
Optimization of Nutrition after Brain Injury: Mechanistic and Therapeutic Considerations
 ,
,
Abstract
:1. Introduction
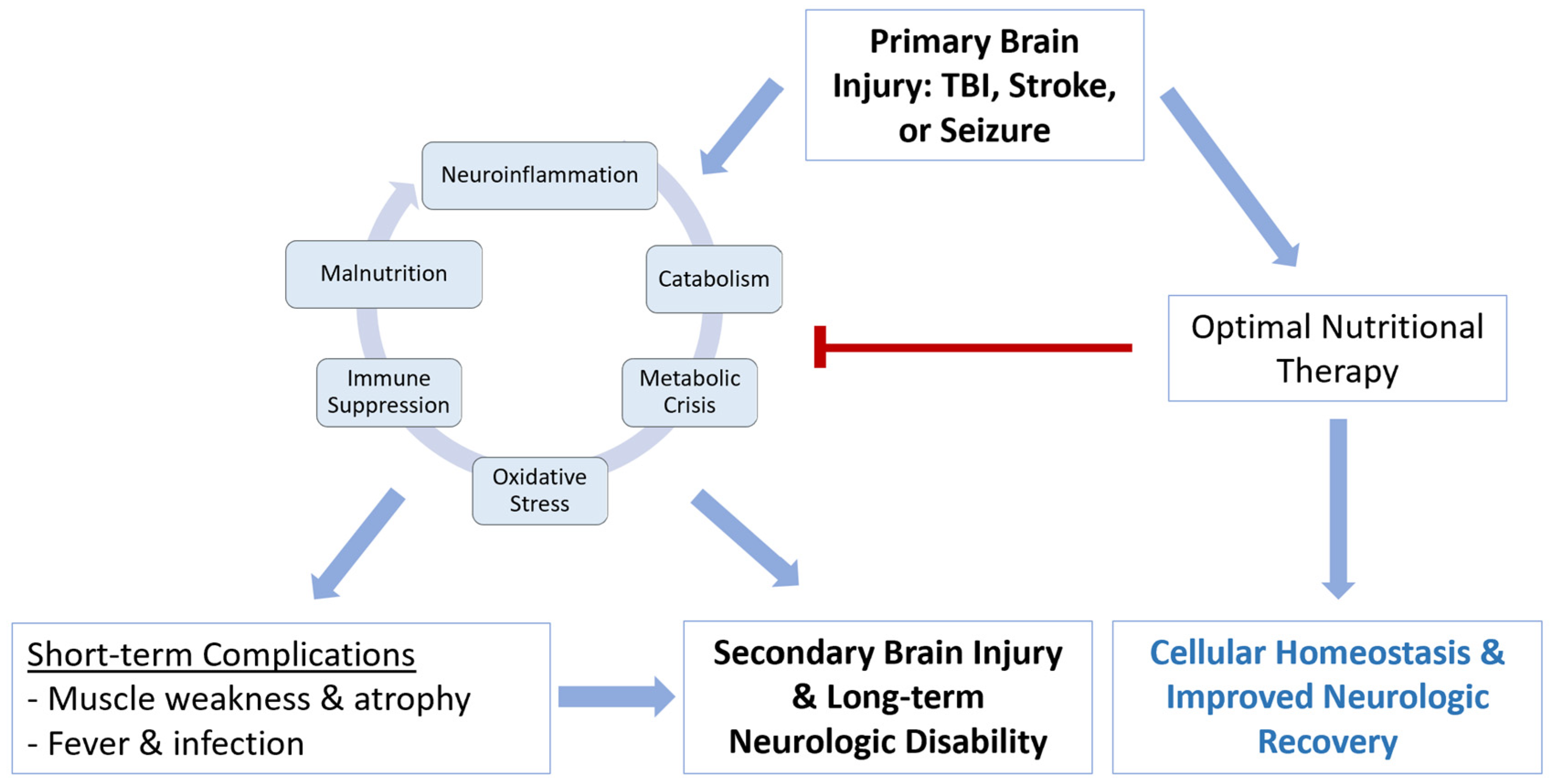
2. Recognizing Malnutrition and Clinical Importance after Acute Brain Injury
3. Pathophysiology of Malnutrition after Acute Brain Injury
3.1. Malnutrition in Specific Acute Neurologic Diseases
3.1.1. Traumatic Brain Injury
3.1.2. Acute Ischemic Stroke
3.1.3. Acute Hemorrhagic Stroke
3.1.4. Anoxic Brain Injury
3.1.5. Status Epilepticus
3.2. Considerations in the Pediatric Population
3.2.1. Differences in Metabolism during Neurodevelopment
3.2.2. Genetic Factors
3.2.3. Estimating Nutritional Requirements in Pediatric Disease
4. Emerging Nutritional Interventions to Promote Early Brain Recovery
4.1. Determining Nutritional Needs to Support Metabolic Demands in Brain-Injured Patients
4.2. Current Guidelines and Controversies
4.2.1. Early Versus Late Feeding
4.2.2. Parenteral Nutrition
4.2.3. Glucose Metabolism and Control
4.2.4. Recommendations for Nutritional Support in Pediatric Patients
4.3. Emerging Nutritional Therapies to Support Neurologic Recovery
4.3.1. Ketogenic Diet
4.3.2. Omega-3 Supplementation
4.3.3. Other Immunonutrition
4.3.4. Other Dietary Components
4.4. Advanced Diagnostics for Nutritional Assessment with Investigations in Brain Injury
4.4.1. Novel Neuroimaging Modalities
4.4.2. Cerebral Microdialysis
5. Conclusions & Future Aims
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thayabaranathan, T.; Kim, J.; Cadilhac, D.A.; Thrift, A.G.; Donnan, G.A.; Howard, G.; Howard, V.J.; Rothwell, P.M.; Feigin, V.; Norrving, B.; et al. Global stroke statistics 2022. Int. J. Stroke 2022, 17, 946–956. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 1, 1–18. [Google Scholar] [CrossRef]
- Feigin, V.L.; Brainin, M.; Norrving, B.; Martins, S.; Sacco, R.L.; Hacke, W.; Fisher, M.; Pandian, J.; Lindsay, P. World Stroke Organization (WSO): Global Stroke Fact. Sheet 2022. Int. J. Stroke 2022, 17, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Fuentes Padilla, P.; Martinez, G.; Vernooij, R.W.; Urrutia, G.; Roque, I.F.M.; Bonfill Cosp, X. Early enteral nutrition (within 48 hours) versus delayed enteral nutrition (after 48 hours) with or without supplemental parenteral nutrition in critically ill adults. Cochrane Database Syst. Rev. 2019, 2019, CD012340. [Google Scholar] [CrossRef] [PubMed]
- Guenter, P.; Abdelhadi, R.; Anthony, P.; .Blackmer, A.; Malone, A.; Mirtallo, J.M.; Phillips, W.; Resnick, H.E. Malnutrition diagnoses and associated outcomes in hospitalized patients: United States, 2018. Nutr. Clin. Pract. 2021, 36, 957–969. [Google Scholar] [CrossRef]
- Lew, C.C.H.; Yandell, R.; Fraser, R.J.L.; Chua, A.P.; Chong, M.F.F.; Miller, M. Association Between Malnutrition and Clinical Outcomes in the Intensive Care Unit: A Systematic Review. J. Parenter. Enter. Nutr. 2017, 41, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Puthucheary, Z.A.; Rawal, J.; McPhail, M.; Connolly, B.; Ratnayake, G.; Chan, P.; Hopkinson, N.S.; Phadke, R.; Dew, T.; Sidhu, P.S.; et al. Acute skeletal muscle wasting in critical illness. JAMA 2013, 310, 1591–1600. [Google Scholar] [CrossRef]
- Chiang, Y.; Chao, D.; Chu, S.; Lin, H.; Huang, S.; Yeh, Y.; Lui, T.; Binns, C.W.; Chiu, W. Early enteral nutrition and clinical outcomes of severe traumatic brain injury patients in acute stage: A multi-center cohort study. J. Neurotrauma 2012, 29, 75–80. [Google Scholar] [CrossRef]
- Butterworth, C. The skeleton in the hospital closet. Nutr. Today 1974, 9, 4–8. [Google Scholar] [CrossRef]
- White, J.V.; Guenter, P.; Jensen, G.; Malone, A.; Schofield, M.; Academy Malnutrition Work Group, American Society for Parenteral and Enteral Nutrition; Malnutrition Task Force, American Society for Parenteral and Enteral Nutrition; Board of Directors. Consensus statement: Academy of nutrition and dietetics and American Society for Parenteral and Enteral Nutrition: Characteristics recommended for the identification and documentation of adult malnutrition (undernutrition). J. Parenter. Enter. Nutr. 2012, 36, 275–283. [Google Scholar]
- Cederholm, T.; Barazzoni, R.; Austin, P.; Ballmer, P.; Biolo, G.; Bischoff, S.C.; Compher, C.; Correia, I.; Higashiguchi, T.; Holst, M.; et al. ESPEN guidelines on definitions and terminology of clinical nutrition. Clin. Nutr. 2017, 36, 49–64. [Google Scholar] [CrossRef]
- Jensen, G.L.; Cederholm, T.A.; Correia, M.I.; Gonzalez, M.C.; Fukushima, R.; Higashiguchi, T.; Adrianza de Baptista, G.; Barazzoni, R.; Blaauw, R.; Coats, A.J.S.; et al. The GLIM criteria for the diagnosis of malnutrition—A consensus report from the global clinical nutrition community. J. Parenter. Enter. Nutr. 2019, 43, 32–40. [Google Scholar] [CrossRef]
- Mogensen, K.M.; Malone, A.; Becker, P.; Cutrell, S.; Frank, L.; Gonzales, K.; Hudson, L.; Miller, S.; Guenter, P.; Malnutrition Committee of the American Society for Parenteral and Enteral Nutrition (ASPEN). Academy of Nutrition and Dietetics/American Society for Parenteral and Enteral Nutrition Consensus malnutrition characteristics: Usability and association with outcomes. Nutr. Clin. Pract. 2019, 34, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Corkins, M.R.; Guenter, P.; DiMaria-Ghalili, R.A.; Jensen, G.L.; Malone, A.; Miller, S.; Patel, V.; Plogsted, S.; Resnick, H.E.; American Society for Parenteral and Enteral Nutrition. Malnutrition diagnoses in hospitalized patients: United States, 2010. JPEN J. Parenter. Enter. Nutr. 2014, 38, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.; Chittams, J.; Griffith, C.; Compher, C. Malnutrition identified by Academy of Nutrition and Dietetics/American Society for Parenteral and Enteral Nutrition is associated with more 30-day readmissions, greater hospital mortality, and longer hospital stays: A retrospective analysis of nutrition assessment data in a major medical center. J. Parenter. Enter. Nutr. 2018, 42, 892–897. [Google Scholar]
- Correia, M.I.T.D.; Perman, M.I.; Pradelli, L.; Omaralsaleh, A.J.; Waitzberg, D.L. Economic burden of hospital malnutrition and the cost-benefit of supplemental parenteral nutrition in critically ill patients in Latin America. J. Med. Econ. 2018, 21, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Kuo, G.Y.; Tarzi, F.; Louie, S.; Poblete, R. Neuroinflammation in Traumatic Brain Injury; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]
