
The Potential Influence of Uremic Toxins on the Homeostasis of Bones and Muscles in Chronic Kidney Disease
 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Pathophysiology of PBUTs: IS and pCS
2.1. Metabolism of PBUTs
2.2. Pathogenesis of PBUTs
2.2.1. AhR Signaling
2.2.2. PBUTs Enhance ROS
2.2.3. PBUTs Diminish the Synthesis of Nitric Oxide (NO)
2.2.4. The Epigenetic Effects of PBUTs
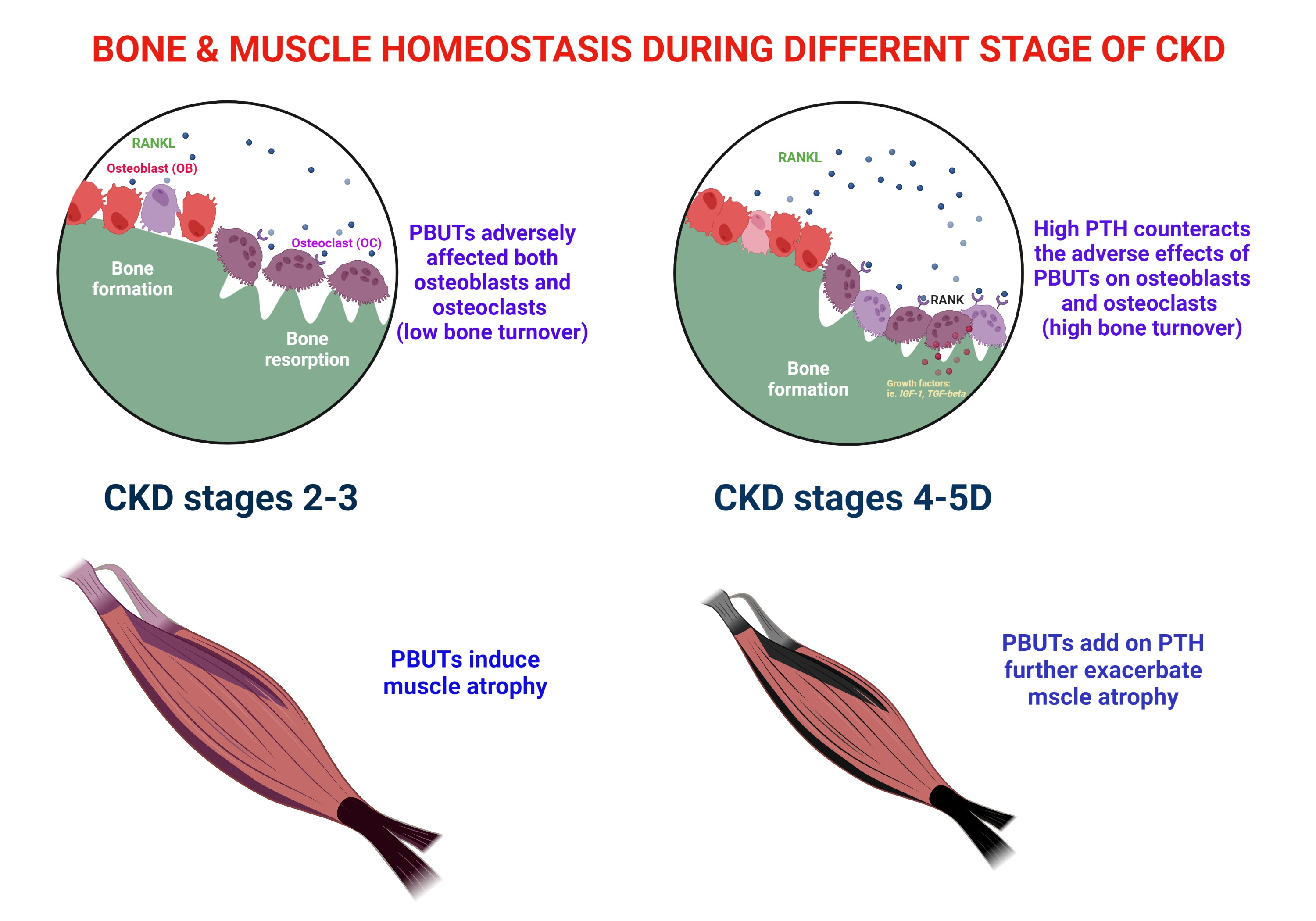
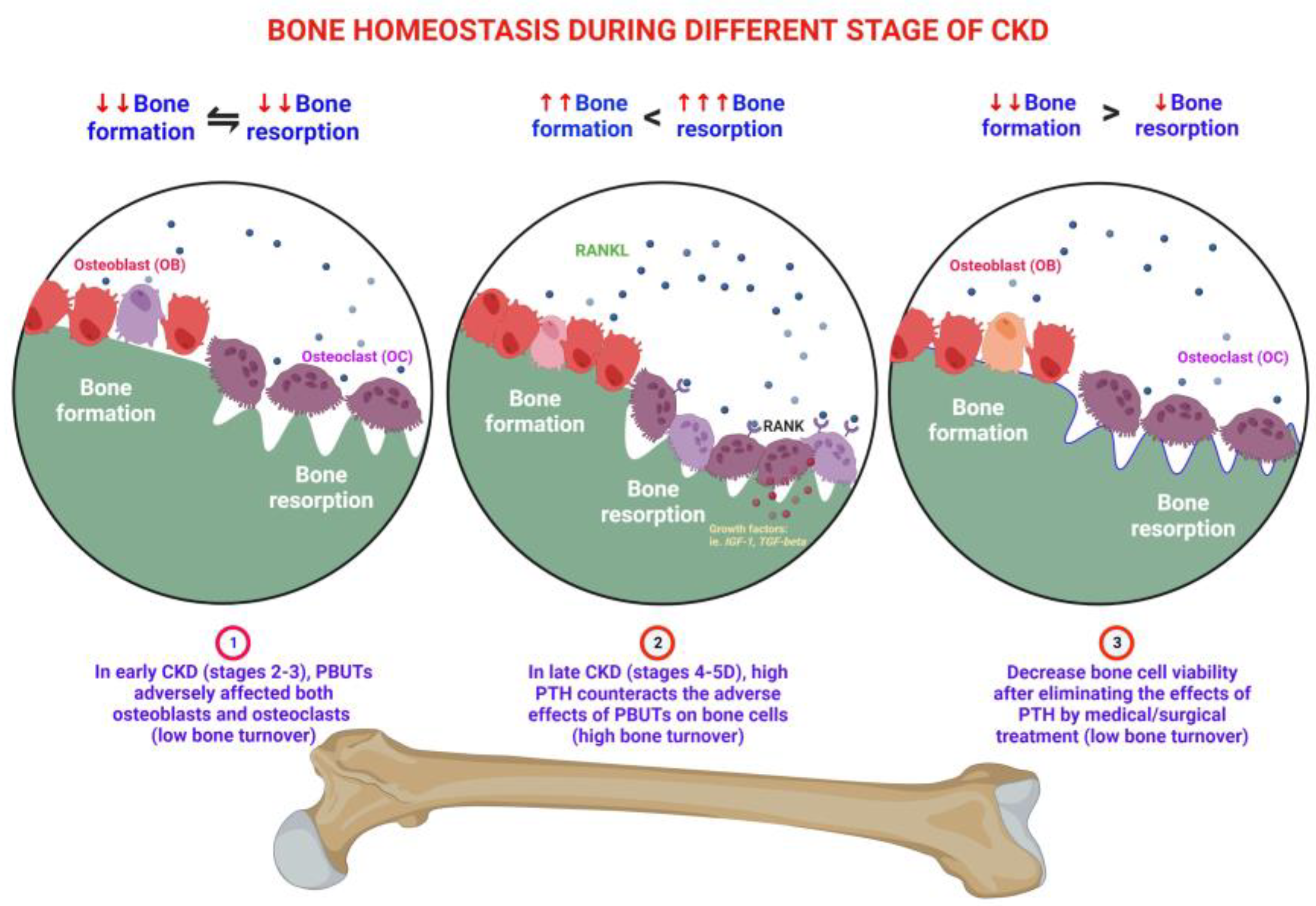
3. Effects of PBUTs on Bone
3.1. PBUTs Influence Bone Metabolism
3.1.1. Uremic Toxin Exposure Affects Osteoclastogenesis
3.1.2. PBUTs Impair Osteoblastogenesis
3.1.3. PBUTs Reduce Bone Mass
3.1.4. PBUTs Reduce the Bone Quality
3.1.5. PBUTs Induce Bone Resistance to PTH
3.1.6. PBUTs Disturb the Synthesis of Vitamin D
3.1.7. PBUTs Affect the Differentiation of T Cells
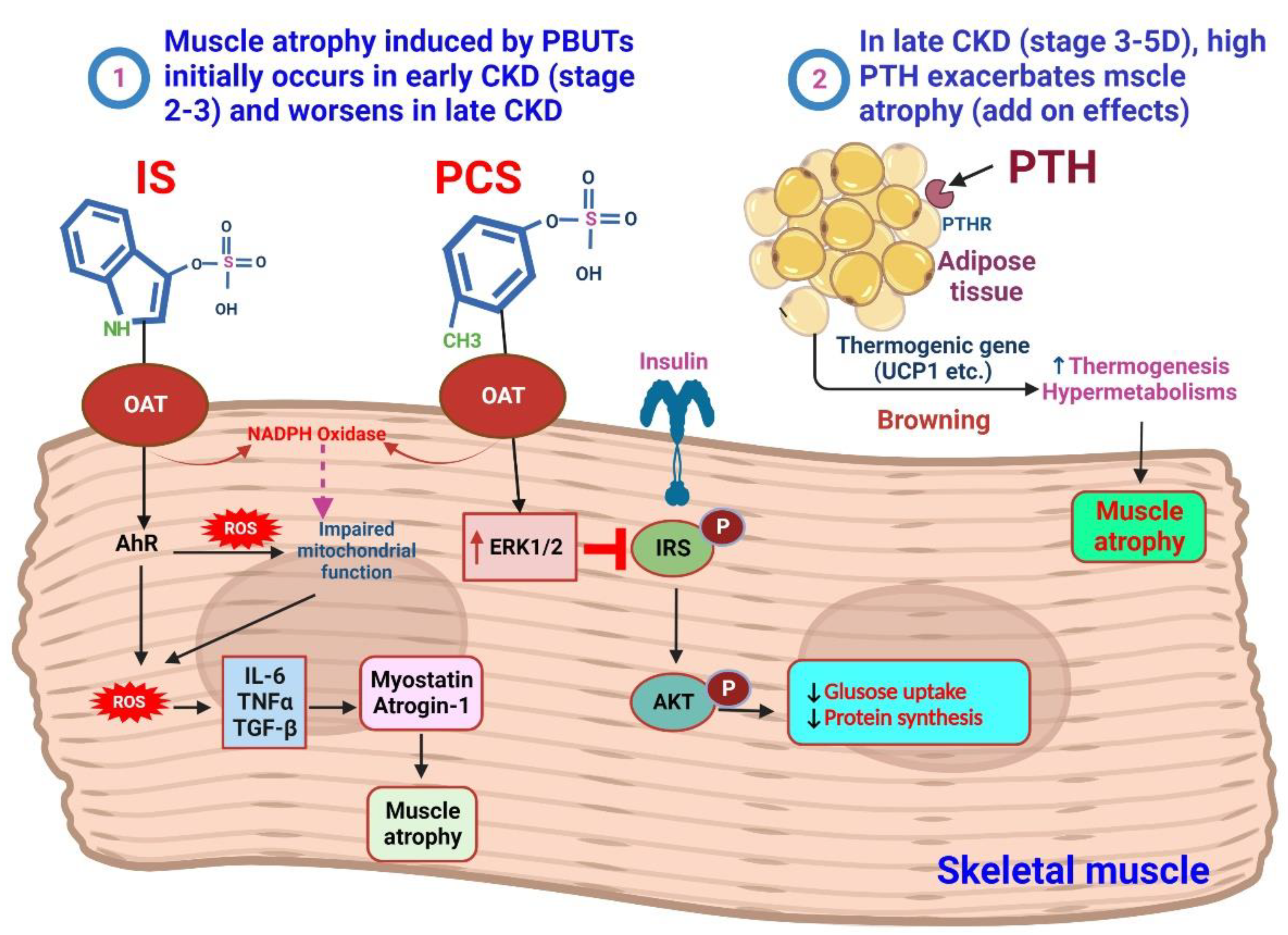
4. IS and pCS on the Muscle
4.1. Sarcopenia in CKD
4.2. PTH and Muscle Atrophy
5. Crosslinks between Bones and Muscles
5.1. Osteokines and Muscle Atrophy
Osteocalcin (OCN)
5.2. Myokines Affect Bones
6. Possible Therapeutic Considerations for Bone and Muscular Health in CKD
Removal or Decrease of PBUT Precursor from the Intestinal Tract
7. Other Possible Therapeutic Strategies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGEs | advanced glycation end products |
| AhR | aryl hydrocarbon receptor |
| Akt | protein kinase B |
| ALK3 | activin receptor-like kinase 3 |
| ARNT | aryl hydrocarbon receptor nuclear translocator |
| BaP | benzo(a)pyrene |
| BAIBA | β-aminoisobutyric acid |
| BDNF | brain-derived neurotrophic factor |
| BMD | bone mineral density |
| BMP | bone morphogenetic protein |
| BMPR1α | BMP receptor 1α |
| cAMP | cyclic adenosine monophosphate |
| cGMP | cyclic guanosine monophosphate |
| CKD | chronic kidney disease |
| CKD–MBD | chronic kidney disease—mineral and bone disorder |
| CYP1A1 | cytochrome P450, family 1, member 1A |
| CYP1B1 | cytochrome P450, family 1, member 1B |
| DKK1 | Dickkopf-1 |
| DNA | deoxyribonucleic acid |
| DNMT | DNA methyltransferase |
| EHSI | elderly hemodialysis sarcopenia index |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| ESRD | end-stage renal disease |
| EWGSOP2 | European Working Group on Sarcopenia in Older People |
| fbox32 | B F-box protein 32 |
| FGF | fibroblast growth factor |
| FoxO | forkhead box protein O |
| GLUT4 | glucose transporter 4 |
| GPRC6A | G protein-coupled receptor class C group 6 member A |
| IAA | indole-3-acetic acid |
| IGF-I | insulin-like growth factor I |
| INF-γ | interferon gamma |
| IL | interleukin |
| IRS | regulation of insulin receptor substrate |
| IS | indoxyl sulfate |
| JNK | Jun NH2-terminal kinase |
| K/DOQI | National Kidney Foundation Kidney Disease Outcomes Quality Initiative |
| KYN | kynurenine |
| KLF6 | Kruppel-like transcription factor 6 |
| MAPK | mitogen-activated protein kinase |
| MC3T3-E1 | mouse calvaria preosteoblastic cells |
| mTOR | mammalian target of rapamycin |
| MCP-1 | monocyte chemoattractant protein-1 |
| MuRF1 | muscle RING-finger 1 |
| NFATc1 | nuclear factor of activated T cells 1 |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NO | nitric oxide |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| OAT | organic anion transporter |
| OB | osteoblast |
| OC | osteoclast |
| OCN | osteocalcin |
| OPG | osteoprotegerin |
| PAI-1 | plasminogen activation inhibitor 1 |
| PBUTs | protein-bound uremic toxins |
| pCS | p-cresyl sulfate |
| PGE2 | prostaglandin E2 |
| PTH | parathyroid hormone |
| PTHR | parathyroid hormone receptor |
| PTHrP | parathyroid hormone-related peptide |
| RANKL | receptor activator of nuclear factor-kappa B ligand |
| Ras–MEK | ERK kinase |
| ROS | reactive oxygen species |
| Runx2 | runt-related transcription factor 2 |
| sFRP | frizzled-related protein |
| SHPT | secondary hyperparathyroidism |
| SOST | sclerostin |
| RSV | resveratrol |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TGF-β | transforming growth factor beta |
| TGF-1 | transforming growth factor-1 |
| Th17 | T-helper 17 |
| TIMP-1 | tissue inhibitor of metalloproteinase-1 |
| TNF-α | tumor necrosis factor-α |
| Treg cells | regulatory T cells |
| TRIM63 | tripartite motif containing 63 |
| TRPV1 | transient receptor potential vanilloid 1 |
| UCP1 | uncoupling protein 1 |
| UPS | ubiquitin–proteasome system |
| Wnt | wingless |
| WHO | World Health Organization |
| XRE | xenobiotic response element |
References
- Vanholder, R.; Van Laecke, S.; Glorieux, G. What is new in uremic toxicity? Pediatr. Nephrol. 2008, 23, 1211–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNerny, E.M.B.; Nickolas, T.L. Bone Quality in Chronic Kidney Disease: Definitions and Diagnostics. Curr. Osteoporos. Rep. 2017, 15, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Kazama, J.J.; Iwasaki, Y.; Fukagawa, M. Uremic osteoporosis. Kidney Int. Suppl. 2013, 3, 446–450. [Google Scholar] [CrossRef] [Green Version]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Holle, J.; Querfeld, U.; Kirchner, M.; Anninos, A.; Okun, J.; Thurn-Valsassina, D.; Bayazit, A.; Niemirska, A.; Canpolat, N.; Bulut, I.K.; et al. Indoxyl sulfate associates with cardiovascular phenotype in children with chronic kidney disease. Pediatr. Nephrol. 2019, 34, 2571–2582. [Google Scholar] [CrossRef]
- Holle, J.; Kirchner, M.; Okun, J.; Bayazit, A.K.; Obrycki, L.; Canpolat, N.; Bulut, I.K.; Azukaitis, K.; Duzova, A.; Ranchin, B.; et al. Serum indoxyl sulfate concentrations associate with progression of chronic kidney disease in children. PLoS ONE 2020, 15, e0240446. [Google Scholar] [CrossRef]
