
How Many Alzheimer–Perusini’s Atypical Forms Do We Still Have to Discover?
 , ,
, ,  and
and
Abstract
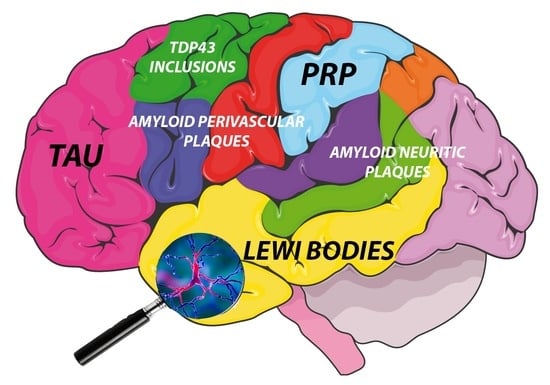
:
1. Introduction
2. A Sea of Atypical Forms Classification: Let Us Dive in!
2.1. Classification Based on Regional NFT Density
2.2. Classifications Based on Neuroimaging Studies
2.3. Other Particular AD Atypical Forms
2.4. Classifications Based on Clinical Manifestations
2.5. PCA
2.5.1. PCA Main and More Infrequent Subtypes
2.5.2. Symptomatology and Clinical Features
- Identify whether it is a neurodegenerative pathology.
- Determine whether it is a posteriorly based cortical entity.
- Discern whether it is pure PCA or PCA in conjunction with Alzheimer’s disease (AD), Lewy body dementia (LBD), Corticobasal Degeneration (CBD), or prion lesion.
- Evaluate whether any relevant biomarkers such as cerebrospinal fluid (CSF) Aβ 1–42, Tau, p-Tau for AD, and 14-3-3 protein for prion disease are present.
2.6. LvPPA
2.6.1. Classifications of LvPPA and Their Symptomatology
2.6.2. Specific Cognitive Tests for Various PPA Forms
2.6.3. Neuroimaging Findings
2.7. Behavioral Variant/Dysexecutive Variant Alzheimer’s Disease
2.7.1. The Definition History Helps to Clarify Clinical and Neuropathological Features
2.7.2. Details about Population Distribution, Clinical Signs and Symptomatology
2.7.3. Neuroimaging Findings Permit a More Specific Clusterizations of Patients
2.8. AD with Corticobasal Syndrome (CBS)
2.8.1. Definition and Milestone Studies
2.8.2. Clinical Features
2.8.3. Neuroimaging Findings
3. A Sea of Atypical Form Classifications: Let Us Learn to Swim in It!
4. Transforming the Sea into a Bathtub: A Genetic–Molecular Attempt
5. Transforming the Sea into a Bathtub: An Immune Attempt
- 5.
- And If It Was Just a Glass, Would It Be Half Full or Half Empty? Closing the Circle
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pomponi, M.; Marta, M. “On the suggestion of Dr. Alzheimer I examined the following four cases.” Dedicated to Gaetano Perusini. Aging Clin. Exp. Res. 1993, 5, 135–139. [Google Scholar] [CrossRef]
- Lucci, B. The contribution of Gaetano Perusini to the definition of Alzheimer’s disease. Ital. J. Neurol. Sci. 1998, 19, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Italy Ministry of Health. Demenze-Dati Epidemiologici. Available online: https://www.salute.gov.it/portale/demenze/dettaglioContenutiDemenze.jsp?lingua=italiano&id=2402&area=demenze&menu=vuoto (accessed on 15 June 2023).
- Kandel, E. Principi di Neuroscienze, 4th ed.; Ambrosiana, C., Ed.; Casa Editrice Ambrosiana: Rozzano, Rozzano, 2015. (In Italian) [Google Scholar]
- Jack, C.R., Jr.; Therneau, T.M.; Weigand, S.D.; Wiste, H.J.; Knopman, D.S.; Vemuri, P.; Lowe, V.J.; Mielke, M.M.; Roberts, R.O.; Machulda, M.M.; et al. Prevalence of Biologically vs. Clinically Defined Alzheimer Spectrum Entities Using the National Institute on Aging-Alzheimer’s Association Research Framework. JAMA Neurol. 2019, 76, 1174–1183. [Google Scholar] [CrossRef] [Green Version]
- Italian Ministry of Health Commissione Unica del Farmaco. I Farmaci Anticolinesterasici per Il Trattamento Sintomatico Della Demenza di Alzheimer; Italian Ministry of Health: Rome, Italy, 1999; pp. 3–18.
