, S.; Bhattacharya, R.; Chowdhury, G. Senescence-Induced Chemoresistance in Triple Negative Breast Cancer and Evolution-Based Treatment Strategies. Front. Oncol. 2021, 11, 674354. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Wang, Z.; Lu, X. Impact of immunosenescence and inflammaging on the effects of immune checkpoint inhibitors. Cancer Pathog. Ther. 2023, 1, E032. [Google Scholar] [CrossRef]
- Liu, Z.; Liang, Q.; Ren, Y.; Guo, C.; Ge, X.; Wang, L.; Cheng, Q.; Luo, P.; Zhang, Y.; Han, X. Immunosenescence: Molecular mechanisms and diseases. Signal Transduct. Target. Ther. 2023, 8, 200. [Google Scholar] [CrossRef]
- Volponi, C.; Gazzillo, A.; Bonavita, E. The Tumor Microenvironment of Hepatocellular Carcinoma: Untying an Intricate Immunological Network. Cancers 2022, 14, 6151. [Google Scholar] [CrossRef]
- Nakagawa, H. Inflammation- and stress-related signaling pathways in hepatocarcinogenesis. World J. Gastroenterol. 2012, 18, 4071. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2018, 12, 153–186. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, P.; Kulik, L.M. Hepatocellular Carcinoma. Clin. Liver Dis. 2023, 27, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, L.; Vetrano, E.; Rinaldi, B.; Galiero, R.; Caturano, A.; Salvatore, T.; Sasso, F.C. HCC and Molecular Targeting Therapies: Back to the Future. Biomedicines 2021, 9, 1345. [Google Scholar] [CrossRef] [PubMed]
- Gilles, H.; Garbutt, T.; Landrum, J. Hepatocellular Carcinoma. Crit. Care Nurs. Clin. N. Am. 2022, 34, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Guerra, N.; Fessas, P.; Murphy, R.; Mineo, T.; Mauri, F.A.; Mukherjee, S.K.; Thursz, M.; Wong, C.N.; Sharma, R.; et al. Immune-based therapies for hepatocellular carcinoma. Oncogene 2020, 39, 3620–3637. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Sangro, B. Targets for immunotherapy of liver cancer. J. Hepatol. 2018, 68, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef]
- Jiang, Y.; Que, W.; Zhu, P.; Li, X.K. The Role of Diverse Liver Cells in Liver Transplantation Tolerance. Front. Immunol. 2020, 11, 1203. [Google Scholar] [CrossRef]
- Marin, J.J.; Macias, R.I.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1663. [Google Scholar] [CrossRef]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. UHRF1 Overexpression Drives DNA Hypomethylation and Hepatocellular Carcinoma. Cancer Cell 2014, 25, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Feng, D.; Mathews, S.; Gao, B. Hepatoprotective and anti-fibrotic functions of interleukin-22: Therapeutic potential for the treatment of alcoholic liver disease. J. Gastroenterol. Hepatol. 2013, 28, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Tordella, L.; Khan, S.; Hohmeyer, A.; Banito, A.; Klotz, S.; Raguz, S.; Martin, N.; Dhamarlingam, G.; Carroll, T.; Meljem, J.M.G.; et al. SWI/SNF regulates a transcriptional program that induces senescence to prevent liver cancer. Genes Dev. 2016, 30, 2187–2198. [Google Scholar] [CrossRef]
- Zhu, R.; Mok, M.T.; Kang, W.; Lau, S.S.; Yip, W.-K.; Chen, Y.; Lai, P.B.; Wong, V.W.; To, K.-F.; Sung, J.J.; et al. Truncated HBx-dependent silencing of GAS2 promotes hepatocarcinogenesis through deregulation of cell cycle, senescence and p53-mediated apoptosis. J. Pathol. 2015, 237, 38–49. [Google Scholar] [CrossRef]