- Flanagan, S.R. Invited Commentary on “Centers for Disease Control and Prevention Report to Congress: Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation”. Arch. Phys. Med. Rehabil. 2015, 96, 1753–1755. [Google Scholar] [CrossRef]
- Dijkink, S.; Meier, K.; Krijnen, P.; Yeh, D.D.; Velmahos, G.C.; Schipper, I.B. Malnutrition and its effects in severely injured trauma patients. Eur. J. Trauma Emerg. Surg. 2020, 46, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Hartl, R.; Gerber, L.M.; Ni, Q.; Ghajar, J. Effect of early nutrition on deaths due to severe traumatic brain injury. J. Neurosurg. 2008, 109, 50–56. [Google Scholar] [CrossRef]
- Yang, L.; Liao, D.; Hou, X.; Wang, Y.; Yang, C. Systematic review and meta-analysis of the effect of nutritional support on the clinical outcome of patients with traumatic brain injury. Ann. Palliat. Med. 2021, 10, 11960–11969. [Google Scholar] [CrossRef]
- Bao, L.; Chen, D.; Ding, L.; Ling, W.; Xu, F. Fever burden is an independent predictor for prognosis of traumatic brain injury. PLoS ONE 2014, 9, e90956. [Google Scholar] [CrossRef]
- Selassie, A.W.; Fakhry, S.M.; Ford, D.W. Population-based study of the risk of in-hospital death after traumatic brain injury: The role of sepsis. J. Trauma 2011, 71, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, L.; Evans, J.E.; Ferguson, S.; Mouzon, B.; Montague, H.; Reed, J.; Crynen, G.; Emmerich, T.; Crocker, M.; Pelot, R.; et al. Lipidomic analyses identify injury-specific phospholipid changes 3 mo after traumatic brain injury. FASEB J. 2014, 28, 5311–5321. [Google Scholar] [CrossRef]
- Baskaya, M.K.; Rao, A.M.; Dogan, A.; Donaldson, D.; Dempsey, R.J. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci. Lett. 1997, 226, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Ekdahl, K.N.; Teramura, Y.; Hamad, O.A.; Asif, S.; Duehrkop, C.; Fromell, K.; Gustafson, E.; Hong, J.; Kozarcanin, H.; Magnusson, P.U.; et al. Dangerous liaisons: Complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol. Rev. 2016, 274, 245–269. [Google Scholar] [CrossRef]
- Kumar Sahel, D.; Kaira, M.; Raj, K.; Sharma, S.; Singh, S. Mitochondrial dysfunctioning and neuroinflammation: Recent highlights on the possible mechanisms involved in Traumatic Brain Injury. Neurosci. Lett. 2019, 710, 134347. [Google Scholar] [CrossRef]
- Lamade, A.M.; Anthonymuthu, T.S.; Hier, Z.E.; Gao, Y.; Kagan, V.E.; Bayır, H. Mitochondrial damage & lipid signaling in traumatic brain injury. Exp. Neurol. 2020, 329, 113307. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, S.M.; Leclercq, P.D.; Moyes, L.; Graham, D.I.; Smith, C.; Griffen, W.S.T.; Nicoll, J.A.R. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic. Sci. Int. 2004, 146, 97–104. [Google Scholar] [CrossRef]
- Collins-Praino, L.E.; Corrigan, F. Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav. Immun. 2017, 60, 369–382. [Google Scholar] [CrossRef]
- Sribnick, E.A.; Popovich, P.G.; Hall, M.W. Central nervous system injury-induced immune suppression. Neurosurg. Focus 2022, 52, E10. [Google Scholar] [CrossRef]
- Marshall, W.A.; Adams, L.M.; Weaver, J.L. The Brain–Gut Axis in Traumatic Brain Injury: Implications for Nutrition Support. Curr. Surg. Rep. 2022, 10, 172–179. [Google Scholar] [CrossRef]
- Painter, T.J.; Rickerds, J.; Alban, R.F. Immune enhancing nutrition in traumatic brain injury—A preliminary study. Int. J. Surg. 2015, 21, 70–74. [Google Scholar] [CrossRef]
- Mehta, S.L.; Manhas, N.; Raghubir, R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. 2007, 54, 34–66. [Google Scholar] [CrossRef]
- Brouns, R.; Sheorajpanday, R.; Wauters, A.; De Surgeloose, D.; Mariën, P.; De Deyn, P.P. Evaluation of lactate as a marker of metabolic stress and cause of secondary damage in acute ischemic stroke or TIA. Clin. Chim. Acta 2008, 397, 27–31. [Google Scholar] [CrossRef]
- Bustamante, A.; Simats, A.; Vilar-Bergua, A.; Garcia-Berrocoso, T.; Montaner, J. Blood/Brain Biomarkers of Inflammation after Stroke and Their Association with Outcome: From C-Reactive Protein to Damage-Associated Molecular Patterns. Neurotherapeutics 2016, 13, 671–684. [Google Scholar] [CrossRef]
- Liesz, A.; Dalpke, A.; Mracsko, E.; Antoine, D.J.; Roth, S.; Zhou, W.; Yang, H.; Na, S.; Akhisaroglu, M.; Fleming, T.; et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J. Neurosci. 2015, 35, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Hu, X.; Kleinschnitz, C.; Offner, H. Functional role of regulatory lymphocytes in stroke: Facts and controversies. Stroke 2015, 46, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, A.; Amaro, S.; Vargas, M.; Obach, V.; Cervera, A.; Gomez-Choco, M.; Torres, F.; Planas, A.M. Catecholamines, infection, and death in acute ischemic stroke. J. Neurol. Sci. 2007, 252, 29–35. [Google Scholar] [CrossRef]
- Haeusler, K.G.; Schmidt, W.U.; Fohring, F.; Meisel, C.; Helms, T.; Jungehulsing, G.J.; Nolte, C.H.; Schmolke, K.; Wegner, B.; Meisel, A.; et al. Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc. Dis. 2008, 25, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Sabbouh, T.; Torbey, M.T. Malnutrition in stroke patients: Risk factors, assessment, and management. Neurocrit. Care 2018, 29, 374–384. [Google Scholar] [CrossRef]
- Dávalos, A.; Ricart, W.; Gonzalez-Huix, F.; Soler, S.; Marrugat, J.; Molins, A.; Suñer, R.; Genís, D. Effect of malnutrition after acute stroke on clinical outcome. Stroke 1996, 27, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Ciancarelli, I.; Morone, G.; Iosa, M.; Cerasa, A.; Calabrò, R.S.; Iolascon, G.; Gimigliano, F.; Tonin, P.; Tozzi Ciancarelli, M.G. Influence of Oxidative Stress and Inflammation on Nutritional Status and Neural Plasticity: New Perspectives on Post-Stroke Neurorehabilitative Outcome. Nutrients 2022, 15, 108. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.J.; Rowe, S.J.; McLaren, L.M. Mobility status during inpatient rehabilitation: A comparison of patients with stroke and traumatic brain injury. Arch. Phys. Med. Rehabil. 2002, 83, 483–490. [Google Scholar] [CrossRef] [PubMed]