- Koch, G.; Motta, C.; Bonni, S.; Pellicciari, M.C.; Picazio, S.; Casula, E.P.; Maiella, M.; Di Lorenzo, F.; Ponzo, V.; Ferrari, C.; et al. Effect of Rotigotine vs. Placebo on Cognitive Functions among Patients with Mild to Moderate Alzheimer Disease: A Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e2010372. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettcher, B.M.; Tansey, M.G.; Dorothee, G.; Heneka, M.T. Peripheral and central immune system crosstalk in Alzheimer disease—A research prospectus. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Aman, Y.; Ng, C.T.; Chau, W.H.; Zhang, Z.; Yue, M.; Bohm, C.; Jia, Y.; Li, S.; et al. Amyloid-beta toxicity modulates tau phosphorylation through the PAX6 signalling pathway. Brain 2021, 144, 2759–2770. [Google Scholar] [CrossRef]
- Donato, L.; Alibrandi, S.; Scimone, C.; Rinaldi, C.; Dascola, A.; Calamuneri, A.; D’Angelo, R.; Sidoti, A. The impact of modifier genes on cone-rod dystrophy heterogeneity: An explorative familial pilot study and a hypothesis on neurotransmission impairment. PLoS ONE 2022, 17, e0278857. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e8. [Google Scholar] [CrossRef] [Green Version]
- Ellison, E.M.; Abner, E.L.; Lovell, M.A. Multiregional analysis of global 5-methylcytosine and 5-hydroxymethylcytosine throughout the progression of Alzheimer’s disease. J. Neurochem. 2017, 140, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Fotuhi, S.N.; Khalaj-Kondori, M.; Hoseinpour Feizi, M.A.; Talebi, M. Long Non-coding RNA BACE1-AS May Serve as an Alzheimer’s Disease Blood-Based Biomarker. J. Mol. Neurosci. 2019, 69, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Khodayi, M.; Khalaj-Kondori, M.; Hoseinpour Feizi, M.A.; Jabarpour Bonyadi, M.; Talebi, M. Plasma lncRNA profiling identified BC200 and NEAT1 lncRNAs as potential blood-based biomarkers for late-onset Alzheimer’s disease. EXCLI J. 2022, 21, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Cummings, J.; Jack, C.R., Jr.; Morris, J.C.; Sperling, R.; Frolich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimers Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
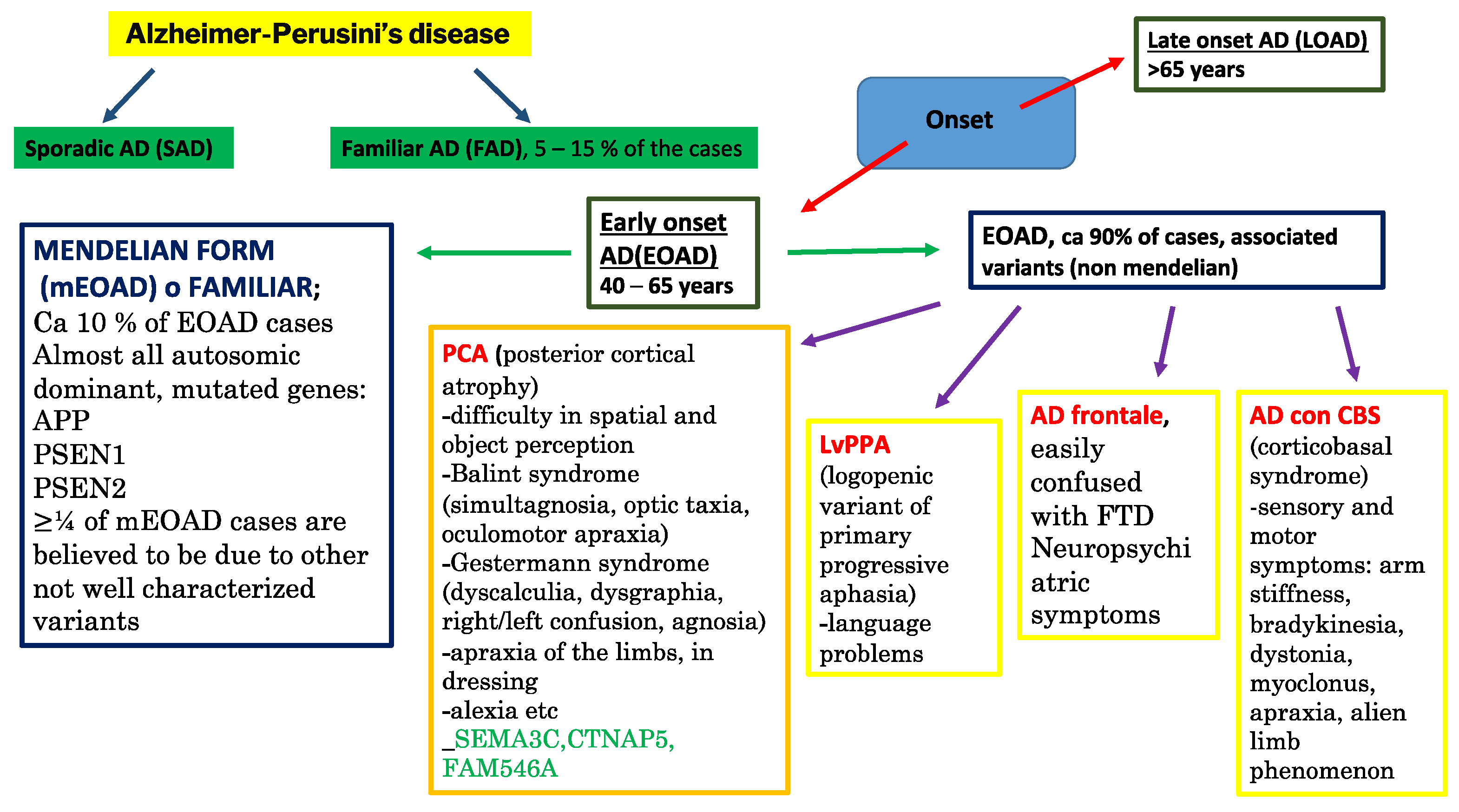
- Sirkis, D.W.; Bonham, L.W.; Johnson, T.P.; La Joie, R.; Yokoyama, J.S. Dissecting the clinical heterogeneity of early-onset Alzheimer’s disease. Mol. Psychiatry 2022, 27, 2674–2688. [Google Scholar] [CrossRef]
- Polsinelli, A.J.; Apostolova, L.G. Atypical Alzheimer Disease Variants. Contin. Lifelong Learn. Neurol. 2022, 28, 676–701. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimers Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef]
- Walsh, S.; Merrick, R.; Milne, R.; Brayne, C. Aducanumab for Alzheimer’s disease? BMJ 2021, 374, n1682. [Google Scholar] [CrossRef]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443. [Google Scholar] [CrossRef]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients with Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Humpel, C. Intranasal neprilysin rapidly eliminates amyloid-beta plaques, but causes plaque compensations: The explanation why the amyloid-beta cascade may fail? Neural Regen. Res. 2022, 17, 1881–1884. [Google Scholar] [CrossRef]