- Pacheco-Rivera, R.; Fattel-Fazenda, S.; Arellanes-Robledo, J.; Silva-Olivares, A.; Alemán-Lazarini, L.; Rodríguez-Segura, M.; Pérez-Carreón, J.; Villa-Treviño, S.; Shibayama, M.; Serrano-Luna, J. Double staining of β-galactosidase with fibrosis and cancer markers reveals the chronological appearance of senescence in liver carcinogenesis induced by diethylnitrosamine. Toxicol. Lett. 2016, 241, 19–31. [Google Scholar] [CrossRef]
- Chrysavgis, L.; Papadopoulos, G.; Chatzigeorgiou, A. Τhe Role of Senescence in NASH-Related HCC; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar] [CrossRef]
- Li, J.; Tan, R.; Wu, J.; Guo, W.; Wang, Y.; You, G.; Zhang, Y.; Yu, Z.; Geng, Y.; Zan, J.; et al. A cellular senescence-related genes model allows for prognosis and treatment stratification of hepatocellular carcinoma: A bioinformatics analysis and experimental verification. Front. Genet. 2023, 13, 1099148. [Google Scholar] [CrossRef]
- Song, M.; Pan, Q.; Yang, J.; He, J.; Zeng, J.; Cheng, S.; Huang, Y.; Zhou, Z.-Q.; Zhu, Q.; Yang, C.; et al. Galectin-3 favours tumour metastasis via the activation of β-catenin signalling in hepatocellular carcinoma. Br. J. Cancer 2020, 123, 1521–1534. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, S.; Wu, H.; Lu, J.; Lu, Y.; Wang, F.; Zhao, W.; Zhan, P.; Lu, J.; Fang, Q.; et al. Deubiquitinase PSMD14 enhances hepatocellular carcinoma growth and metastasis by stabilizing GRB2. Cancer Lett. 2020, 469, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Karabicici, M.; Alptekin, S.; Fırtına Karagonlar, Z.; Erdal, E. Doxorubicin-induced senescence promotes stemness and tumorigenicity in EpCAM−/CD133− nonstem cell population in hepatocellular carcinoma cell line, HuH-7. Mol. Oncol. 2021, 15, 2185–2202. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, P.; Yi, W.; Ruan, W.; Xiong, X. Identification of cell senescence molecular subtypes in prediction of the prognosis and immunotherapy of hepatitis B virus-related hepatocellular carcinoma. Front. Immunol. 2022, 13, 1029872. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, X.; Meng, Y.; Shao, C.; Liao, J.; Li, F.; Li, R.; Jing, Y.; Huang, A. The hepatic senescence-associated secretory phenotype promotes hepatocarcinogenesis through Bcl3-dependent activation of macrophages. Cell Biosci. 2021, 11, 173. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Xie, Z.-Y.; Yan, L.-Y.; Liu, X.-F.; Zhang, Y.; Wang, D.-A.; Dong, J.; Sun, H.-T. The correlation of EZH2 expression with the progression and prognosis of hepatocellular carcinoma. BMC Immunol. 2022, 23, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, X.; Liu, D.; Yang, Y.; Zhang, Y.; Wang, G. Construction of a competing endogenous RNA network to analyse glucose-6-phosphate dehydrogenase dysregulation in hepatocellular carcinoma. Biosci. Rep. 2022, 42, BSR20220674. [Google Scholar] [CrossRef] [PubMed]
- Petrara, M.R.; Shalaby, S.; Ruffoni, E.; Taborelli, M.; Carmona, F.; Giunco, S.; Del Bianco, P.; Piselli, P.; Serraino, D.; Cillo, U.; et al. Immune Activation, Exhaustion and Senescence Profiles as Possible Predictors of Cancer in Liver Transplanted Patients. Front. Oncol. 2022, 12, 899170. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, Z.; Ning, K.; Wu, Z.; Chen, Z.; Wu, Z.; Yin, X. Comprehensive Analysis of Cellular Senescence-Related Genes in Prognosis, Molecular Characterization and Immunotherapy of Hepatocellular Carcinoma. Biol. Proced. Online 2022, 24, 24. [Google Scholar] [CrossRef]