- Nii-Kono, T.; Iwasaki, Y.; Uchida, M.; Fujieda, A.; Hosokawa, A.; Motojima, M.; Yamato, H.; Kurokawa, K.; Fukagawa, M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007, 71, 738–743. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.C.; Wu, C.C.; Lim, P.S.; Chien, S.W.; Hou, Y.C.; Zheng, C.M.; Shyu, J.F.; Lin, Y.F.; Lu, K.C. Effect of uremic toxin-indoxyl sulfate on the skeletal system. Clin. Chim. Acta 2018, 484, 197–206. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kwak, K.A.; Gil, H.W.; Song, H.Y.; Hong, S.Y. Indoxyl sulfate promotes apoptosis in cultured osteoblast cells. BMC Pharmacol. Toxicol. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, Y.; Yamato, H.; Nii-Kono, T.; Fujieda, A.; Uchida, M.; Hosokawa, A.; Motojima, M.; Fukagawa, M. Administration of oral charcoal adsorbent (AST-120) suppresses low-turnover bone progression in uraemic rats. Nephrol. Dial. Transplant. 2006, 21, 2768–2774. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J. Metabolic Acidosis in Chronic Kidney Disease: Pathogenesis, Clinical Consequences, and Treatment. Electrolyte Blood Press. 2021, 19, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.M.; Zheng, J.Q.; Wu, C.C.; Lu, C.L.; Shyu, J.F.; Yung-Ho, H.; Wu, M.Y.; Chiu, I.J.; Wang, Y.H.; Lin, Y.F.; et al. Bone loss in chronic kidney disease: Quantity or quality? Bone 2016, 87, 57–70. [Google Scholar] [CrossRef]
- Drueke, T.B.; Massy, Z.A. Changing bone patterns with progression of chronic kidney disease. Kidney Int. 2016, 89, 289–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.C.; Sakata, T.; Pfleger, L.L.; Bencsik, M.; Halloran, B.P.; Bikle, D.D.; Nissenson, R.A. PTH differentially regulates expression of RANKL and OPG. J. Bone Miner. Res. 2004, 19, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Simões e Silva, A.C.; Oliveira, E.A.; Cheung, W.W.; Mak, R.H. Redox Signaling in Chronic Kidney Disease-Associated Cachexia. Antioxidants 2023, 12, 945. [Google Scholar] [CrossRef]
- Umakanthan, M.; Li, J.W.; Sud, K.; Duque, G.; Guilfoyle, D.; Cho, K.; Brown, C.; Boersma, D.; Gangadharan Komala, M. Prevalence and Factors Associated with Sarcopenia in Patients on Maintenance Dialysis in Australia-A Single Centre, Cross-Sectional Study. Nutrients 2021, 13, 3284. [Google Scholar] [CrossRef]
- Adhikari, A.; Mondal, S.; Chatterjee, T.; Das, M.; Biswas, P.; Ghosh, R.; Darbar, S.; Alessa, H.; Althakafy, J.T.; Sayqal, A.; et al. Redox nanomedicine ameliorates chronic kidney disease (CKD) by mitochondrial reconditioning in mice. Commun. Biol. 2021, 4, 1013. [Google Scholar] [CrossRef]
- Cheung, W.W.; Zheng, R.; Hao, S.; Wang, Z.; Gonzalez, A.; Zhou, P.; Hoffman, H.M.; Mak, R.H. The role of IL-1 in adipose browning and muscle wasting in CKD-associated cachexia. Sci. Rep. 2021, 11, 15141. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E.; Price, S.R. Pathophysiological mechanisms leading to muscle loss in chronic kidney disease. Nat. Rev. Nephrol. 2022, 18, 138–152. [Google Scholar] [CrossRef]
- Enoki, Y.; Watanabe, H.; Arake, R.; Sugimoto, R.; Imafuku, T.; Tominaga, Y.; Ishima, Y.; Kotani, S.; Nakajima, M.; Tanaka, M.; et al. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress-mediated expression of myostatin and atrogin-1. Sci. Rep. 2016, 6, 32084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoli, C.; Zonefrati, R.; Lucattelli, E.; Innocenti, M.; Civinini, R.; Iantomasi, T.; Brandi, M.L. In Vitro Effects of PTH (1-84) on Human Skeletal Muscle-Derived Satellite Cells. Biomedicines 2023, 11, 1017. [Google Scholar] [CrossRef] [PubMed]
- Bislev, L.S.; Langagergaard Rodbro, L.; Sikjaer, T.; Rejnmark, L. Effects of Elevated Parathyroid Hormone Levels on Muscle Health, Postural Stability and Quality of Life in Vitamin D-Insufficient Healthy Women: A Cross-Sectional Study. Calcif. Tissue Int. 2019, 105, 642–650. [Google Scholar] [CrossRef]
- Patten, B.M.; Bilezikian, J.P.; Mallette, L.E.; Prince, A.; Engel, W.K.; Aurbach, G.D. Neuromuscular disease in primary hyperparathyroidism. Ann. Intern. Med. 1974, 80, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Reppe, S.; Stilgren, L.; Abrahamsen, B.; Olstad, O.K.; Cero, F.; Brixen, K.; Nissen-Meyer, L.S.; Gautvik, K.M. Abnormal muscle and hematopoietic gene expression may be important for clinical morbidity in primary hyperparathyroidism. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1465–E1473. [Google Scholar] [CrossRef] [Green Version]
- Minisola, S.; Arnold, A.; Belaya, Z.; Brandi, M.L.; Clarke, B.L.; Hannan, F.M.; Hofbauer, L.C.; Insogna, K.L.; Lacroix, A.; Liberman, U.; et al. Epidemiology, Pathophysiology, and Genetics of Primary Hyperparathyroidism. J. Bone Miner. Res. 2022, 37, 2315–2329. [Google Scholar] [CrossRef]
- de Souza Genaro, P.; de Medeiros Pinheiro, M.; Szejnfeld, V.L.; Martini, L.A. Secondary hyperparathyroidism and its relationship with sarcopenia in elderly women. Arch. Gerontol. Geriatr. 2015, 60, 349–353. [Google Scholar] [CrossRef]
- Visser, M.; Deeg, D.J.; Lips, P.; Longitudinal Aging Study, A. Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (sarcopenia): The Longitudinal Aging Study Amsterdam. J. Clin. Endocrinol. Metab. 2003, 88, 5766–5772. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.S.; Mitch, W.E. Parathyroid hormone stimulates adipose tissue browning: A pathway to muscle wasting. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, C.; Brandi, M.L. Muscle Physiopathology in Parathyroid Hormone Disorders. Front. Med. 2021, 8, 764346. [Google Scholar] [CrossRef]