- Hoy, S.M. Lecanemab: First Approval. Drugs 2023, 83, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Recent update on the heterogeneity of the Alzheimer’s disease spectrum. J. Neural Transm. 2022, 129, 1–24. [Google Scholar] [CrossRef]
- McFarland, K.N.; Chakrabarty, P. Microglia in Alzheimer’s Disease: A Key Player in the Transition Between Homeostasis and Pathogenesis. Neurotherapeutics 2022, 19, 186–208. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Frisoni, G.B.; Teipel, S.J.; Grinberg, L.T.; Amaro, E., Jr.; Heinsen, H.; Thompson, P.M.; Hampel, H. Staging Alzheimer’s disease progression with multimodality neuroimaging. Prog. Neurobiol. 2011, 95, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, N.; Ossenkoppele, R.; Smith, R.; Strandberg, O.; Ohlsson, T.; Jogi, J.; Palmqvist, S.; Stomrud, E.; Hansson, O. Greater tau load and reduced cortical thickness in APOE epsilon4-negative Alzheimer’s disease: A cohort study. Alzheimers Res. Ther. 2018, 10, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A. Neuropathological subtypes of Alzheimer’s disease. Acta Neuropathol. 2012, 123, 153–154. [Google Scholar] [CrossRef]
- Sintini, I.; Graff-Radford, J.; Senjem, M.L.; Schwarz, C.G.; Machulda, M.M.; Martin, P.R.; Jones, D.T.; Boeve, B.F.; Knopman, D.S.; Kantarci, K.; et al. Longitudinal neuroimaging biomarkers differ across Alzheimer’s disease phenotypes. Brain 2020, 143, 2281–2294. [Google Scholar] [CrossRef] [PubMed]
- Dong, A.; Toledo, J.B.; Honnorat, N.; Doshi, J.; Varol, E.; Sotiras, A.; Wolk, D.; Trojanowski, J.Q.; Davatzikos, C.; Alzheimer’s Disease Neuroimaging Initiative. Heterogeneity of neuroanatomical patterns in prodromal Alzheimer’s disease: Links to cognition, progression and biomarkers. Brain 2017, 140, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Golde, T.E. Harnessing Immunoproteostasis to Treat Neurodegenerative Disorders. Neuron 2019, 101, 1003–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppedal, K.; Ferreira, D.; Cavallin, L.; Lemstra, A.W.; Ten Kate, M.; Padovani, A.; Rektorova, I.; Bonanni, L.; Wahlund, L.O.; Engedal, K.; et al. A signature pattern of cortical atrophy in dementia with Lewy bodies: A study on 333 patients from the European DLB consortium. Alzheimers Dement. 2019, 15, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulakis, K.; Pereira, J.B.; Mecocci, P.; Vellas, B.; Tsolaki, M.; Kloszewska, I.; Soininen, H.; Lovestone, S.; Simmons, A.; Wahlund, L.O.; et al. Heterogeneous patterns of brain atrophy in Alzheimer’s disease. Neurobiol. Aging 2018, 65, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Kim, C.M.; Jeon, S.; Lee, J.M.; Hong, Y.J.; Roh, J.H.; Lee, J.H.; Koh, J.Y.; Na, D.L.; Alzheimer’s Disease Neuroimaging Initiative. Prediction of Alzheimer’s disease pathophysiology based on cortical thickness patterns. Alzheimers Dement. 2016, 2, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Noh, Y.; Jeon, S.; Lee, J.M.; Seo, S.W.; Kim, G.H.; Cho, H.; Ye, B.S.; Yoon, C.W.; Kim, H.J.; Chin, J.; et al. Anatomical heterogeneity of Alzheimer disease: Based on cortical thickness on MRIs. Neurology 2014, 83, 1936–1944. [Google Scholar] [CrossRef] [Green Version]