- Qu, K.; Lin, T.; Wang, Z.; Liu, S.; Chang, H.; Xu, X.; Meng, F.; Zhou, L.; Wei, J.; Tai, M.; et al. Reactive oxygen species generation is essential for cisplatininduced accelerated senescence in hepatocellular carcinoma. Front. Med. 2014, 8, 227–235. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, X.; Yu, M.; Lin, B.; Yu, C. Radiation-induced liver injury and hepatocyte senescence. Cell Death Discov. 2021, 7, 244. [Google Scholar] [CrossRef]
- Le, O.N.L.; Rodier, F.; Fontaine, F.; Coppe, J.; Campisi, J.; DeGregori, J.; Laverdière, C.; Kokta, V.; Haddad, E.; Beauséjour, C.M. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef]
- Serra, M.P.; Marongiu, F.; Sini, M.; Marongiu, M.; Contini, A.; Wolff, H.; Rave-Frank, M.; Krause, P.; Laconi, E.; Koenig, S. Hepatocyte senescence induced by radiation and partial hepatectomy in rat liver. Int. J. Radiat. Biol. 2014, 90, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Sarcognato, S.; Sacchi, D.; Fassan, M.; Fabris, L.; Cadamuro, M.; Zanus, G.; Cataldo, I.; Capelli, P.; Baciorri, F.; Cacciatore, M.; et al. Cholangiocarcinoma. Pathologica 2021, 113, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Elvevi, A.; Laffusa, A.; Scaravaglio, M.; Rossi, R.E.; Longarini, R.; Stagno, A.M.; Cristoferi, L.; Ciaccio, A.; Cortinovis, D.L.; Invernizzi, P.; et al. Clinical treatment of cholangiocarcinoma: An updated comprehensive review. Ann. Hepatol. 2022, 27, 100737. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhang, B.; Li, C.; Zhang, X. Overcome Drug Resistance in Cholangiocarcinoma: New Insight into Mechanisms and Refining the Preclinical Experiment Models. Front. Oncol. 2022, 12, 850732. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Olaizola, P.; Paiva, N.A.; Olaizola, I.; Agirre-Lizaso, A.; Landa, A.; Bujanda, L.; Perugorria, M.J.; Banales, J.M. Pathogenesis of Cholangiocarcinoma. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 433–463. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Vita, F.; Olaizola, I.; Amato, F.; Rae, C.; Marco, S.; Banales, J.M.; Braconi, C. Heterogeneity of Cholangiocarcinoma Immune Biology. Cells 2023, 12, 846. [Google Scholar] [CrossRef]
- Fabris, L.; Perugorria, M.J.; Mertens, J.; Björkström, N.K.; Cramer, T.; Lleo, A.; Solinas, A.; Sänger, H.; Lukacs-Kornek, V.; Moncsek, A.; et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int. 2019, 39, 63–78. [Google Scholar] [CrossRef]
- Tamma, R.; Annese, T.; Ruggieri, S.; Brunetti, O.; Longo, V.; Cascardi, E.; Mastropasqua, M.G.; Maiorano, E.; Silvestris, N.; Ribatti, D. Inflammatory cells infiltrate and angiogenesis in locally advanced and metastatic cholangiocarcinoma. Eur. J. Clin. Investig. 2019, 49, e13087. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhu, L.; Wang, T.; Chen, J. Immune microenvironment of cholangiocarcinoma: Biological concepts and treatment strategies. Front. Immunol. 2023, 14, 1037945. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cai, Q.; Chen, Y.; Shi, T.; Liu, W.; Mao, L.; Deng, B.; Ying, Z.; Gao, Y.; Luo, H.; et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology 2022, 75, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Affo, S.; Nair, A.; Brundu, F.; Ravichandra, A.; Bhattacharjee, S.; Matsuda, M.; Chin, L.; Filliol, A.; Wen, W.; Song, X.; et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 2021, 39, 866–882.e11. [Google Scholar] [CrossRef]