- Allison, S.J. Chronic kidney disease: Role of PTH/PTHrP receptor in cachexia. Nat. Rev. Nephrol. 2016, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Komaba, H.; Garcia, A.P.; Economopoulos, K.P.; Liu, W.; Lanske, B.; Hodin, R.A.; Spiegelman, B.M. PTH/PTHrP Receptor Mediates Cachexia in Models of Kidney Failure and Cancer. Cell. Metab. 2016, 23, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Chiang, C.K. Uremic Toxins and Protein-Bound Therapeutics in AKI and CKD: Up-to-Date Evidence. Toxins 2021, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.T.; Lin, S.H. Uremic Toxins and Frailty in Patients with Chronic Kidney Disease: A Molecular Insight. Int. J. Mol. Sci. 2021, 22, 6270. [Google Scholar] [CrossRef]
- Deguchi, T.; Kusuhara, H.; Takadate, A.; Endou, H.; Otagiri, M.; Sugiyama, Y. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 2004, 65, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.Y.; Hsu, H.H.; Wu, M.S. p-Cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol. Dial. Transplant. 2013, 28, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.L.; Liao, C.H.; Lu, K.C.; Ma, M.C. TRPV1 Hyperfunction Involved in Uremic Toxin Indoxyl Sulfate-Mediated Renal Tubular Damage. Int. J. Mol. Sci. 2020, 21, 6212. [Google Scholar] [CrossRef]
- Lee, W.J.; Liu, S.H.; Chiang, C.K.; Lin, S.Y.; Liang, K.W.; Chen, C.H.; Tien, H.R.; Chen, P.H.; Wu, J.P.; Tsai, Y.C.; et al. Aryl Hydrocarbon Receptor Deficiency Attenuates Oxidative Stress-Related Mesangial Cell Activation and Macrophage Infiltration and Extracellular Matrix Accumulation in Diabetic Nephropathy. Antioxid. Redox Signal. 2016, 24, 217–231. [Google Scholar] [CrossRef]
- Yavuz, A.; Tetta, C.; Ersoy, F.F.; D’Intini, V.; Ratanarat, R.; De Cal, M.; Bonello, M.; Bordoni, V.; Salvatori, G.; Andrikos, E.; et al. Uremic toxins: A new focus on an old subject. Semin. Dial. 2005, 18, 203–211. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [Green Version]
- De Smet, R.; Van Kaer, J.; Van Vlem, B.; De Cubber, A.; Brunet, P.; Lameire, N.; Vanholder, R. Toxicity of free p-cresol: A prospective and cross-sectional analysis. Clin. Chem. 2003, 49, 470–478. [Google Scholar] [CrossRef]
- Bammens, B.; Evenepoel, P.; Verbeke, K.; Vanrenterghem, Y. Removal of middle molecules and protein-bound solutes by peritoneal dialysis and relation with uremic symptoms. Kidney Int. 2003, 64, 2238–2243. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Claes, K.; Bammens, B.; de Loor, H.; Viaene, L.; Verbeke, K.; Kuypers, D.; Vanrenterghem, Y.; Evenepoel, P. p-Cresol and cardiovascular risk in mild-to-moderate kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1182–1189. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Viaene, L.; Verbeke, K.; Augustijns, P.; Bammens, B.; Claes, K.; Kuypers, D.; Evenepoel, P.; Meijers, B. Cardiovascular disease relates to intestinal uptake of p-cresol in patients with chronic kidney disease. BMC Nephrol. 2014, 15, 87. [Google Scholar] [CrossRef] [Green Version]
- Korkalainen, M.; Kallio, E.; Olkku, A.; Nelo, K.; Ilvesaro, J.; Tuukkanen, J.; Mahonen, A.; Viluksela, M. Dioxins interfere with differentiation of osteoblasts and osteoclasts. Bone 2009, 44, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.J.; De Castro, K.P.; Joshi, A.D.; Elferink, C.J. Canonical and non-canonical aryl hydrocarbon receptor signaling pathways. Curr. Opin. Toxicol. 2017, 2, 87–92. [Google Scholar] [CrossRef]
- Alhamad, D.W.; Bensreti, H.; Dorn, J.; Hill, W.D.; Hamrick, M.W.; McGee-Lawrence, M.E. Aryl hydrocarbon receptor (AhR)-mediated signaling as a critical regulator of skeletal cell biology. J. Mol. Endocrinol. 2022, 69, R109–R124. [Google Scholar] [CrossRef]
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy States. Int. J. Tryptophan Res. 2009, 2, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Fukagawa, M. Uremic Toxicity and Bone in CKD. J. Nephrol. 2017, 30, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Lu, Z.; Wang, L.; Ji, C.; Zou, C.; Liu, X. The Aryl Hydrocarbon Receptor in Chronic Kidney Disease: Friend or Foe? Front. Cell. Dev. Biol. 2020, 8, 589752. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Tomino, Y.; Lu, K.C. Impacts of Indoxyl Sulfate and p-Cresol Sulfate on Chronic Kidney Disease and Mitigating Effects of AST-120. Toxins 2018, 10, 367. [Google Scholar] [CrossRef] [Green Version]
- Monnouchi, S.; Maeda, H.; Yuda, A.; Serita, S.; Wada, N.; Tomokiyo, A.; Akamine, A. Benzo[a]pyrene/aryl hydrocarbon receptor signaling inhibits osteoblastic differentiation and collagen synthesis of human periodontal ligament cells. J. Periodontal Res. 2016, 51, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Herlin, M.; Finnila, M.A.; Zioupos, P.; Aula, A.; Risteli, J.; Miettinen, H.M.; Jamsa, T.; Tuukkanen, J.; Korkalainen, M.; Hakansson, H.; et al. New insights to the role of aryl hydrocarbon receptor in bone phenotype and in dioxin-induced modulation of bone microarchitecture and material properties. Toxicol. Appl. Pharmacol. 2013, 273, 219–226. [Google Scholar] [CrossRef]
- Jamsa, T.; Viluksela, M.; Tuomisto, J.T.; Tuomisto, J.; Tuukkanen, J. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on bone in two rat strains with different aryl hydrocarbon receptor structures. J. Bone Miner. Res. 2001, 16, 1812–1820. [Google Scholar] [CrossRef]
- Yu, H.; Du, Y.; Zhang, X.; Sun, Y.; Li, S.; Dou, Y.; Li, Z.; Yuan, H.; Zhao, W. The aryl hydrocarbon receptor suppresses osteoblast proliferation and differentiation through the activation of the ERK signaling pathway. Toxicol. Appl. Pharmacol. 2014, 280, 502–510. [Google Scholar] [CrossRef]