- Planche, V.; Coupe, P.; Helmer, C.; Le Goff, M.; Amieva, H.; Tison, F.; Dartigues, J.F.; Catheline, G. Evolution of brain atrophy subtypes during aging predicts long-term cognitive decline and future Alzheimer’s clinical syndrome. Neurobiol. Aging 2019, 79, 22–29. [Google Scholar] [CrossRef]
- Zhang, B.; Lin, L.; Wu, S.; Al-Masqari, Z. Multiple Subtypes of Alzheimer’s Disease Base on Brain Atrophy Pattern. Brain Sci. 2021, 11, 278. [Google Scholar] [CrossRef]
- Ten Kate, M.; Dicks, E.; Visser, P.J.; van der Flier, W.M.; Teunissen, C.E.; Barkhof, F.; Scheltens, P.; Tijms, B.M.; Alzheimer’s Disease Neuroimaging Initiative. Atrophy subtypes in prodromal Alzheimer’s disease are associated with cognitive decline. Brain 2018, 141, 3443–3456. [Google Scholar] [CrossRef]
- Rauchmann, B.S.; Ersoezlue, E.; Stoecklein, S.; Keeser, D.; Brosseron, F.; Buerger, K.; Dechent, P.; Dobisch, L.; Ertl-Wagner, B.; Fliessbach, K.; et al. Resting-State Network Alterations Differ between Alzheimer’s Disease Atrophy Subtypes. Cereb. Cortex 2021, 31, 4901–4915. [Google Scholar] [CrossRef]
- Levine, D.N.; Lee, J.M.; Fisher, C.M. The visual variant of Alzheimer’s disease: A clinicopathologic case study. Neurology 1993, 43, 305–313. [Google Scholar] [CrossRef]
- Jeon, S.; Kang, J.M.; Seo, S.; Jeong, H.J.; Funck, T.; Lee, S.Y.; Park, K.H.; Lee, Y.B.; Yeon, B.K.; Ido, T.; et al. Topographical Heterogeneity of Alzheimer’s Disease Based on MR Imaging, Tau PET, and Amyloid PET. Front. Aging Neurosci. 2019, 11, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiraboschi, P.; Sabbagh, M.N.; Hansen, L.A.; Salmon, D.P.; Merdes, A.; Gamst, A.; Masliah, E.; Alford, M.; Thal, L.J.; Corey-Bloom, J. Alzheimer disease without neocortical neurofibrillary tangles: “A second look”. Neurology 2004, 62, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Zangrossi, A.; Montemurro, S.; Altoe, G.; Mondini, S. Heterogeneity and Factorial Structure in Alzheimer’s Disease: A Cognitive Perspective. J. Alzheimers Dis. 2021, 83, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Pillai, J.A.; Appleby, B.S.; Safar, J.; Leverenz, J.B. Rapidly Progressive Alzheimer’s Disease in Two Distinct Autopsy Cohorts. J. Alzheimers Dis. 2018, 64, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Wolff, M.; Weitz, M.; Bartlau, T.; Korth, C.; Zerr, I. Rapidly progressive Alzheimer disease. Arch. Neurol. 2011, 68, 1124–1130. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Haik, S.; Satoh, K.; Rabano, A.; Martinez-Martin, P.; Roeber, S.; Brandel, J.P.; Calero-Lara, M.; de Pedro-Cuesta, J.; Laplanche, J.L.; et al. Rapidly progressive Alzheimer’s disease: A multicenter update. J. Alzheimers Dis. 2012, 30, 751–756. [Google Scholar] [CrossRef]