- Fabris, L.; Sato, K.; Alpini, G.; Strazzabosco, M. The Tumor Microenvironment in Cholangiocarcinoma Progression. Hepatology. 2021, 73, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Alvisi, G.; Termanini, A.; Soldani, C.; Portale, F.; Carriero, R.; Pilipow, K.; Costa, G.; Polidoro, M.; Franceschini, B.; Malenica, I.; et al. Multimodal single-cell profiling of intrahepatic cholangiocarcinoma defines hyperactivated Tregs as a potential therapeutic target. J. Hepatol. 2022, 77, 1359–1372. [Google Scholar] [CrossRef]
- Polidoro, M.A.; Ferrari, E.; Soldani, C.; Franceschini, B.; Saladino, G.; Rosina, A.; Mainardi, A.; D’autilia, F.; Pugliese, N.; Costa, G.; et al. Cholangiocarcinoma-on-a-chip: A human 3D platform for personalized medicine. JHEP Rep. 2024, 6, 100910. [Google Scholar] [CrossRef]
- Sasaki, M.; Nakanuma, Y. New concept: Cellular senescence in pathophysiology of cholangiocarcinoma. Expert. Rev. Gastroenterol. Hepatol. 2016, 10, 625–638. [Google Scholar] [CrossRef]
- Sasaki, M.; Yamaguchi, J.; Itatsu, K.; Ikeda, H.; Nakanuma, Y. Over-expression of polycomb group protein EZH2 relates to decreased expression of p16 INK4a in cholangiocarcinogenesis in hepatolithiasis. J. Pathol. 2008, 215, 175–183. [Google Scholar] [CrossRef]
- Sasaki, M.; Ikeda, H.; Itatsu, K.; Yamaguchi, J.; Sawada, S.; Minato, H.; Ohta, T.; Nakanuma, Y. The overexpression of polycomb group proteins Bmi1 and EZH2 is associated with the progression and aggressive biological behavior of hepatocellular carcinoma. Lab. Investig. 2008, 88, 873–882. [Google Scholar] [CrossRef]
- Rosenberg, N.; Van Haele, M.; Lanton, T.; Brashi, N.; Bromberg, Z.; Adler, H.; Giladi, H.; Peled, A.; Goldenberg, D.S.; Axelrod, J.H.; et al. Combined hepatocellular-cholangiocarcinoma derives from liver progenitor cells and depends on senescence and IL-6 trans-signaling. J. Hepatol. 2022, 77, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Arras, D.; Galun, E.; Rose-John, S. The two facets of gp130 signalling in liver tumorigenesis. Semin. Immunopathol. 2021, 43, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-Associated Macrophages Produce Interleukin 6 and Signal via STAT3 to Promote Expansion of Human Hepatocellular Carcinoma Stem Cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Ma, L.; Tang, W.; Huang, P.; Yang, B.; Wang, L.; Chen, S.; Gao, Q.; Zhang, S.; Xia, J. FXR Acts as a Metastasis Suppressor in Intrahepatic Cholangiocarcinoma by Inhibiting IL-6-Induced Epithelial-Mesenchymal Transition. Cell Physiol. Biochem. 2018, 48, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Kittirat, Y.; Suksawat, M.; Thongchot, S.; Padthaisong, S.; Phetcharaburanin, J.; Wangwiwatsin, A.; Klanrit, P.; Sangkhamanon, S.; Titapun, A.; Loilome, W.; et al. Interleukin-6-derived cancer-associated fibroblasts activate STAT3 pathway contributing to gemcitabine resistance in cholangiocarcinoma. Front. Pharmacol. 2022, 13, 897368. [Google Scholar] [CrossRef]
- Jia, X.; Lu, S.; Zeng, Z.; Liu, Q.; Dong, Z.; Chen, Y.; Zhu, Z.; Hong, Z.; Zhang, T.; Du, G.; et al. Characterization of Gut Microbiota, Bile Acid Metabolism, and Cytokines in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 71, 893–906. [Google Scholar] [CrossRef]
- Kleinegger, F.; Hofer, E.; Wodlej, C.; Golob-Schwarzl, N.; Birkl-Toeglhofer, A.M.; Stallinger, A.; Petzold, J.; Orlova, A.; Krassnig, S.; Reihs, R.; et al. Pharmacologic IL-6Rα inhibition in cholangiocarcinoma promotes cancer cell growth and survival. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2019, 1865, 308–321. [Google Scholar] [CrossRef]