- McCrea, M.A.; Giacino, J.T.; Barber, J.; Temkin, N.R.; Nelson, L.D.; Levin, H.S.; Dikmen, S.; Stein, M.; Bodien, Y.G.; Boase, K.; et al. Functional Outcomes Over the First Year After Moderate to Severe Traumatic Brain Injury in the Prospective, Longitudinal TRACK-TBI Study. JAMA Neurol. 2021, 78, 982–992. [Google Scholar] [CrossRef]
- Pinho, J.; Costa, A.S.; Araújo, J.M.; Amorim, J.M.; Ferreira, C. Intracerebral hemorrhage outcome: A comprehensive update. J. Neurol. Sci. 2019, 398, 54–66. [Google Scholar] [CrossRef]
- Koukiasa, P.; Bitzani, M.; Papaioannou, V.; Pnevmatikos, I. Resting Energy Expenditure in Critically Ill Patients With Spontaneous Intracranial Hemorrhage. J. Parenter. Enter. Nutr. 2015, 39, 917–921. [Google Scholar] [CrossRef]
- Badjatia, N.; Fernandez, L.; Schlossberg, M.J.; Schmidt, J.M.; Claassen, J.; Lee, K.; Connolly, E.S.; Mayer, S.A.; Rosenbaum, M. Relationship between energy balance and complications after subarachnoid hemorrhage. J. Parenter. Enter. Nutr. 2010, 34, 64–69. [Google Scholar] [CrossRef]
- Shiga, Y.; Nezu, T.; Shimomura, R.; Sato, K.; Himeno, T.; Terasawa, Y.; Aoki, S.; Hosomi, N.; Kohriyama, T.; Maruyama, H. Various effects of nutritional status on clinical outcomes after intracerebral hemorrhage. Intern. Emerg. Med. 2022, 17, 1043–1052. [Google Scholar] [CrossRef]
- Chalkias, A.; Xanthos, T. Post-cardiac arrest brain injury: Pathophysiology and treatment. J. Neurol. Sci. 2012, 315, 1–8. [Google Scholar] [CrossRef]
- Perkins, G.D.; Callaway, C.W.; Haywood, K.; Neumar, R.W.; Lilja, G.; Rowland, M.J.; Sawyer, K.N.; Skrifvars, M.B.; Nolan, J.P. Brain injury after cardiac arrest. Lancet 2021, 398, 1269–1278. [Google Scholar] [CrossRef]
- Fehler, P.; Zielińska, M.; Uchmanowicz, B.; Juárez-Vela, R.; Lewandowski, Ł.; Zieliński, S.; Czapla, M. Do Body Mass Index and Nutritional Risk Score 2002 Influence the In-Hospital Mortality of Patients Following Cardiac Arrest? Nutrients 2023, 15, 436. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Carlson, C.; Kalra, R.; Elliott, A.M.; Yannopoulos, D.; Bartos, J.A. Outcomes associated with delayed enteral feeding after cardiac arrest treated with veno-arterial extracorporeal membrane oxygenation and targeted temperature management. Resuscitation 2021, 164, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, M.S.; Ainslie, P.N.; Griesdale, D.E. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: A “two-hit” model. Crit. Care 2017, 21, 90. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Bradley, J.L.; Zheng, G.; Ge, W.; Xu, J.; Hu, J.; He, F.; Shabnam, R.; Peberdy, M.A.; Ornato, J.P.; et al. Cerebral and myocardial mitochondrial injury differ in a rat model of cardiac arrest and cardiopulmonary resuscitation. Biomed. Pharmacother. 2021, 140, 111743. [Google Scholar] [CrossRef] [PubMed]
- Vereczki, V.; Martin, E.; Rosenthal, R.E.; Hof, P.R.; Hoffman, G.E.; Fiskum, G. Normoxic resuscitation after cardiac arrest protects against hippocampal oxidative stress, metabolic dysfunction, and neuronal death. J. Cereb. Blood Flow. Metab. 2006, 26, 821–835. [Google Scholar] [CrossRef]
- Brandt, M.J.V.; Nijboer, C.H.; Nessel, I.; Mutshiya, T.R.; Michael-Titus, A.T.; Counotte, D.S.; Schipper, L.; van der Aa, N.E.; Benders, M.J.N.L.; de Theije, C.G.M. Nutritional Supplementation Reduces Lesion Size and Neuroinflammation in a Sex-Dependent Manner in a Mouse Model of Perinatal Hypoxic-Ischemic Brain Injury. Nutrients 2022, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, X.; Yang, W.; Gao, Y.; Chen, J. Omega-3 polyunsaturated fatty acid supplementation confers long-term neuroprotection against neonatal hypoxic–ischemic brain injury through anti-inflammatory actions. Stroke 2010, 41, 2341–2347. [Google Scholar] [CrossRef]
- Khairallah, R.J.; Kim, J.; O’Shea, K.M.; O’Connell, K.A.; Brown, B.H.; Galvao, T.; Daneault, C.; Des Rosiers, C.; Polster, B.M.; Hoppel, C.L.; et al. Improved mitochondrial function with diet-induced increase in either docosahexaenoic acid or arachidonic acid in membrane phospholipids. PLoS ONE 2012, 7, e34402. [Google Scholar] [CrossRef]
- Betjemann, J.P.; Lowenstein, D.H. Status epilepticus in adults. Lancet Neurol. 2015, 14, 615–624. [Google Scholar] [CrossRef]
- Crepin, S.; Godet, B.; Chassain, B.; Preux, P.M.; Desport, J.C. Malnutrition and epilepsy: A two-way relationship. Clin. Nutr. 2009, 28, 219–225. [Google Scholar] [CrossRef]
- Palencia, G.; Calvillo, M.; Sotelo, J. Chronic malnutrition caused by a corn-based diet lowers the threshold for pentylenetetrazol-induced seizures in rats. Epilepsia 1996, 37, 583–586. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Uma Devi, P.; Pillai, K.K.; Vohora, D. Modulation of pentylenetetrazole-induced seizures and oxidative stress parameters by sodium valproate in the absence and presence of N-acetylcysteine. Fundam. Clin. Pharmacol. 2006, 20, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Testai, L.; Colletti, A.; Cicero, A.F.G. Coenzyme Q10: Clinical Applications in Cardiovascular Diseases. Antioxidants 2020, 9, 341. [Google Scholar] [CrossRef]
- Tawfik, M.K. Coenzyme Q10 enhances the anticonvulsant effect of phenytoin in pilocarpine-induced seizures in rats and ameliorates phenytoin-induced cognitive impairment and oxidative stress. Epilepsy Behav. 2011, 22, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Chen, S.D.; Lin, K.J.; Chuang, Y.C. Seizure-Induced Oxidative Stress in Status Epilepticus: Is Antioxidant Beneficial? Antioxidants 2020, 9, 1029. [Google Scholar] [CrossRef]
- de Aquino, C.C.; Leitão, R.A.; Oliveira Alves, L.A.; Coelho-Santos, V.; Guerrant, R.L.; Ribeiro, C.F.; Malva, J.O.; Silva, A.P.; Oriá, R.B. Effect of Hypoproteic and High-Fat Diets on Hippocampal Blood-Brain Barrier Permeability and Oxidative Stress. Front. Nutr. 2019, 5, 131. [Google Scholar] [CrossRef] [PubMed]
- Koppel, S.J.; Swerdlow, R.H. Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem. Int. 2018, 117, 114–125. [Google Scholar] [CrossRef]
- Bauernfeind, A.L.; Babbitt, C.C. The appropriation of glucose through primate neurodevelopment. J. Hum. Evol. 2014, 77, 132–140. [Google Scholar] [CrossRef]
- Goyal, M.S.; Raichle, M.E. Glucose Requirements of the Developing Human Brain. J. Pediatr. Gastroenterol. Nutr. 2018, 66, S46–S49. [Google Scholar] [CrossRef]