- Tosto, G.; Gasparini, M.; Brickman, A.M.; Letteri, F.; Renie, R.; Piscopo, P.; Talarico, G.; Canevelli, M.; Confaloni, A.; Bruno, G. Neuropsychological predictors of rapidly progressive Alzheimer’s disease. Acta Neurol. Scand. 2015, 132, 417–422. [Google Scholar] [CrossRef]
- Drummond, E.; Nayak, S.; Faustin, A.; Pires, G.; Hickman, R.A.; Askenazi, M.; Cohen, M.; Haldiman, T.; Kim, C.; Han, X.; et al. Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer’s disease. Acta Neuropathol. 2017, 133, 933–954. [Google Scholar] [CrossRef]
- Zafar, S.; Shafiq, M.; Younas, N.; Schmitz, M.; Ferrer, I.; Zerr, I. Prion Protein Interactome: Identifying Novel Targets in Slowly and Rapidly Progressive Forms of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 59, 265–275. [Google Scholar] [CrossRef]
- Shafiq, M.; Zafar, S.; Younas, N.; Noor, A.; Puig, B.; Altmeppen, H.C.; Schmitz, M.; Matschke, J.; Ferrer, I.; Glatzel, M.; et al. Prion protein oligomers cause neuronal cytoskeletal damage in rapidly progressive Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 11. [Google Scholar] [CrossRef]
- Younas, N.; Zafar, S.; Shafiq, M.; Noor, A.; Siegert, A.; Arora, A.S.; Galkin, A.; Zafar, A.; Schmitz, M.; Stadelmann, C.; et al. SFPQ and Tau: Critical factors contributing to rapid progression of Alzheimer’s disease. Acta Neuropathol. 2020, 140, 317–339. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Attems, J. Neuropathological evaluation of mixed dementia. J. Neurol. Sci. 2007, 257, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Weigand, A.J.; Bangen, K.J.; Thomas, K.R.; Delano-Wood, L.; Gilbert, P.E.; Brickman, A.M.; Bondi, M.W.; Alzheimer’s Disease Neuroimaging Initiative. Is tau in the absence of amyloid on the Alzheimer’s continuum? A study of discordant PET positivity. Brain Commun. 2020, 2, fcz046. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Murray, M.E.; Tosakulwong, N.; Whitwell, J.L.; Knopman, D.S.; Machulda, M.M.; Weigand, S.D.; Boeve, B.F.; Kantarci, K.; Petrucelli, L.; et al. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: A clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol. 2017, 133, 705–715. [Google Scholar] [CrossRef]
- Ruiz-Gabarre, D.; Carnero-Espejo, A.; Avila, J.; Garcia-Escudero, V. What’s in a Gene? The Outstanding Diversity of MAPT. Cells 2022, 11, 840. [Google Scholar] [CrossRef]
- Bancher, C.; Egensperger, R.; Kosel, S.; Jellinger, K.; Graeber, M.B. Low prevalence of apolipoprotein E epsilon 4 allele in the neurofibrillary tangle predominant form of senile dementia. Acta Neuropathol. 1997, 94, 403–409. [Google Scholar] [CrossRef]
- Jellinger, K.A. Different patterns of hippocampal tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 2018, 136, 811–813. [Google Scholar] [CrossRef]
- Walker, J.M.; Fudym, Y.; Farrell, K.; Iida, M.A.; Bieniek, K.F.; Seshadri, S.; White, C.L.; Crary, J.F.; Richardson, T.E. Asymmetry of Hippocampal Tau Pathology in Primary Age-Related Tauopathy and Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2021, 80, 436–445. [Google Scholar] [CrossRef]