- Moris, D.; Palta, M.; Kim, C.; Allen, P.J.; Morse, M.A.; Lidsky, M.E. Advances in the treatment of intrahepatic cholangiocarcinoma: An overview of the current and future therapeutic landscape for clinicians. CA Cancer J. Clin. 2023, 73, 198–222. [Google Scholar] [CrossRef]
- Greten, T.F.; Schwabe, R.; Bardeesy, N.; Ma, L.; Goyal, L.; Kelley, R.K.; Wang, X.W. Immunology and immunotherapy of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 349–365. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Baba, T.; Mukaida, N. Gemcitabine induces cell senescence in human pancreatic cancer cell lines. Biochem. Biophys. Res. Commun. 2016, 477, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Piyawajanusorn, C.; Kittirat, Y.; Sa-Ngiamwibool, P.; Titapun, A.; Loilome, W.; Namwat, N. PRIMA-1MET Induces Cellular Senescence and Apoptotic Cell Death in Cholangiocarcinoma Cells. Cancer Genom. Proteom. 2019, 16, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Caliò, A.; Zamò, A.; Ponzoni, M.; Zanolin, M.E.; Ferreri, A.J.; Pedron, S.; Montagna, L.; Parolini, C.; Fraifeld, V.E.; Wolfson, M.; et al. Cellular Senescence Markers p16INK4a and p21CIP1/WAF Are Predictors of Hodgkin Lymphoma Outcome. Clin. Cancer Res. 2015, 21, 5164–5172. [Google Scholar] [CrossRef] [PubMed]
- Moolmuang, B.; Singhirunnusorn, P.; Ruchirawat, M. Effects of 5-Aza-2′-Deoxycytidine, Bromodeoxyuridine, Interferons and Hydrogen Peroxide on Cellular Senescence in Cholangiocarcinoma Cells. Asian Pac. J. Cancer Prev. 2016, 17, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Baidoun, F.; Elshiwy, K.; Elkeraie, Y.; Merjaneh, Z.; Khoudari, G.; Sarmini, M.T.; Gad, M.; Al-Husseini, M.; Saad, A. Colorectal Cancer Epidemiology: Recent Trends and Impact on Outcomes. Curr. Drug Targets 2021, 22, 998–1009. [Google Scholar] [CrossRef]
- Buzzelli, J.N.; Ouaret, D.; Brown, G.; Allen, P.D.; Muschel, R.J. Colorectal cancer liver metastases organoids retain characteristics of original tumor and acquire chemotherapy resistance. Stem Cell Res. 2018, 27, 109–120. [Google Scholar] [CrossRef]
- Wang, C.; Fakih, M. Targeting MSS colorectal cancer with immunotherapy: Are we turning the corner? Expert. Opin. Biol. Ther. 2021, 21, 1347–1357. [Google Scholar] [CrossRef]
- Engstrand, J.; Nilsson, H.; Strömberg, C.; Jonas, E.; Freedman, J. Colorectal cancer liver metastases—A population-based study on incidence, management and survival. BMC Cancer 2018, 18, 78. [Google Scholar] [CrossRef]
- Zhao, W.; Dai, S.; Yue, L.; Xu, F.; Gu, J.; Dai, X.; Qian, X. Emerging mechanisms progress of colorectal cancer liver metastasis. Front. Endocrinol. 2022, 13, 1081585. [Google Scholar] [CrossRef]
- Gazzillo, A.; Polidoro, M.A.; Soldani, C.; Franceschini, B.; Lleo, A.; Donadon, M. Relationship between Epithelial-to-Mesenchymal Transition and Tumor-Associated Macrophages in Colorectal Liver Metastases. Int. J. Mol. Sci. 2022, 23, 16197. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Babaei, G.; Aziz, S.G.G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Reynolds, A.R. Vessel co-option and resistance to anti-angiogenic therapy. Angiogenesis 2020, 23, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2022, 12, 134–153. [Google Scholar] [CrossRef]