- Kuzawa, C.W.; Chugani, H.T.; Grossman, L.I.; Lipovich, L.; Muzik, O.; Hof, P.R.; Wildman, D.E.; Sherwood, C.C.; Leonard, W.R.; Lange, N. Metabolic costs and evolutionary implications of human brain development. Proc. Natl. Acad. Sci. USA 2014, 111, 13010–13015. [Google Scholar] [CrossRef]
- Schirmbeck, G.H.; Sizonenko, S.; Sanches, E.F. Neuroprotective Role of Lactoferrin during Early Brain Development and Injury through Lifespan. Nutrients 2022, 14, 2923. [Google Scholar] [CrossRef]
- Group, N.H.W.; Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; et al. The NIH human microbiome project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef]
- Paoli, A.; Mancin, L.; Bianco, A.; Thomas, E.; Mota, J.F.; Piccini, F. Ketogenic Diet and Microbiota: Friends or Enemies? Genes 2019, 10, 534. [Google Scholar] [CrossRef]
- Brichtová, E.; Kozák, L. Apolipoprotein E genotype and traumatic brain injury in children--association with neurological outcome. Childs Nerv. Syst. 2008, 24, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.M.; Taylor, H.G.; Ganesalingam, K.; Gastier-Foster, J.M.; Frick, J.; Bangert, B.; Dietrich, A.; Nuss, K.E.; Rusin, J.; Wright, M.; et al. Apolipoprotein E4 as a predictor of outcomes in pediatric mild traumatic brain injury. J. Neurotrauma. 2009, 26, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Reuter-Rice, K.; Regier, M.; Bennett, E.; Laskowitz, D. The Effect of the Relationship of APOE Polymorphisms and Cerebral Vasospasm on Functional Outcomes in Children With Traumatic Brain Injury. Biol. Res. Nurs. 2018, 20, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.M.; Conley, Y.P.; Wagner, A.K.; Jha, R.M.; Clark, R.S.; Poloyac, S.M.; Kochanek, P.M.; Empey, P.E. The pharmacogenomics of severe traumatic brain injury. Pharmacogenomics 2017, 18, 1413–1425. [Google Scholar] [CrossRef]
- Havalad, S.; Quaid, M.; Sapiega, V. Energy Expenditure in Children with Severe Head Injury: Lack of Agreement between Measured and Estimated Energy Expenditure. Nutr. Clin. Pract. 2006, 21, 175–181. [Google Scholar] [CrossRef]
- Mtaweh, H.; Smith, R.; Kochanek, P.M.; Wisniewski, S.R.; Fabio, A.; Vavilala, M.S.; Adelson, P.D.; Toney, N.A.; Bell, M.J. Energy expenditure in children after severe traumatic brain injury. Pediatr. Crit. Care Med. 2014, 15, 242–249. [Google Scholar] [CrossRef]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.; Hawryluk, G.W.J.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the management of severe traumatic brain injury 4th edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.E.; McClave, S.A.; Martindale, R.G.; Warren, M.M.; Johnson, D.R.; Braunschweig, C.; McCarthy, M.S.; Davanos, E.; Rice, T.W.; Cresci, G.A.; et al. Guidelines for the Provision and Assessment of Nutrition Support Therapy in the Adult Critically Ill Patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.). Crit. Care Med. 2016, 44, 390–438. [Google Scholar] [CrossRef]
- Patel, J.J.; Mundi, M.S.; Taylor, B.; McClave, S.A.; Mechanick, J.I. Casting Light on the Necessary, Expansive, and Evolving Role of the Critical Care Dietitian: An Essential Member of the Critical Care Team. Crit. Care Med. 2022, 50, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Arribas-López, E.; Zand, N.; Ojo, O.; Snowden, M.J.; Kochhar, T. The Effect of Amino Acids on Wound Healing: A Systematic Review and Meta-Analysis on Arginine and Glutamine. Nutrients 2021, 13, 2498. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.J.; Fettes, S.B.; Jewkes, C.; Nelson, R.J. Prospective, randomized, controlled trial to determine the effect of early enhanced enteral nutrition on clinical outcome in mechanically ventilated patients suffering head injury. Crit. Care Med. 1999, 27, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Chapple, L.S.; Deane, A.M.; Heyland, D.K.; Lange, K.; Kranz, A.J.; Williams, L.T.; Chapman, M.J. Energy and protein deficits throughout hospitalization in patients admitted with a traumatic brain injury. Clin. Nutr. 2016, 35, 1315–1322. [Google Scholar] [CrossRef]
- Quintard, H.; Ichai, C. Nutritional and metabolic supplementation for the injured brain: An update. Curr. Opin. Crit. Care 2019, 25, 126–131. [Google Scholar] [CrossRef]
- Singer, P.; Blaser, A.R.; Berger, M.M.; Alhazzani, W.; Calder, P.C.; Casaer, M.P.; Hiesmayr, M.; Mayer, K.; Montejo, J.C.; Pichard, C.; et al. ESPEN guideline on clinical nutrition in the intensive care unit. Clin. Nutr. 2019, 38, 48–79. [Google Scholar] [CrossRef]
- Compher, C.; Bingham, A.L.; McCall, M.; Patel, J.; Rice, T.W.; Braunschweig, C.; McKeever, L. Guidelines for the provision of nutrition support therapy in the adult critically ill patient: The American Society for Parenteral and Enteral Nutrition. J. Parenter. Enter. Nutr. 2022, 46, 12–41. [Google Scholar] [CrossRef]
- Tavarez, T.; Roehl, K.; Koffman, L. Nutrition in the Neurocritical Care Unit: A New Frontier. Curr. Treat Options Neurol. 2021, 23, 16–18. [Google Scholar] [CrossRef]
- Burgos, R.; Bretón, I.; Cereda, E.; Desport, J.C.; Dziewas, R.; Genton, L.; Gomes, F.; Jesus, P.; Leischker, A.; Muscaritoli, M.; et al. ESPEN guideline clinical nutrition in neurology. Clin. Nutr. 2018, 37, 354–396. [Google Scholar] [CrossRef]
- Brain Trauma Foundation; American Association of Neurological Surgeons; Congress of Neurological Surgeons; Joint Section on Neurotrauma and Critical Care, AANS/CNS; Bratton, S.; Chestnut, R.M.; Ghajar, J.; McConnell Hammond, F.F.; Harris, O.A.; Hartl, R.; et al. Guidelines for the management of severe traumatic brain injury: XII. Nutrition. J. Neurotrauma 2007, 24 (Suppl. 1), S77–S82. [Google Scholar] [CrossRef] [PubMed]
- Casaer, M.P.; Mesotten, D.; Hermans, G.; Wouters, P.J.; Schetz, M.; Meyfroidt, G.; Cromphaut, S.V.; Ingels, C.; Meersseman, P.; Muller, J.; et al. Early versus late parenteral nutrition in critically ill adults. N. Engl. J. Med. 2011, 365, 506–517. [Google Scholar] [CrossRef]