- Zheng, C.; Xu, R. Molecular subtyping of Alzheimer’s disease with consensus non-negative matrix factorization. PLoS ONE 2021, 16, e0250278. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, B.; Wang, X.; Lu, H.; Shao, F.; Rozemuller, A.J.M.; Liang, H.; Liu, C.; Chen, J.; Huang, M.; et al. Phosphorylated TDP-43 Staging of Primary Age-Related Tauopathy. Neurosci. Bull. 2019, 35, 183–192. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Del Tredici, K.; Thomas, T.L.; Braak, H.; Diamond, M.I. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 2018, 136, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groot, C.; Dore, V.; Robertson, J.; Burnham, S.C.; Savage, G.; Ossenkoppele, R.; Rowe, C.C.; Villemagne, V.L. Mesial temporal tau is related to worse cognitive performance and greater neocortical tau load in amyloid-beta-negative cognitively normal individuals. Neurobiol. Aging 2021, 97, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Besser, L.M.; Teylan, M.A.; Nelson, P.T. Limbic Predominant Age-Related TDP-43 Encephalopathy (LATE): Clinical and Neuropathological Associations. J. Neuropathol. Exp. Neurol. 2020, 79, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [Green Version]
- Tome, S.O.; Vandenberghe, R.; Ospitalieri, S.; Van Schoor, E.; Tousseyn, T.; Otto, M.; von Arnim, C.A.F.; Thal, D.R. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: Relationship with clinical phenotypes. Acta Neuropathol. Commun. 2020, 8, 61. [Google Scholar] [CrossRef]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.S.; Yu, L.; White, C.C.; Chibnik, L.B.; Chhatwal, J.P.; Sperling, R.A.; Bennett, D.A.; Schneider, J.A.; De Jager, P.L. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE epsilon4 haplotype status: A community-based cohort study. Lancet Neurol. 2018, 17, 773–781. [Google Scholar] [CrossRef]
- Bayram, E.; Shan, G.; Cummings, J.L. Associations between Comorbid TDP-43, Lewy Body Pathology, and Neuropsychiatric Symptoms in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 69, 953–961. [Google Scholar] [CrossRef]
- Agrawal, S.; Yu, L.; Nag, S.; Arfanakis, K.; Barnes, L.L.; Bennett, D.A.; Schneider, J.A. The association of Lewy bodies with limbic-predominant age-related TDP-43 encephalopathy neuropathologic changes and their role in cognition and Alzheimer’s dementia in older persons. Acta Neuropathol. Commun. 2021, 9, 156. [Google Scholar] [CrossRef]
- Teylan, M.; Besser, L.M.; Crary, J.F.; Mock, C.; Gauthreaux, K.; Thomas, N.M.; Chen, Y.C.; Kukull, W.A. Clinical diagnoses among individuals with primary age-related tauopathy versus Alzheimer’s neuropathology. Lab. Investig. 2019, 99, 1049–1055. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerstein, O.; Parand, L.; Liang, L.J.; Isaac, A.; Mendez, M.F. Benson’s Disease or Posterior Cortical Atrophy, Revisited. J. Alzheimers Dis. 2021, 82, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F.; Ghajarania, M.; Perryman, K.M. Posterior cortical atrophy: Clinical characteristics and differences compared to Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2002, 14, 33–40. [Google Scholar] [CrossRef] [PubMed]