- Iqbal, J.; Sun, L.; Cao, J.; Yuen, T.; Lu, P.; Bab, I.; Leu, N.A.; Srinivasan, S.; Wagage, S.; Hunter, C.A.; et al. Smoke carcinogens cause bone loss through the aryl hydrocarbon receptor and induction of Cyp1 enzymes. Proc. Natl. Acad. Sci. USA 2013, 110, 11115–11120. [Google Scholar] [CrossRef]
- Voronov, I.; Heersche, J.N.; Casper, R.F.; Tenenbaum, H.C.; Manolson, M.F. Inhibition of osteoclast differentiation by polycyclic aryl hydrocarbons is dependent on cell density and RANKL concentration. Biochem. Pharmacol. 2005, 70, 300–307. [Google Scholar] [CrossRef]
- Eskenazi, B.; Warner, M.; Brambilla, P.; Signorini, S.; Ames, J.; Mocarelli, P. The Seveso accident: A look at 40 years of health research and beyond. Environ. Int. 2018, 121 Pt 1, 71–84. [Google Scholar] [CrossRef]
- Alaluusua, S.; Calderara, P.; Gerthoux, P.M.; Lukinmaa, P.L.; Kovero, O.; Needham, L.; Patterson, D.G., Jr.; Tuomisto, J.; Mocarelli, P. Developmental dental aberrations after the dioxin accident in Seveso. Environ. Health Perspect. 2004, 112, 1313–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskenazi, B.; Warner, M.; Sirtori, M.; Fuerst, T.; Rauch, S.A.; Brambilla, P.; Mocarelli, P.; Rubinacci, A. Serum dioxin concentrations and bone density and structure in the Seveso Women’s Health Study. Environ. Health Perspect. 2014, 122, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milbrath, M.O.; Wenger, Y.; Chang, C.W.; Emond, C.; Garabrant, D.; Gillespie, B.W.; Jolliet, O. Apparent half-lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast-feeding. Environ. Health Perspect. 2009, 117, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaska, B.; Pawlak, K.; Domaniewski, T.; Oksztulska-Kolanek, E.; Znorko, B.; Roszczenko, A.; Rogalska, J.; Brzoska, M.M.; Lipowicz, P.; Doroszko, M.; et al. Elevated Levels of Peripheral Kynurenine Decrease Bone Strength in Rats with Chronic Kidney Disease. Front. Physiol. 2017, 8, 836. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Oh, Y.; Jo, S.; Ji, J.D.; Kim, T.H. Inhibition of Human Osteoclast Differentiation by Kynurenine through the Aryl-Hydrocarbon Receptor Pathway. Cells 2021, 10, 3498. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.Y.; Pang, W.J.; Yang, G.S. Aryl hydrocarbon receptors in osteoclast lineage cells are a negative regulator of bone mass. PLoS ONE 2015, 10, e0117112. [Google Scholar] [CrossRef] [Green Version]
- Brito, J.S.; Borges, N.A.; Esgalhado, M.; Magliano, D.C.; Soulage, C.O.; Mafra, D. Aryl Hydrocarbon Receptor Activation in Chronic Kidney Disease: Role of Uremic Toxins. Nephron 2017, 137, 1–7. [Google Scholar] [CrossRef]
- Niwa, T. Indoxyl sulfate is a nephro-vascular toxin. J. Ren. Nutr. 2010, 20, S2–S6. [Google Scholar] [CrossRef]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-kappaB and free radical in proximal tubular cells. Kidney Int. 2003, 63, 1671–1680. [Google Scholar] [CrossRef] [Green Version]
- Stockler-Pinto, M.B.; Saldanha, J.F.; Yi, D.; Mafra, D.; Fouque, D.; Soulage, C.O. The uremic toxin indoxyl sulfate exacerbates reactive oxygen species production and inflammation in 3T3-L1 adipose cells. Free. Radic. Res. 2016, 50, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Borges, N.A.; Barros, A.F.; Nakao, L.S.; Dolenga, C.J.; Fouque, D.; Mafra, D. Protein-Bound Uremic Toxins from Gut Microbiota and Inflammatory Markers in Chronic Kidney Disease. J. Ren. Nutr. 2016, 26, 396–400. [Google Scholar] [CrossRef]
- Gelasco, A.K.; Raymond, J.R. Indoxyl sulfate induces complex redox alterations in mesangial cells. Am. J. Physiol. Renal Physiol. 2006, 290, F1551–F1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owada, S.; Goto, S.; Bannai, K.; Hayashi, H.; Nishijima, F.; Niwa, T. Indoxyl sulfate reduces superoxide scavenging activity in the kidneys of normal and uremic rats. Am. J. Nephrol. 2008, 28, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Takayanagi, K.; Kojima, M.; Taguchi, K.; Kobayashi, T. Correction to: Indoxyl sulfate enhances endothelin-1-induced contraction via impairment of NO/cGMP signaling in rat aorta. Pflug. Arch. 2021, 473, 1329. [Google Scholar] [CrossRef]
- Lu, Z.; Lu, F.; Zheng, Y.; Zeng, Y.; Zou, C.; Liu, X. Grape seed proanthocyanidin extract protects human umbilical vein endothelial cells from indoxyl sulfate-induced injury via ameliorating mitochondrial dysfunction. Ren. Fail. 2016, 38, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, K.; Itoya, M.; Takemoto, N.; Matsuura, Y.; Tawa, M.; Matsumura, Y.; Ohkita, M. Indoxyl sulfate induces ROS production via the aryl hydrocarbon receptor-NADPH oxidase pathway and inactivates NO in vascular tissues. Life Sci. 2021, 265, 118807. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Klotho and chronic kidney disease. Contrib. Nephrol. 2013, 180, 47–63. [Google Scholar]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012, 81, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Dogan, F.; Forsyth, N.R. Telomerase Regulation: A Role for Epigenetics. Cancers 2021, 13, 1213. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Klotho as a regulator of oxidative stress and senescence. Biol. Chem. 2008, 389, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Haruna, Y.; Kashihara, N.; Satoh, M.; Tomita, N.; Namikoshi, T.; Sasaki, T.; Fujimori, T.; Xie, P.; Kanwar, Y.S. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc. Natl. Acad. Sci. USA 2007, 104, 2331–2336. [Google Scholar] [CrossRef]
- Tabibzadeh, S. Signaling pathways and effectors of aging. Front. Biosci. 2021, 26, 50–96. [Google Scholar] [CrossRef]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Klotho and kidney disease. J. Nephrol. 2010, 23 (Suppl. S16), S136–S144. [Google Scholar]