- Elke, G.; van Zanten, A.R.H.; Lemieux, M.; McCall, M.; Jeejeebhoy, K.N.; Kott, M.; Jiang, X.; Day, A.G.; Heyland, D.K. Enteral versus parenteral nutrition in critically ill patients: An updated systematic review and meta-analysis of randomized controlled trials. Crit. Care 2016, 20, 117. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.A.; Teckman, J.H. Controversies in the Mechanism of Total Parenteral Nutrition Induced Pathology. Children 2015, 2, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Roh, E.; Song, D.; Kim, M.S. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp. Mol. Med. 2016, 48, e216. [Google Scholar] [CrossRef] [PubMed]
- Gore, D.C.; Chinkes, D.L.; Hart, D.W.; Wolf, S.E.; Herndon, D.N.; Sanford, A.P. Hyperglycemia exacerbates muscle protein catabolism in burn-injured patients. Crit. Care Med. 2002, 30, 2438–2442. [Google Scholar] [CrossRef]
- Cole, N.W.; Weaver, K.R.; Walcher, B.N.; Adams, Z.F.; Miller, R.R., Jr. Hyperglycemia-induced membrane lipid peroxidation and elevated homocysteine levels are poorly attenuated by exogenous folate in embryonic chick brains. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 150, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, J.; Pasonen, S.; Yrjänheikki, J.; Chan, P.H. Spreading depression-induced gene expression is regulated by plasma glucose. Stroke 1999, 30, 114–119. [Google Scholar] [CrossRef]
- Li, W.; Roy Choudhury, G.; Winters, A.; Prah, J.; Lin, W.; Liu, R.; Yang, S.H. Hyperglycemia Alters Astrocyte Metabolism and Inhibits Astrocyte Proliferation. Aging Dis. 2018, 9, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G.; Wilmer, A.; Hermans, G.; Meersseman, W.; Wouters, P.J.; Milants, I.; Van Wijngaerden, E.; Bobbaers, H.; Bouillon, R. Intensive insulin therapy in the medical ICU. N. Engl. J. Med. 2006, 354, 449–461. [Google Scholar] [CrossRef] [PubMed]
- NICE-SUGAR Study Investigators. Intensive versus conventional glucose control in critically ill patients. N. Engl. J. Med. 2009, 360, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.C.; Bruno, A.; Pauls, Q.; Hall, C.E.; Barrett, K.M.; Barsan, W.; Fansler, A.; Van de Bruinhorst, K.; Janis, S.; Durkalski-Mauldin, V.L.; et al. Intensive vs. Standard Treatment of Hyperglycemia and Functional Outcome in Patients With Acute Ischemic Stroke: The SHINE Randomized Clinical Trial. JAMA 2019, 322, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P.; Boonyaputthikul, R.; McArthur, D.L.; Miller, C.; Etchepare, M.; Bergsneider, M.; Glenn, T.; Martin, N.; Hovda, D. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit. Care Med. 2006, 34, 850–856. [Google Scholar] [CrossRef]
- Hermanides, J.; Plummer, M.P.; Finnis, M.; Deane, A.M.; Coles, J.P.; Menon, D.K. Glycaemic control targets after traumatic brain injury: A systematic review and meta-analysis. Crit. Care 2018, 22, 11. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Tasker, R.C.; Carney, N.; Totten, A.M.; Adelson, P.D.; Selden, N.R.; Davis-O’Reilly, C.; Hart, E.L.; Bell, M.J.; Bratton, S.L.; et al. Guidelines for the management of pediatric severe traumatic brain injury, third edition: Update of the brain trauma foundation guidelines. Pediat. Crit. Care Med. 2019, 20, 1–82. [Google Scholar] [CrossRef]
- Briassoulis, G.; Filippou, O.; Kanariou, M.; Papassotiriou, I.; Hatzis, T. Temporal nutritional and inflammatory changes in children with severe head injury fed a regular or an immune-enhancing diet: A randomized, controlled trial. Pediatr. Crit. Care Med. 2006, 7, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Fivez, T.; Kerklaan, D.; Mesotten, D.; Verbruggen, S.; Wouters, P.J.; Vanhorebeek, I.; Debaveye, Y.; Vlasselaers, D.; Desmet, L.; Casaer, M.P.; et al. Early versus Late Parenteral Nutrition in Critically Ill Children. N. Engl. J. Med. 2016, 374, 1111–1122. [Google Scholar] [CrossRef]
- Michaud, L.J.; Rivara, F.P.; Longstreth, W.T., Jr.; Grady, M.S. Elevated initial blood glucose levels and poor outcome following severe brain injuries in children. J. Trauma. 1991, 31, 1356–1362. [Google Scholar] [CrossRef]
- Cochran, A.; Scaife, E.R.; Hansen, K.W.; Downey, E.C. Hyperglycemia and outcomes from pediatric traumatic brain injury. J. Trauma. 2003, 55, 1035–1038. [Google Scholar] [CrossRef]
- Chiaretti, A.; Piastra, M.; Pulitano, S.; Pietrini, D.; De Rosa, G.; Barbaro, R.; Di Rocco, C. Prognostic factors and outcome of children with severe head injury: An 8-year experience. Childs Nerv. Syst. 2002, 18, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Tam, E.W.Y.; Kamino, D.; Shatil, A.S.; Chau, V.; Moore, A.M.; Brant, R.; Widjaja, E. Hyperglycemia associated with acute brain injury in neonatal encephalopathy. Neuroimage Clin. 2021, 32, 102835. [Google Scholar] [CrossRef]
- Lee, I.C.; Yang, J.J.; Liou, Y.M. Early Blood Glucose Level Post-Admission Correlates with the Outcomes and Oxidative Stress in Neonatal Hypoxic-Ischemic Encephalopathy. Antioxidants 2021, 11, 39. [Google Scholar] [CrossRef]
- Muller-Schwarze, A.B.; Tandon, P.; Liu, Z.; Yang, Y.; Holmes, G.L.; Stafstrom, C.E. Ketogenic diet reduces spontaneous seizures and mossy fiber sprouting in the kainic acid model. Neuroreport 1999, 10, 1517–1522. [Google Scholar] [CrossRef]
- Noh, H.S.; Kim, Y.S.; Lee, H.P.; Chung, K.M.; Kim, D.W.; Kang, S.S.; Cho, G.J.; Choi, W.S. The protective effect of a ketogenic diet on kainic acid-induced hippocampal cell death in the male ICR mice. Epilepsy Res. 2003, 53, 119–128. [Google Scholar] [CrossRef]
- Tai, K.K.; Nguyen, N.; Pham, L.; Truong, D.D. Ketogenic diet prevents cardiac arrest-induced cerebral ischemic neurodegeneration. J. Neural. Transm. 2008, 115, 1011–1017. [Google Scholar] [CrossRef]
- Yamada, K.A.; Rensing, N.; Thio, L.L. Ketogenic diet reduces hypoglycemia-induced neuronal death in young rats. Neurosci. Lett. 2005, 385, 210–214. [Google Scholar] [CrossRef]
- Appelberg, K.S.; Hovda, D.A.; Prins, M.L. The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. J. Neurotrauma. 2009, 26, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Streijger, F.; Plunet, W.T.; Lee, J.H.; Liu, J.; Lam, C.K.; Park, S.; Hilton, B.J.; Fransen, B.L.; Matheson, K.A.; Assinck, P.; et al. Ketogenic diet improves forelimb motor function after spinal cord injury in rodents. PLoS ONE 2013, 8, e78765. [Google Scholar] [CrossRef] [PubMed]
- Haces, M.L.; Hernández-Fonseca, K.; Medina-Campos, O.N.; Montiel, T.; Pedraza-Chaverri, J.; Massieu, L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 2008, 211, 85–96. [Google Scholar] [CrossRef]