- McMonagle, P.; Deering, F.; Berliner, Y.; Kertesz, A. The cognitive profile of posterior cortical atrophy. Neurology 2006, 66, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Tang-Wai, D.F.; Graff-Radford, N.R.; Boeve, B.F.; Dickson, D.W.; Parisi, J.E.; Crook, R.; Caselli, R.J.; Knopman, D.S.; Petersen, R.C. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004, 63, 1168–1174. [Google Scholar] [CrossRef]
- Beh, S.C.; Muthusamy, B.; Calabresi, P.; Hart, J.; Zee, D.; Patel, V.; Frohman, E. Hiding in plain sight: A closer look at posterior cortical atrophy. Pract. Neurol. 2015, 15, 5–13. [Google Scholar] [CrossRef]
- Altabakhi, I.W.; Liang, J.W. Gerstmann Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- Shir, D.; Graff-Radford, J.; Machulda, M.M.; Pham, N.T.T.; Jack, C.R., Jr.; Lowe, V.J.; Whitwell, J.L.; Josephs, K.A. Posterior cortical atrophy: Primary occipital variant. Eur. J. Neurol. 2022, 29, 2138–2143. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef]
- Crutch, S.J.; Schott, J.M.; Rabinovici, G.D.; Murray, M.; Snowden, J.S.; van der Flier, W.M.; Dickerson, B.C.; Vandenberghe, R.; Ahmed, S.; Bak, T.H.; et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017, 13, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Rajaram Manoharan, S.V.R.; Munakomi, S. Posterior Cortical Atrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Maia da Silva, M.N.; Millington, R.S.; Bridge, H.; James-Galton, M.; Plant, G.T. Visual Dysfunction in Posterior Cortical Atrophy. Front. Neurol. 2017, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.; Higginbotham, L.; Bagchi, P.; Watson, C.M.; Zhang, T.; Levey, A.I.; Rangaraju, S.; Seyfried, N.T. Systems-based proteomics to resolve the biology of Alzheimer’s disease beyond amyloid and tau. Neuropsychopharmacology 2021, 46, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Pham, N.T.T.; Graff-Radford, J.; Machulda, M.M.; Lowe, V.J.; Whitwell, J.L. Medial Temporal Atrophy in Posterior Cortical Atrophy and Its Relationship to the Cingulate Island Sign. J. Alzheimers Dis. 2022, 86, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Arani, A.; Graff-Radford, J.; Senjem, M.L.; Martin, P.R.; Machulda, M.M.; Schwarz, C.G.; Shu, Y.; Cogswell, P.M.; Knopman, D.S.; et al. Distinct brain iron profiles associated with logopenic progressive aphasia and posterior cortical atrophy. Neuroimage Clin. 2022, 36, 103161. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Brioschi, S. Neuroinflammation and neurodegeneration in human brain at single-cell resolution. Nat. Rev. Immunol. 2020, 20, 81–82. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Mesulam, M.M.; Coventry, C.A.; Bigio, E.H.; Sridhar, J.; Gill, N.; Fought, A.J.; Zhang, H.; Thompson, C.K.; Geula, C.; Gefen, T.; et al. Neuropathological fingerprints of survival, atrophy and language in primary progressive aphasia. Brain 2022, 145, 2133–2148. [Google Scholar] [CrossRef]