- Cortese, N.; Soldani, C.; Franceschini, B.; Barbagallo, M.; Marchesi, F.; Torzilli, G.; Donadon, M. Macrophages in Colorectal Cancer Liver Metastases. Cancers 2019, 11, 633. [Google Scholar] [CrossRef]
- Sathe, A.; Mason, K.; Grimes, S.M.; Zhou, Z.; Lau, B.T.; Bai, X.; Su, A.; Tan, X.; Lee, H.; Suarez, C.J.; et al. Colorectal Cancer Metastases in the Liver Establish Immunosuppressive Spatial Networking between Tumor-Associated SPP1 + Macrophages and Fibroblasts. Clin. Cancer Res. 2023, 29, 244–260. [Google Scholar] [CrossRef]
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Benner, A.; Spille, A.; Pommerencke, T.; von Knebel, D.M.; Folprecht, G.; Luber, B.; et al. Localization and Density of Immune Cells in the Invasive Margin of Human Colorectal Cancer Liver Metastases Are Prognostic for Response to Chemotherapy. Cancer Res. 2011, 71, 5670–5677. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Q.; Xing, B.; Luo, N.; Gao, R.; Yu, K.; Hu, X.; Bu, Z.; Peng, J.; Ren, X.; et al. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell 2022, 40, 424–437.e5. [Google Scholar] [CrossRef]
- Donadon, M.; Cortese, N.; Marchesi, F.; Cimino, M.; Mantovani, A.; Torzilli, G. Hepatobiliary surgeons meet immunologists: The case of colorectal liver metastases patients. Hepatobiliary Surg. Nutr. 2019, 8, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Akgül, Ö. Role of surgery in colorectal cancer liver metastases. World J. Gastroenterol. 2014, 20, 6113. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Berber, E. Role of thermal ablation in the management of colorectal liver metastasis. Hepatobiliary Surg. Nutr. 2020, 9, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, Z.; Wang, Y.; Wen, X.; Amador, E.H.; Yuan, L.; Ran, X.; Xiong, L.; Ran, Y.; Chen, W.; et al. Colorectal liver metastasis: Molecular mechanism and interventional therapy. Signal Transduct. Target. Ther. 2022, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sandhu, J.; Ouyang, C.; Ye, J.; Lee, P.P.; Fakih, M. Clinical Response to Immunotherapy Targeting Programmed Cell Death Receptor 1/Programmed Cell Death Ligand 1 in Patients with Treatment-Resistant Microsatellite Stable Colorectal Cancer with and without Liver Metastases. JAMA Netw. Open 2021, 4, e2118416. [Google Scholar] [CrossRef]
- Panczyk, M. Pharmacogenetics research on chemotherapy resistance in colorectal cancer over the last 20 years. World J. Gastroenterol. 2014, 20, 9775. [Google Scholar] [CrossRef]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef]
- Haugstetter, A.M.; Loddenkemper, C.; Lenze, D.; Gröne, J.; Standfuß, C.; Petersen, I.; Dörken, B.; Schmitt, C.A. Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br. J. Cancer 2010, 103, 505–509. [Google Scholar] [CrossRef]
- Choi, Y.W.; Kim, Y.H.; Oh, S.Y.; Suh, K.W.; Lee, G.; Yoon, J.E.; Park, S.S.; Lee, Y.; Park, Y.J.; Kim, H.S.; et al. Senescent Tumor Cells Build a Cytokine Shield in Colorectal Cancer. Adv. Sci. 2021, 8, 2002497. [Google Scholar] [CrossRef]
- Braumüller, H.; Mauerer, B.; Berlin, C.; Plundrich, D.; Marbach, P.; Cauchy, P.; Laessle, C.; Biesel, E.; Holzner, P.A.; Kesselring, R. Senescent Tumor Cells in the Peritoneal Carcinomatosis Drive Immunosenescence in the Tumor Microenvironment. Front. Immunol. 2022, 13, 908449. [Google Scholar] [CrossRef]