- Gómora-García, J.C.; Montiel, T.; Hüttenrauch, M.; Salcido-Gómez, A.; García-Velázquez, L.; Ramiro-Cortés, Y.; Gomora, J.C.; Castro-Obregón, S.; Massieu, L. Effect of the Ketone Body, D-β-Hydroxybutyrate, on Sirtuin2-Mediated Regulation of Mitochondrial Quality Control and the Autophagy-Lysosomal Pathway. Cells 2023, 12, 486. [Google Scholar] [CrossRef]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2022, 12, 825816, Erratum Front. Physiol. 2022, 13, 867827. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.; Blázquez, C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol. Metab. 2001, 12, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Erecinska, M.; Nelson, D.; Daikhin, Y.; Yudkoff, M. Regulation of GABA level in rat brain synaptosomes: Fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J. Neurochem. 1996, 67, 2325–2334. [Google Scholar] [CrossRef]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic control of vesicular glutamate transport and release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef]
- Noviawaty, I.; Olaru, E.; Rondello, C.; Fitzsimmons, B.; Raghavan, M. Clinical Reasoning: Ketogenic diet in adult super-refractory status epilepticus. Neurology 2020, 94, 541–546. [Google Scholar] [CrossRef]
- Mahmoud, S.H.; Ho-Huang, E.; Buhler, J. Systematic review of ketogenic diet use in adult patients with status epilepticus. Epilepsia Open. 2019, 5, 10–21. [Google Scholar] [CrossRef]
- Gaspard, N.; Hirsch, L.J.; Sculier, C.; Loddenkemper, T.; van Baalen, A.; Lancrenon, J.; Emmery, M.; Specchio, N.; Farias-Moeller, R.; Wong, N.; et al. New-onset refractory status epilepticus (NORSE) and febrile infection-related epilepsy syndrome (FIRES): State of the art and perspectives. Epilepsia 2018, 59, 745–752. [Google Scholar] [CrossRef]
- Lin, J.J.; Lin, K.L.; Chan, O.W.; Hsia, S.H.; Wang, H.S.; CHEESE Study Group. Intravenous ketogenic diet therapy for treatment of the acute stage of super-refractory status epilepticus in a pediatric patient. Pediatr. Neurol. 2015, 52, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Demirel, A.; Li, J.; Morrow, C.; Barnes, S.; Jansen, J.; Gower, B.; Kirksey, K.; Redden, D.; Yarar-Fisher, C. Evaluation of a ketogenic diet for improvement of neurological recovery in individuals with acute spinal cord injury: Study protocol for a randomized controlled trial. Trials 2020, 21, 372. [Google Scholar] [CrossRef]
- Arora, N.; Litofsky, N.S.; Golzy, M.; Aneja, R.; Staudenmyer, D.; Qualls, K.; Patil, S. Phase I single center trial of ketogenic diet for adults with traumatic brain injury. Clin. Nutr. ESPEN 2022, 47, 339–345. [Google Scholar] [CrossRef]
- Rippee, M.A.; Chen, J.; Taylor, M.K. The Ketogenic Diet in the Treatment of Post-concussion Syndrome-A Feasibility Study. Front. Nutr. 2020, 7, 160. [Google Scholar] [CrossRef]
- Deng-Bryant, Y.; Prins, M.L.; Hovda, D.A.; Harris, N.G. Ketogenic Diet Prevents Alterations in Brain Metabolism in Young but not Adult Rats after Traumatic Brain Injury. J. Neurotrauma. 2011, 28, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Bernini, A.; Masoodi, M.; Solari, D.; Miroz, J.P.; Carteron, L.; Christinat, N.; Morelli, P.; Beaumont, M.; Abed-Maillard, S.; Hartweg, M.; et al. Modulation of cerebral ketone metabolism following traumatic brain injury in humans. J. Cereb. Blood Flow Metab. 2020, 40, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Eiden, M.; Christinat, N.; Chakrabarti, A.; Sonnay, S.; Miroz, J.P.; Cuenoud, B.; Oddo, M.; Masoodi, M. Discovery and validation of temporal patterns involved in human brain ketometabolism in cerebral microdialysis fluids of traumatic brain injury patients. EBioMedicine 2019, 44, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T. Lipid mediators in health and disease: Enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu. Rev. Pharmacol Toxicol. 2009, 49, 123–150. [Google Scholar] [CrossRef]
- Musto, A.E.; Walker, C.P.; Petasis, N.A.; Bazan, N.G. Hippocampal neuro-networks and dendritic spine perturbations in epileptogenesis are attenuated by neuroprotectin d1. PLoS ONE 2015, 10, e0116543. (In English) [Google Scholar] [CrossRef]
- Medeiros, R.; Kitazawa, M.; Passos, G.F.; Baglietto-Vargas, D.; Cheng, D.; Cribbs, D.H.; LaFerla, F.M. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am. J. Pathol. 2013, 182, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Hu, Q.; Xu, L.; Guo, Z.; Ou, Y.; He, Y.; Yin, C.; Sun, X.; Tang, J.; Zhang, J.H. Lipoxin A4 Reduces Inflammation Through Formyl Peptide Receptor 2/p38 MAPK Signaling Pathway in Subarachnoid Hemorrhage Rats. Stroke 2016, 47, 490–497. [Google Scholar] [CrossRef]
- Bazan, N.G.; Eady, T.N.; Khoutorova, L.; Atkins, K.D.; Hong, S.; Lu, Y.; Zhang, C.; Jun, B.; Obenaus, A.; Fredman, G.; et al. Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp. Neurol. 2012, 236, 122–130. [Google Scholar] [CrossRef]
- Poblete, R.A.; Arenas, M.; Sanossian, N.; Hong, Y.; Freeman, W.D.; Lyden, P.D.; Louie, S.G. Pro-resolving lipid mediators in traumatic brain injury: Emerging concepts and translational approach. Am. J. Transl Res. 2022, 14, 1482–1494. [Google Scholar]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J. Neurotrauma. 2004, 21, 1457–1467. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Omega-3 fatty acids supplementation restores mechanisms that maintain brain homeostasis in traumatic brain injury. J. Neurotrauma. 2007, 24, 1587–1595. [Google Scholar] [CrossRef]
- Mills, J.D.; Hadley, K.; Bailes, J.E. Dietary supplementation with the omega-3 fatty acid docosahexaenoic acid in traumatic brain injury. Neurosurgery 2011, 68, 474–481; discussion 481. [Google Scholar] [CrossRef]
- Chang, C.Y.; Kuan, Y.H.; Li, J.R.; Chen, W.Y.; Ou, Y.C.; Pan, H.C.; Liao, S.L.; Raung, S.L.; Chang, C.J.; Chen, C.J. Docosahexaenoic acid reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. J. Nutr. Biochem. 2013, 24, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hébert, M.; Julien, C.; Cordeau, P.; Bohacek, I.; Weng, Y.C.; Calon, F.; Kriz, J. Accumulation of dietary docosahexaenoic acid in the brain attenuates acute immune response and development of postischemic neuronal damage. Stroke 2011, 42, 2903–2909. [Google Scholar] [CrossRef] [PubMed]