- Kuro-o, M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr. Opin. Nephrol. Hypertens. 2006, 15, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef] [PubMed]
- Roshanravan, B.; Gamboa, J.; Wilund, K. Exercise and CKD: Skeletal Muscle Dysfunction and Practical Application of Exercise to Prevent and Treat Physical Impairments in CKD. Am. J. Kidney Dis. 2017, 69, 837–852. [Google Scholar] [CrossRef]
- Severinsen, M.C.K.; Pedersen, B.K. Muscle-Organ Crosstalk: The Emerging Roles of Myokines. Endocr. Rev. 2020, 41, 594–609. [Google Scholar] [CrossRef]
- Kirk, B.; Feehan, J.; Lombardi, G.; Duque, G. Muscle, Bone, and Fat Crosstalk: The Biological Role of Myokines, Osteokines, and Adipokines. Curr. Osteoporos. Rep. 2020, 18, 388–400. [Google Scholar] [CrossRef]
- Lorenzo, J. The many ways of osteoclast activation. J. Clin. Investig. 2017, 127, 2530–2532. [Google Scholar] [CrossRef] [Green Version]
- Luecke-Johansson, S.; Gralla, M.; Rundqvist, H.; Ho, J.C.; Johnson, R.S.; Gradin, K.; Poellinger, L. A Molecular Mechanism To Switch the Aryl Hydrocarbon Receptor from a Transcription Factor to an E3 Ubiquitin Ligase. Mol. Cell. Biol. 2017, 37, e00630-16. [Google Scholar] [CrossRef] [Green Version]
- Mozar, A.; Louvet, L.; Godin, C.; Mentaverri, R.; Brazier, M.; Kamel, S.; Massy, Z.A. Indoxyl sulphate inhibits osteoclast differentiation and function. Nephrol. Dial. Transplant. 2012, 27, 2176–2181. [Google Scholar] [CrossRef] [Green Version]
- Park, R.; Madhavaram, S.; Ji, J.D. The Role of Aryl-Hydrocarbon Receptor (AhR) in Osteoclast Differentiation and Function. Cells 2020, 9, 2294. [Google Scholar] [CrossRef] [PubMed]
- Wejheden, C.; Brunnberg, S.; Larsson, S.; Lind, P.M.; Andersson, G.; Hanberg, A. Transgenic mice with a constitutively active aryl hydrocarbon receptor display a gender-specific bone phenotype. Toxicol. Sci. 2010, 114, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tominari, T.; Hirata, M.; Matsumoto, C.; Hirata, J.; Murphy, G.; Nagase, H.; Miyaura, C.; Inada, M. Indoxyl sulfate, a uremic toxin in chronic kidney disease, suppresses both bone formation and bone resorption. FEBS Open. Bio 2017, 7, 1178–1185. [Google Scholar] [CrossRef]
- Tampe, B.; Tampe, D.; Nyamsuren, G.; Klopper, F.; Rapp, G.; Kauffels, A.; Lorf, T.; Zeisberg, E.M.; Muller, G.A.; Kalluri, R.; et al. Pharmacological induction of hypoxia-inducible transcription factor ARNT attenuates chronic kidney failure. J. Clin. Investig. 2018, 128, 3053–3070. [Google Scholar] [CrossRef] [Green Version]
- Tou, L.; Quibria, N.; Alexander, J.M. Transcriptional regulation of the human Runx2/Cbfa1 gene promoter by bone morphogenetic protein-7. Mol. Cell. Endocrinol. 2003, 205, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Takahashi, T.; Tsujisawa, T.; Ariyoshi, W.; Nishihara, T. Mechanical stress-mediated Runx2 activation is dependent on Ras/ERK1/2 MAPK signaling in osteoblasts. J. Cell. Biochem. 2007, 101, 1266–1277. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Kazama, J.J.; Yamato, H.; Matsugaki, A.; Nakano, T.; Fukagawa, M. Altered material properties are responsible for bone fragility in rats with chronic kidney injury. Bone 2015, 81, 247–254. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Yamato, H.; Nii-Kono, T.; Fujieda, A.; Uchida, M.; Hosokawa, A.; Motojima, M.; Fukagawa, M. Insufficiency of PTH action on bone in uremia. Kidney Int. Suppl. 2006, 70, S34–S36. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Miyawaki, N.; Moon, J.; Kasselman, L.J.; Voloshyna, I.; D’Avino, R., Jr.; De Leon, J. CKD, arterial calcification, atherosclerosis and bone health: Inter-relationships and controversies. Atherosclerosis 2018, 278, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Kazama, J.J.; Yamato, H.; Shimoda, H.; Fukagawa, M. Accumulated uremic toxins attenuate bone mechanical properties in rats with chronic kidney disease. Bone 2013, 57, 477–483. [Google Scholar] [CrossRef]
- West, S.L.; Patel, P.; Jamal, S.A. How to predict and treat increased fracture risk in chronic kidney disease. J. Intern. Med. 2015, 278, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, P.D. Bone disease in CKD: A focus on osteoporosis diagnosis and management. Am. J. Kidney Dis. 2014, 64, 290–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Canziani, M.E.; Tomiyama, C.; Higa, A.; Mozar, A.; Glorieux, G.; Vanholder, R.; Massy, Z.; de Carvalho, A.B. Association between indoxyl sulfate and bone histomorphometry in pre-dialysis chronic kidney disease patients. J. Bras. Nefrol. 2014, 36, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Kidney Foundation. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am. J. Kidney Dis. 2003, 42 (Suppl. S3), S1–S201. [Google Scholar] [CrossRef]
- Iwasaki-Ishizuka, Y.; Yamato, H.; Nii-Kono, T.; Kurokawa, K.; Fukagawa, M. Downregulation of parathyroid hormone receptor gene expression and osteoblastic dysfunction associated with skeletal resistance to parathyroid hormone in a rat model of renal failure with low turnover bone. Nephrol. Dial. Transplant. 2005, 20, 1904–1911. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.C.; Wu, C.C.; Hung, Y.M.; Liao, M.T.; Shyu, J.F.; Lin, Y.F.; Lu, K.C.; Yeh, K.C. Pleiotropic effects of vitamin D in chronic kidney disease. Clin. Chim. Acta 2016, 453, 1–12. [Google Scholar] [CrossRef]