- Alkhamesi, N.A.; Ziprin, P.; Pfistermuller, K.; Peck, D.H.; Darzi, A.W. ICAM-1 Mediated Peritoneal Carcinomatosis, A Target for Therapeutic Intervention. Clin. Exp. Metastasis 2005, 22, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Mikuła-Pietrasik, J.; Sosińska, P.; Maksin, K.; Kucińska, M.; Piotrowska, H.; Murias, M.; Woźniak, A.; Szpurek, D.; Książek, K. Colorectal cancer-promoting activity of the senescent peritoneal mesothelium. Oncotarget 2015, 6, 29178–29195. [Google Scholar] [CrossRef] [PubMed]
- Garbarino, O.; Lambroia, L.; Basso, G.; Marrella, V.; Franceschini, B.; Soldani, C.; Pasqualini, F.; Giuliano, D.; Costa, G.; Peano, C.; et al. Spatial resolution of cellular senescence dynamics in human colorectal liver metastasis. Aging Cell 2023, 22, e13853. [Google Scholar] [CrossRef] [PubMed]
- Bruni, E.; Cazzetta, V.; Donadon, M.; Cimino, M.; Torzilli, G.; Spata, G.; Leonardi, G.; Dieli, F.; Mikulak, J.; Mavilio, D. Chemotherapy accelerates immune-senescence and functional impairments of Vδ2pos T cells in elderly patients affected by liver metastatic colorectal cancer. J. Immunother. Cancer 2019, 7, 347. [Google Scholar] [CrossRef] [PubMed]
- Tato-Costa, J.; Casimiro, S.; Pacheco, T.; Pires, R.; Fernandes, A.; Alho, I.; Pereira, P.; Costa, P.; Castelo, H.B.; Ferreira, J.; et al. Therapy-Induced Cellular Senescence Induces Epithelial-to-Mesenchymal Transition and Increases Invasiveness in Rectal Cancer. Clin. Colorectal Cancer 2016, 15, 170–178.e3. [Google Scholar] [CrossRef] [PubMed]
- Short, S.; Fielder, E.; Miwa, S.; Von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef]
- Li, F.; Huangyang, P.; Burrows, M.; Guo, K.; Riscal, R.; Godfrey, J.; Lee, K.E.; Lin, N.; Lee, P.; Blair, I.A.; et al. FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat. Cell Biol. 2020, 22, 728–739. [Google Scholar] [CrossRef]
- Thadathil, N.; Selvarani, R.; Mohammed, S.; Nicklas, E.H.; Tran, A.L.; Kamal, M.; Luo, W.; Brown, J.L.; Lawrence, M.M.; Borowik, A.K.; et al. Senolytic treatment reduces cell senescence and necroptosis in Sod1 knockout mice that is associated with reduced inflammation and hepatocellular carcinoma. Aging Cell 2022, 21, e13676. [Google Scholar] [CrossRef]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib + Quercetin (D + Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front. Oncol. 2018, 8, 459. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.-K.; Chiu, D.K.-C.; Yuen, V.W.-H.; Law, C.-T.; Wong, B.P.-Y.; Thu, K.L.; Cescon, D.W.; Soria-Bretones, I.; Cheu, J.W.-S.; Lee, D.; et al. CFI-402257, a TTK inhibitor, effectively suppresses hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2022, 119, e2119514119. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazzillo, A.; Volponi, C.; Soldani, C.; Polidoro, M.A.; Franceschini, B.; Lleo, A.; Bonavita, E.; Donadon, M. Cellular Senescence in Liver Cancer: How Dying Cells Become “Zombie” Enemies. Biomedicines 2024, 12, 26. https://doi.org/10.3390/biomedicines12010026
Gazzillo A, Volponi C, Soldani C, Polidoro MA, Franceschini B, Lleo A, Bonavita E, Donadon M. Cellular Senescence in Liver Cancer: How Dying Cells Become “Zombie” Enemies. Biomedicines. 2024; 12(1):26. https://doi.org/10.3390/biomedicines12010026
Chicago/Turabian StyleGazzillo, Aurora, Camilla Volponi, Cristiana Soldani, Michela Anna Polidoro, Barbara Franceschini, Ana Lleo, Eduardo Bonavita, and Matteo Donadon. 2024. "Cellular Senescence in Liver Cancer: How Dying Cells Become “Zombie” Enemies" Biomedicines 12, no. 1: 26. https://doi.org/10.3390/biomedicines12010026