- Mayurasakorn, K.; Williams, J.J.; Ten, V.S.; Deckelbaum, R.J. Docosahexaenoic acid: Brain accretion and roles in neuroprotection after brain hypoxia and ischemia. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Wurtman, R.J.; Cansev, M.; Sakamoto, T.; Ulus, I.H. Use of phosphatide precursors to promote synaptogenesis. Annu. Rev. Nutr. 2009, 29, 59–87. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, J.; Hu, X.; Li, P.; Leak, R.K.; Gao, Y.; Chen, J. n-3 Polyunsaturated Fatty Acids Reduce Neonatal Hypoxic/Ischemic Brain Injury by Promoting Phosphatidylserine Formation and Akt Signaling. Stroke 2015, 46, 2943–2950. [Google Scholar] [CrossRef]
- Lewis, M.; Ghassemi, P.; Hibbeln, J. Therapeutic use of omega-3 fatty acids in severe head trauma. Am. J. Emerg. Med. 2013, 31, 273.e5–273.e8. [Google Scholar] [CrossRef]
- Bailes, J.E.; Abusuwwa, R.; Arshad, M.; Chowdhry, S.A.; Schleicher, D.; Hempeck, N.; Gandhi, Y.N.; Jaffa, Z.; Bokhari, F.; Karahalios, D.; et al. Omega-3 fatty acid supplementation in severe brain trauma: Case for a large multicenter trial. J. Neurosurg. 2020, 133, 1–5. [Google Scholar] [CrossRef]
- Agostoni, C.; Bresson, J.L.; Fairweather Tait, S.; Flynn, A.; Golly, I.; Korhonen, H.; Lagiou, P.; Løvik, M.; Marchelli, R.; Martin, A.; et al. Scientific opinion on the tolerable upper intake level of eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and docosapentaenoic acid (DPA): EFSA panel on dietetic products, nutrition and allergies (NDA). EFSA J. 2012, 10, 1–48. [Google Scholar]
- Grau-Carmona, T.; Bonet-Saris, A.; Garcia-de-Lorenzo, A.; Sánchez-Alvarez, C.; Rodríguez-Pozo, A.; Acosta-Escribano, J.; Miñambres, E.; Herrero-Meseguer, J.I.; Mesejo, A. Influence of n-3 polyunsaturated fatty acids enriched lipid emulsions on nosocomial infections and clinical outcomes in critically ill patients: ICU lipids study. Crit. Care Med. 2015, 43, 31–39. [Google Scholar] [CrossRef]
- Pradelli, L.; Klek, S.; Mayer, K.; Alsaleh, A.J.O.; Rosenthal, M.D.; Heller, A.R.; Muscaritoli, M. Omega-3 fatty acid-containing parenteral nutrition in ICU patients: Systematic review with meta-analysis and cost-effectiveness analysis. Crit. Care. 2020, 24, 634. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak-Siedlecka, K.; Daca, A.; Folwarski, M.; Makarewicz, W.; Lebiedzińska, A. Immunonutritional support as an important part of multidisciplinary anti-cancer therapy. Cent. Eur. J. Immunol. 2020, 45, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Zheng, X.; Wang, G.; Liu, M.; Li, Y.; Yu, P.; Yang, M.; Guo, N.; Ma, X.; Bu, Y.; et al. Immunonutrition vs Standard Nutrition for Cancer Patients: A Systematic Review and Meta-Analysis (Part 1). J. Parenter. Enter. Nutr. 2020, 44, 742–767. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.S.; Morgan, B.B.; Heineman, J.T.; Martindale, R.G. Nutritional armor for the injured warfighter: Omega-3 fatty acids in surgery, trauma, and intensive care. Mil. Med. 2014, 179 (Suppl. 11), 88–94. [Google Scholar] [CrossRef]
- Kavalukas, S.; McClave, S.A. Immunonutrition vs standard nutrition for patients with cancer. Nutr. Clin. Pract. 2023, 38, 924–931. [Google Scholar] [CrossRef]
- Levenson, C.W. Zinc and Traumatic Brain Injury: From Chelation to Supplementation. Med. Sci. 2020, 8, 36. [Google Scholar] [CrossRef]
- Vonder Haar, C.; Peterson, T.C.; Martens, K.M.; Hoane, M.R. Vitamins and nutrients as primary treatments in experimental brain injury: Clinical implications for nutraceutical therapies. Brain Res. 2016, 1640 (Pt A), 114–129. [Google Scholar] [CrossRef]
- Ortiz, J.F.; Ruxmohan, S.; Saxena, A.; Cox, A.M.; Bashir, F.; Tambo, W.; Ghani, M.R.; Moya, G.; Cordova, I. Minocycline and Magnesium as Neuroprotective Agents for Ischemic Stroke: A Systematic Review. Cureus 2020, 12, e12339. [Google Scholar] [CrossRef]
- Huang, W.; Zhu, L.; Song, W.; Zhang, M.; Teng, L.; Wu, M. Crosstalk between the Gut and Brain in Ischemic Stroke: Mechanistic Insights and Therapeutic Options. Mediat. Inflamm. 2022, 2022, 6508046. [Google Scholar] [CrossRef] [PubMed]
- Sorboni, S.G.; Moghaddam, H.S.; Jafarzadeh-Esfehani, R.; Soleimanpour, S. A Comprehensive Review on the Role of the Gut Microbiome in Human Neurological Disorders. Clin. Microbiol. Rev. 2022, 35, e0033820. [Google Scholar] [CrossRef] [PubMed]
- Dani, K.A.; An, L.; Henning, E.C.; Shen, J.; Warach, S.; National Institute of Neurological Disorders and Stroke Natural History of Stroke Investigators. Multivoxel MR spectroscopy in acute ischemic stroke: Comparison to the stroke protocol MRI. Stroke 2012, 43, 2962–2967. [Google Scholar] [CrossRef] [PubMed]
- Amyot, F.; Arciniegas, D.B.; Brazaitis, M.P.; Curley, K.C.; Diaz-Arrastia, R.; Gandjbakhche, A.; Herscovitch, P.; Hinds, S.R., 2nd; Manley, G.T.; Pacifico, A.; et al. A Review of the Effectiveness of Neuroimaging Modalities for the Detection of Traumatic Brain Injury. J. Neurotrauma. 2015, 32, 1693–1721. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.; Junqué, C.; Mataró, M.; Poca, M.A.; Bargalló, N.; Olondo, M.; Sahuquillo, J. Neuropsychological correlates of basal ganglia and medial temporal lobe NAA/Cho reductions in traumatic brain injury. Arch. Neurol. 2004, 61, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Longo, D.; Lucignani, M.; Pasquini, L.; Rossi-Espagnet, M.C.; Lucignani, G.; Maiorana, A.; Elia, D.; De Liso, P.; Dionisi-Vici, C.; et al. The Ketogenic Diet Increases In Vivo Glutathione Levels in Patients with Epilepsy. Metabolites 2020, 10, 504. [Google Scholar] [CrossRef] [PubMed]
- Hazany, S.; DeClouette, B.; Lowe, J.; Kim, P.E.; Bluml, S.; Partikian, A. Brain Glutathione Increase and Seizure Burden Decrease in Patients with Intractable Epilepsy on Ketogenic Diet. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Wang, Z.; Mascarenhas, C.; Jia, X. Positron Emission Tomography After Ischemic Brain Injury: Current Challenges and Future Developments. Transl. Stroke Res. 2020, 11, 628–642. [Google Scholar] [CrossRef]
- Schaafsma, A.; de Jong, B.M.; Bams, J.L.; Haaxma-Reiche, H.; Pruim, J.; Zijlstra, J.G. Cerebral perfusion and metabolism in resuscitated patients with severe post-hypoxic encephalopathy. J. Neurol. Sci. 2003, 210, 23–30. [Google Scholar] [CrossRef]