- Tsujimoto, M.; Nagano, Y.; Hosoda, S.; Shiraishi, A.; Miyoshi, A.; Hiraoka, S.; Furukubo, T.; Izumi, S.; Yamakawa, T.; Minegaki, T.; et al. Effects of decreased vitamin D and accumulated uremic toxin on human CYP3A4 activity in patients with end-stage renal disease. Toxins 2013, 5, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Xiang, F.; Cao, X.; Shen, B.; Chen, X.; Guo, M.; Ding, X.; Zou, J. Transcriptome Profiling Reveals Indoxyl Sulfate Should Be Culpable of Impaired T Cell Function in Chronic Kidney Disease. Front. Med. 2020, 7, 178. [Google Scholar] [CrossRef] [PubMed]
- Sallee, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Kimura, A.; Naka, T.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9721–9726. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Zheng, C.M.; Lu, C.L.; Lin, Y.F.; Shyu, J.F.; Wu, C.C.; Lu, K.C. Vitamin D and immune function in chronic kidney disease. Clin. Chim. Acta 2015, 450, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Kraj, P. Bone Morphogenetic Proteins Shape Treg Cells. Front. Immunol. 2022, 13, 865546. [Google Scholar] [CrossRef] [PubMed]
- Bataille, S.; Chauveau, P.; Fouque, D.; Aparicio, M.; Koppe, L. Myostatin and muscle atrophy during chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 1986–1993. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.L.; Zheng, C.M.; Lu, K.C.; Liao, M.T.; Wu, K.L.; Ma, M.C. Indoxyl-Sulfate-Induced Redox Imbalance in Chronic Kidney Disease. Antioxidants 2021, 10, 936. [Google Scholar] [CrossRef]
- Yang, J.; Li, H.; Zhang, C.; Zhou, Y. Indoxyl sulfate reduces Ito,f by activating ROS/MAPK and NF-kappaB signaling pathways. JCI Insight 2022, 7, e145475. [Google Scholar] [CrossRef]
- Sato, E.; Mori, T.; Mishima, E.; Suzuki, A.; Sugawara, S.; Kurasawa, N.; Saigusa, D.; Miura, D.; Morikawa-Ichinose, T.; Saito, R.; et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci. Rep. 2016, 6, 36618. [Google Scholar] [CrossRef] [Green Version]
- Koppe, L.; Pillon, N.J.; Vella, R.E.; Croze, M.L.; Pelletier, C.C.; Chambert, S.; Massy, Z.; Glorieux, G.; Vanholder, R.; Dugenet, Y.; et al. p-Cresyl sulfate promotes insulin resistance associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Anderson, E.M.; Thome, T.; Lu, G.; Salyers, Z.R.; Cort, T.A.; O’Malley, K.A.; Scali, S.T.; Ryan, T.E. Skeletal myopathy in CKD: A comparison of adenine-induced nephropathy and 5/6 nephrectomy models in mice. Am. J. Physiol. Renal Physiol. 2021, 321, F106–F119. [Google Scholar] [CrossRef]
- Mohanasundaram, S.; Fernando, E. Uremic Sarcopenia. Indian. J. Nephrol. 2022, 32, 399–405. [Google Scholar] [PubMed]
- Nishi, H.; Takemura, K.; Higashihara, T.; Inagi, R. Uremic Sarcopenia: Clinical Evidence and Basic Experimental Approach. Nutrients 2020, 12, 1814. [Google Scholar] [CrossRef]
- Fahal, I.H. Uraemic sarcopenia: Aetiology and implications. Nephrol. Dial. Transplant. 2014, 29, 1655–1665. [Google Scholar] [CrossRef] [Green Version]
- Carrero, J.J.; Chmielewski, M.; Axelsson, J.; Snaedal, S.; Heimburger, O.; Barany, P.; Suliman, M.E.; Lindholm, B.; Stenvinkel, P.; Qureshi, A.R. Muscle atrophy, inflammation and clinical outcome in incident and prevalent dialysis patients. Clin. Nutr. 2008, 27, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Martinson, M.; Ikizler, T.A.; Morrell, G.; Wei, G.; Almeida, N.; Marcus, R.L.; Filipowicz, R.; Greene, T.H.; Beddhu, S. Associations of body size and body composition with functional ability and quality of life in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- Hanatani, S.; Izumiya, Y.; Onoue, Y.; Tanaka, T.; Yamamoto, M.; Ishida, T.; Yamamura, S.; Kimura, Y.; Araki, S.; Arima, Y.; et al. Non-invasive testing for sarcopenia predicts future cardiovascular events in patients with chronic kidney disease. Int. J. Cardiol. 2018, 268, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Enoki, Y.; Maruyama, T. Sarcopenia in Chronic Kidney Disease: Factors, Mechanisms, and Therapeutic Interventions. Biol. Pharm. Bull. 2019, 42, 1437–1445. [Google Scholar] [CrossRef] [Green Version]
- Workeneh, B.T.; Mitch, W.E. Review of muscle wasting associated with chronic kidney disease. Am. J. Clin. Nutr. 2010, 91, 1128S–1132S. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Gao, X.L.; Hirschberg, R.; Vadgama, J.V.; Kopple, J.D. Impaired actions of insulin-like growth factor 1 on protein Synthesis and degradation in skeletal muscle of rats with chronic renal failure. Evidence for a postreceptor defect. J. Clin. Investig. 1996, 97, 1064–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, X.H.; Wang, H.; Du, J.; Mitch, W.E. Satellite cell dysfunction and impaired IGF-1 signaling cause CKD-induced muscle atrophy. J. Am. Soc. Nephrol. 2010, 21, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, L.A.; O’Sullivan, T.F.; Robinson, K.A.; Graham-Brown, M.P.M.; Major, R.W.; Ashford, R.U.; Smith, A.C.; Philp, A.; Watson, E.L. Primary skeletal muscle cells from chronic kidney disease patients retain hallmarks of cachexia in vitro. J. Cachexia Sarcopenia Muscle 2022, 13, 1238–1249. [Google Scholar] [CrossRef]
- Eguchi, Y.; Toyoguchi, T.; Inage, K.; Fujimoto, K.; Orita, S.; Suzuki, M.; Kanamoto, H.; Abe, K.; Norimoto, M.; Umimura, T.; et al. Advanced glycation end products are associated with sarcopenia in older women: Aging marker dynamics. J. Women Aging 2021, 33, 328–340. [Google Scholar] [CrossRef]
- Mori, H.; Kuroda, A.; Ishizu, M.; Ohishi, M.; Takashi, Y.; Otsuka, Y.; Taniguchi, S.; Tamaki, M.; Kurahashi, K.; Yoshida, S.; et al. Association of accumulated advanced glycation end-products with a high prevalence of sarcopenia and dynapenia in patients with type 2 diabetes. J. Diabetes Investig. 2019, 10, 1332–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Song, S.H. Sarcopenia in chronic kidney disease: From bench to bedside. Korean J. Intern. Med. 2023, 38, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Bogin, E.; Massry, S.G.; Harary, I. Effect of parathyroid hormone on rat heart cells. J. Clin. Investig. 1981, 67, 1215–1227. [Google Scholar] [CrossRef]