- Rudolf, J.; Ghaemi, M.; Ghaemi, M.; Haupt, W.F.; Szelies, B.; Heiss, W.D. Cerebral glucose metabolism in acute and persistent vegetative state. J. Neurosurg. Anesthesiol. 1999, 11, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, T.; Fukumoto, D.; Kakiuchi, T.; Nishiyama, S.; Yamamoto, S.; Ohba, H.; Tsukada, H.; Ueki, T.; Sato, K.; Ouchi, Y. In vivo tspo and cannabinoid receptor type 2 availability early in post-stroke neuroinflammation in rats: A positron emission tomography study. J. Neuroinflamm. 2017, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yue, X.; Kiesewetter, D.O.; Wang, Z.; Lu, J.; Niu, G.; Teng, G.; Chen, X. [(18)f]dpa-714 pet imaging of amd3100 treatment in a mouse model of stroke. Mol. Pharm. 2014, 11, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, T.; Doche, E.; Philip, M.; Cammilleri, S.; Suissa, L.; Guedj, E. Regional brain glucose metabolism is differentially affected by ketogenic diet: A human semiquantitative positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Ellingson, B.M.; Yao, J.; Raymond, C.; Chakhoyan, A.; Khatibi, K.; Salamon, N.; Villablanca, J.P.; Wanner, I.; Real, C.R.; Laiwalla, A.; et al. pH-weighted molecular MRI in human traumatic brain injury (TBI) using amine proton chemical exchange saturation transfer echoplanar imaging (CEST EPI). Neuroimage Clin. 2019, 22, 101736. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, T.J.M.; Straathof, M.; van der Toorn, A.; Otte, W.M.; Adan, R.A.H.; Dijkhuizen, R.M. Diet as connecting factor: Functional brain connectivity in relation to food intake and sucrose tasting, assessed with resting-state functional MRI in rats. J. Neurosci. Res. 2022, 100, 1182–1190. [Google Scholar] [CrossRef]
- Kurtz, P.; Rocha, E.E.M. Nutrition Therapy, Glucose Control, and Brain Metabolism in Traumatic Brain Injury: A Multimodal Monitoring Approach. Front. Neurosci. 2020, 14, 190. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P.M. The implications of cerebral ischemia and metabolic dysfunction for treatment strategies in neurointensive care. Curr. Opin. Crit. Care 2006, 12, 119–123. [Google Scholar] [CrossRef]
- Vespa, P.M.; Miller, C.; McArthur, D.; Eliseo, M.; Etchepare, M.; Hirt, D.; Glenn, T.C.; Martin, N.; Hovda, D. Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit. Care Med. 2007, 35, 2830–2836. [Google Scholar] [CrossRef]
- Vespa, P.M.; McArthur, D.; O’Phelan, K.; Glenn, T.; Etchepare, M.; Kelly, D.; Bergsneider, M.; Martin, N.A.; Hovda, D.A. Persistently low extracellular glucose correlates with poor outcome 6 months after human traumatic brain injury despite a lack of increased lactate: A microdialysis study. J. Cereb. Blood Flow Metab. 2003, 23, 865–877. [Google Scholar] [CrossRef]
- Vespa, P.; Bergsneider, M.; Hattori, N.; Wu, H.; Huang, S.; Martin, N.A.; Glenn, T.C.; McArthur, D.L.; Hovda, D.A. Metabolic crisis without brain ischemia is common after traumatic brain injury: A combined microdialysis and positron emission tomography study. J. Cereb. Blood Flow Metab. 2005, 25, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Timofeev, I.; Carpenter, K.L.H.; Nortje, J.; Al-Rawi, P.G.; O’Connell, M.T.; Czosnyka, M.; Smielewski, P.; Pickard, J.D.; Menon, D.K.; Kirkpatrick, P.J.; et al. Cerebral extracellular chemistry and outcome following traumatic brain injury: A microdialysis study of 223 patients. Brain 2011, 134, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.M.; Claassen, J.; Ko, S. Nutritional support and brain tissue glucose metabolism in poor-grade SAH: A retrospective observational study. Crit. Care. 2012, 16, R15. [Google Scholar] [CrossRef] [PubMed]
- Kofler, M.; Schiefecker, A.J.; Beer, R.; Gaasch, M.; Rhomberg, P.; Stover, J.; Pfausler, B.; Thomé, C.; Schmutzhard, E.; Helbok, R. Enteral nutrition increases interstitial brain glucose levels in poor-grade subarachnoid hemorrhage patients. J. Cereb. Blood Flow Metab. 2018, 38, 518–527. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Nutrient | Proposed Metabolic or Cellular Benefit |
| Ketone bodies and/or ketogenic diet | Provision of an alternative energy substrate through ketone bodies. Reduced excitotoxicity, reduced oxidative stress, improved mitochondrial and astrocyte ATP production, anti-inflammatory and anti-seizure properties. |
| Omega-3 fatty acids: EPA, DHA, DPA | Promotion of the resolution of inflammation. Attenuated excitotoxicity, cell membrane stabilization, support of neurogenesis and antioxidant activities. |
| Glutamine | Promotion of cell proliferation, ATP biosynthesis, glycogenesis, maintenance of acid-base balance, immunomodulator. |
| Arginine | Synthesis of intracellular proteins, nitric oxide, polyamines, glutamic acid, ornithine, proline and creatine, immunomodulator. |
| Nucleotides | Stimulation of lymphocyte proliferation, modulation of the immune activity of natural killer cells and macrophages. |
| Other amino acids | Support for glutathione biosynthesis for tissue repair, support for catecholamine biosynthesis, ROS scavenging. |
| Mineral elements (ex. zinc, magnesium) | May contribute to regulating antioxidant capabilities. |
| Probiotics and Prebiotics | Support for the production of neuroactive molecules that promote communication between the gut microbiota and brain while maintaining homeostasis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poblete, R.A.; Yaceczko, S.; Aliakbar, R.; Saini, P.; Hazany, S.; Breit, H.; Louie, S.G.; Lyden, P.D.; Partikian, A. Optimization of Nutrition after Brain Injury: Mechanistic and Therapeutic Considerations. Biomedicines 2023, 11, 2551. https://doi.org/10.3390/biomedicines11092551
Poblete RA, Yaceczko S, Aliakbar R, Saini P, Hazany S, Breit H, Louie SG, Lyden PD, Partikian A. Optimization of Nutrition after Brain Injury: Mechanistic and Therapeutic Considerations. Biomedicines. 2023; 11(9):2551. https://doi.org/10.3390/biomedicines11092551
Chicago/Turabian StylePoblete, Roy A., Shelby Yaceczko, Raya Aliakbar, Pravesh Saini, Saman Hazany, Hannah Breit, Stan G. Louie, Patrick D. Lyden, and Arthur Partikian. 2023. "Optimization of Nutrition after Brain Injury: Mechanistic and Therapeutic Considerations" Biomedicines 11, no. 9: 2551. https://doi.org/10.3390/biomedicines11092551