- Kimura, S.; Yoshioka, K. Parathyroid hormone and parathyroid hormone type-1 receptor accelerate myocyte differentiation. Sci. Rep. 2014, 4, 5066. [Google Scholar] [CrossRef] [Green Version]
- Baczynski, R.; Massry, S.G.; Magott, M.; el-Belbessi, S.; Kohan, R.; Brautbar, N. Effect of parathyroid hormone on energy metabolism of skeletal muscle. Kidney Int. 1985, 28, 722–727. [Google Scholar] [CrossRef] [Green Version]
- Rendina-Ruedy, E.; Rosen, C.J. Parathyroid hormone (PTH) regulation of metabolic homeostasis: An old dog teaches us new tricks. Mol. Metab. 2022, 60, 101480. [Google Scholar] [CrossRef]
- Shimonty, A.; Bonewald, L.F.; Huot, J.R. Metabolic Health and Disease: A Role of Osteokines? Calcif. Tissue Int. 2023, 113, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.C.; Liu, Y.M.; Liao, M.T.; Zheng, C.M.; Lu, C.L.; Liu, W.C.; Hung, K.C.; Lin, S.M.; Lu, K.C. Indoxyl sulfate mediates low handgrip strength and is predictive of high hospitalization rates in patients with end-stage renal disease. Front. Med. (Lausanne) 2023, 10, 1023383. [Google Scholar] [CrossRef] [PubMed]
- Cha, R.H.; Kang, S.H.; Han, M.Y.; An, W.S.; Kim, S.H.; Kim, J.C. Effects of AST-120 on muscle health and quality of life in chronic kidney disease patients: Results of RECOVERY study. J. Cachexia Sarcopenia Muscle 2022, 13, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.C.; Shyu, J.F.; Lim, P.S.; Fang, T.C.; Lu, C.L.; Zheng, C.M.; Hou, Y.C.; Wu, C.C.; Lin, Y.F.; Lu, K.C. Concentration and Duration of Indoxyl Sulfate Exposure Affects Osteoclastogenesis by Regulating NFATc1 via Aryl Hydrocarbon Receptor. Int. J. Mol. Sci. 2020, 21, 3486. [Google Scholar] [CrossRef]
- Liu, W.C.; Shyu, J.F.; Lin, Y.F.; Chiu, H.W.; Lim, P.S.; Lu, C.L.; Zheng, C.M.; Hou, Y.C.; Chen, P.H.; Lu, K.C. Resveratrol Rescue Indoxyl Sulfate-Induced Deterioration of Osteoblastogenesis via the Aryl Hydrocarbon Receptor /MAPK Pathway. Int. J. Mol. Sci. 2020, 21, 7483. [Google Scholar] [CrossRef]
- Pathak, J.L.; Bravenboer, N.; Klein-Nulend, J. The Osteocyte as the New Discovery of Therapeutic Options in Rare Bone Diseases. Front. Endocrinol. (Lausanne) 2020, 11, 405. [Google Scholar] [CrossRef]
- Tresguerres, F.G.F.; Torres, J.; Lopez-Quiles, J.; Hernandez, G.; Vega, J.A.; Tresguerres, I.F. The osteocyte: A multifunctional cell within the bone. Ann. Anat. 2020, 227, 151422. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell... and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, X.; Delgado-Calle, J.; Condon, K.W.; Maycas, M.; Zhang, H.; Carlesso, N.; Taketo, M.M.; Burr, D.B.; Plotkin, L.I.; Bellido, T. Osteocytes mediate the anabolic actions of canonical Wnt/beta-catenin signaling in bone. Proc. Natl. Acad. Sci. USA 2015, 112, E478–E486. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Bonewald, L.F. The role of the wnt/beta-catenin signaling pathway in formation and maintenance of bone and teeth. Int. J. Biochem. Cell Biol. 2016, 77 Pt A, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Hauschka, P.V.; Lian, J.B.; Cole, D.E.; Gundberg, C.M. Osteocalcin and matrix Gla protein: Vitamin K-dependent proteins in bone. Physiol. Rev. 1989, 69, 990–1047. [Google Scholar] [CrossRef] [PubMed]
- Delmas, P.D.; Eastell, R.; Garnero, P.; Seibel, M.J.; Stepan, J.; Committee of Scientific Advisors of the International Osteoporosis Foundation. The use of biochemical markers of bone turnover in osteoporosis. Committee of Scientific Advisors of the International Osteoporosis Foundation. Osteoporos. Int. 2000, 11 (Suppl. S6), S2–S17. [Google Scholar] [CrossRef]
- Ducy, P.; Desbois, C.; Boyce, B.; Pinero, G.; Story, B.; Dunstan, C.; Smith, E.; Bonadio, J.; Goldstein, S.; Gundberg, C.; et al. Increased bone formation in osteocalcin-deficient mice. Nature 1996, 382, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Gremminger, V.L.; Phillips, C.L. Impact of Intrinsic Muscle Weakness on Muscle-Bone Crosstalk in Osteogenesis Imperfecta. Int. J. Mol. Sci. 2021, 22, 4963. [Google Scholar] [CrossRef]
- Karsenty, G.; Mera, P. Molecular bases of the crosstalk between bone and muscle. Bone 2018, 115, 43–49. [Google Scholar] [CrossRef]
- Mera, P.; Laue, K.; Wei, J.; Berger, J.M.; Karsenty, G. Osteocalcin is necessary and sufficient to maintain muscle mass in older mice. Mol. Metab. 2016, 5, 1042–1047. [Google Scholar] [CrossRef]
- Chowdhury, S.; Schulz, L.; Palmisano, B.; Singh, P.; Berger, J.M.; Yadav, V.K.; Mera, P.; Ellingsgaard, H.; Hidalgo, J.; Bruning, J.; et al. Muscle-derived interleukin 6 increases exercise capacity by signaling in osteoblasts. J. Clin. Investig. 2020, 130, 2888–2902. [Google Scholar] [CrossRef] [Green Version]
- Lara-Castillo, N.; Johnson, M.L. Bone-Muscle Mutual Interactions. Curr. Osteoporos. Rep. 2020, 18, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Tsubakihara, Y.; Takabatake, Y.; Oka, K.; Shoji, T.; Togawa, M.; Okada, N.; Takahito, I.; Imai, E. Effects of the oral adsorbent AST-120 on tryptophan metabolism in uremic patients. Am. J. Kidney Dis. 2003, 41 (Suppl. S1), S38–S41. [Google Scholar] [CrossRef]
- Niwa, T.; Emoto, Y.; Maeda, K.; Uehara, Y.; Yamada, N.; Shibata, M. Oral sorbent suppresses accumulation of albumin-bound indoxyl sulphate in serum of haemodialysis patients. Nephrol. Dial. Transplant. 1991, 6, 105–109. [Google Scholar] [CrossRef]
- Amador, A.; Campbell, S.; Kazantzis, M.; Lan, G.; Burris, T.P.; Solt, L.A. Distinct roles for REV-ERBalpha and REV-ERBbeta in oxidative capacity and mitochondrial biogenesis in skeletal muscle. PLoS ONE 2018, 13, e0196787. [Google Scholar] [CrossRef] [Green Version]
- Geng, T.; Li, P.; Okutsu, M.; Yin, X.; Kwek, J.; Zhang, M.; Yan, Z. PGC-1alpha plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am. J. Physiol.-Cell Physiol. 2010, 298, C572–C579. [Google Scholar] [CrossRef] [Green Version]
- Enoki, Y.; Watanabe, H.; Arake, R.; Fujimura, R.; Ishiodori, K.; Imafuku, T.; Nishida, K.; Sugimoto, R.; Nagao, S.; Miyamura, S.; et al. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J. Cachexia Sarcopenia Muscle 2017, 8, 735–747. [Google Scholar] [CrossRef]
- Fremont, L. Biological effects of resveratrol. Life Sci. 2000, 66, 663–673. [Google Scholar] [CrossRef]
- Suzuki, T. Regulation of the intestinal barrier by nutrients: The role of tight junctions. Anim. Sci. J. 2020, 91, e13357. [Google Scholar] [CrossRef] [Green Version]
- Saito, H.; Yoshimura, M.; Saigo, C.; Komori, M.; Nomura, Y.; Yamamoto, Y.; Sagata, M.; Wakida, A.; Chuman, E.; Nishi, K.; et al. Hepatic sulfotransferase as a nephropreventing target by suppression of the uremic toxin indoxyl sulfate accumulation in ischemic acute kidney injury. Toxicol. Sci. 2014, 141, 206–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira-Pinto, B.; Costa, L.; Felgueira, E.; Fonseca, B.M.; Rebelo, I. Low Doses of Resveratrol Protect Human Granulosa Cells from Induced-Oxidative Stress. Antioxidants 2021, 10, 561. [Google Scholar] [CrossRef] [PubMed]
- Ragonese, F.; Monarca, L.; De Luca, A.; Mancinelli, L.; Mariani, M.; Corbucci, C.; Gerli, S.; Iannitti, R.G.; Leonardi, L.; Fioretti, B. Resveratrol depolarizes the membrane potential in human granulosa cells and promotes mitochondrial biogenesis. Fertil. Steril. 2021, 115, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.K.; Kim, B.G.; Lee, A.R.; In Choe, Y.; Khan, I.; Moon, K.M.; Jeon, R.H.; Byun, J.H.; Hwang, S.C.; Woo, D.K. Resveratrol can enhance osteogenic differentiation and mitochondrial biogenesis from human periosteum-derived mesenchymal stem cells. J. Orthop. Surg. Res. 2020, 15, 203. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.K.; Jaiswal, N.; Bruder, S.P.; Mbalaviele, G.; Marshak, D.R.; Pittenger, M.F. Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. J. Biol. Chem. 2000, 275, 9645–9652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipska, I.; Winiarska, A.; Knysak, M.; Stompor, T. Contribution of Gut Microbiota-Derived Uremic Toxins to the Cardiovascular System Mineralization. Toxins 2021, 13, 274. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, V.S.; Rao, M.; Menon, V.; Gordon, P.L.; Pilichowska, M.; Castaneda, F.; Castaneda-Sceppa, C. Resistance training increases muscle mitochondrial biogenesis in patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Kojima-Yuasa, A.; Tadano, H.; Mizuno, A.; Kon, A.; Norikura, T. Ursolic acid improves the indoxyl sulfate-induced impairment of mitochondrial biogenesis in C2C12 cells. Nutr. Res. Pract. 2022, 16, 147–160. [Google Scholar] [CrossRef]
- Czaya, B.; Heitman, K.; Campos, I.; Yanucil, C.; Kentrup, D.; Westbrook, D.; Gutierrez, O.; Babitt, J.L.; Jung, G.; Salusky, I.B.; et al. Hyperphosphatemia increases inflammation to exacerbate anemia and skeletal muscle wasting independently of FGF23-FGFR4 signaling. eLife 2022, 11, e74782. [Google Scholar] [CrossRef]
- Lee, S.M.; Jeong, E.G.; Jeong, Y.I.; Rha, S.H.; Kim, S.E.; An, W.S. Omega-3 fatty acid and menaquinone-7 combination are helpful for aortic calcification prevention, reducing osteoclast area of bone and Fox0 expression of muscle in uremic rats. Ren. Fail. 2022, 44, 1873–1885. [Google Scholar] [CrossRef]
- Sanchez-Tocino, M.L.; Mas-Fontao, S.; Gracia-Iguacel, C.; Pereira, M.; Gonzalez-Ibarguren, I.; Ortiz, A.; Arenas, M.D.; Parra, E.G. A Sarcopenia Index Derived from Malnutrition Parameters in Elderly Haemodialysis Patients. Nutrients 2023, 15, 1115. [Google Scholar] [CrossRef]
- Noce, A.; Marrone, G.; Ottaviani, E.; Guerriero, C.; Di Daniele, F.; Pietroboni Zaitseva, A.; Di Daniele, N. Uremic Sarcopenia and Its Possible Nutritional Approach. Nutrients 2021, 13, 147. [Google Scholar] [CrossRef]
- Noor, H.; Reid, J.; Slee, A. Resistance exercise and nutritional interventions for augmenting sarcopenia outcomes in chronic kidney disease: A narrative review. J. Cachexia Sarcopenia Muscle 2021, 12, 1621–1640. [Google Scholar] [CrossRef] [PubMed]
- Chatzipetrou, V.; Begin, M.J.; Hars, M.; Trombetti, A. Sarcopenia in Chronic Kidney Disease: A Scoping Review of Prevalence, Risk Factors, Association with Outcomes, and Treatment. Calcif. Tissue Int. 2022, 110, 1–31. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, K.-C.; Yao, W.-C.; Liu, Y.-L.; Yang, H.-J.; Liao, M.-T.; Chong, K.; Peng, C.-H.; Lu, K.-C. The Potential Influence of Uremic Toxins on the Homeostasis of Bones and Muscles in Chronic Kidney Disease. Biomedicines 2023, 11, 2076. https://doi.org/10.3390/biomedicines11072076
Hung K-C, Yao W-C, Liu Y-L, Yang H-J, Liao M-T, Chong K, Peng C-H, Lu K-C. The Potential Influence of Uremic Toxins on the Homeostasis of Bones and Muscles in Chronic Kidney Disease. Biomedicines. 2023; 11(7):2076. https://doi.org/10.3390/biomedicines11072076
Chicago/Turabian StyleHung, Kuo-Chin, Wei-Cheng Yao, Yi-Lien Liu, Hung-Jen Yang, Min-Tser Liao, Keong Chong, Ching-Hsiu Peng, and Kuo-Cheng Lu. 2023. "The Potential Influence of Uremic Toxins on the Homeostasis of Bones and Muscles in Chronic Kidney Disease" Biomedicines 11, no. 7: 2076. https://doi.org/10.3390/biomedicines11072076