- Matarin, M.; Salih, D.A.; Yasvoina, M.; Cummings, D.M.; Guelfi, S.; Liu, W.; Nahaboo Solim, M.A.; Moens, T.G.; Paublete, R.M.; Ali, S.S.; et al. A genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep. 2015, 10, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Matias-Guiu, J.A.; Grasso, S.M. Primary progressive aphasia: In search of brief cognitive assessments. Brain Commun. 2022, 4, fcac227. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Peterson, K.A.; Ingram, R.U.; Storey, I.; Cappa, S.F.; Catricala, E.; Halai, A.; Patterson, K.E.; Lambon Ralph, M.A.; Rowe, J.B.; et al. A ‘Mini Linguistic State Examination’ to classify primary progressive aphasia. Brain Commun. 2022, 4, fcab299. [Google Scholar] [CrossRef] [PubMed]
- Foxe, D.; Hu, A.; Cheung, S.C.; Ahmed, R.M.; Cordato, N.J.; Devenney, E.; Hwang, Y.T.; Halliday, G.M.; Mueller, N.; Leyton, C.E.; et al. Utility of the Addenbrooke’s Cognitive Examination III online calculator to differentiate the primary progressive aphasia variants. Brain Commun. 2022, 4, fcac161. [Google Scholar] [CrossRef] [PubMed]
- Sellami, L.; Meilleur-Durand, S.; Chouinard, A.M.; Bergeron, D.; Verret, L.; Poulin, S.; Jean, L.; Fortin, M.P.; Nadeau, Y.; Molin, P.; et al. The Depistage Cognitif de Quebec: A New Clinician’s Tool for Early Recognition of Atypical Dementia. Dement. Geriatr. Cogn. Disord. 2018, 46, 310–321. [Google Scholar] [CrossRef]
- Rothman, S.M.; Tanis, K.Q.; Gandhi, P.; Malkov, V.; Marcus, J.; Pearson, M.; Stevens, R.; Gilliland, J.; Ware, C.; Mahadomrongkul, V.; et al. Human Alzheimer’s disease gene expression signatures and immune profile in APP mouse models: A discrete transcriptomic view of Abeta plaque pathology. J. Neuroinflamm. 2018, 15, 256. [Google Scholar] [CrossRef] [Green Version]
- Martersteck, A.; Ayala, I.; Ohm, D.T.; Spencer, C.; Coventry, C.; Weintraub, S.; Bigio, E.H.; Mesulam, M.; Geula, C.; Rogalski, E. Focal amyloid and asymmetric tau in an imaging-to-autopsy case of clinical primary progressive aphasia with Alzheimer disease neuropathology. Acta Neuropathol. Commun. 2022, 10, 111. [Google Scholar] [CrossRef]
- Nicholas, M.; Obler, L.; Albert, M.; Goodglass, H. Lexical retrieval in healthy aging. Cortex 1985, 21, 595–606. [Google Scholar] [CrossRef]
- Cho, S.; Quilico Cousins, K.A.; Shellikeri, S.; Ash, S.; Irwin, D.J.; Liberman, M.Y.; Grossman, M.; Nevler, N. Lexical and Acoustic Speech Features Relating to Alzheimer Disease Pathology. Neurology 2022, 99, e313–e322. [Google Scholar] [CrossRef]
- Da Cunha, E.; Plonka, A.; Arslan, S.; Mouton, A.; Meyer, T.; Robert, P.; Meunier, F.; Manera, V.; Gros, A. Logogenic Primary Progressive Aphasia or Alzheimer Disease: Contribution of Acoustic Markers in Early Differential Diagnosis. Life 2022, 12, 933. [Google Scholar] [CrossRef]
- Whitwell, J.; Martin, P.R.; Graff-Radford, J.; Machulda, M.; Sintini, I.; Buciuc, M.; Senjem, M.L.; Schwarz, C.G.; Botha, H.; Carrasquillo, M.M.; et al. Investigating Heterogeneity and Neuroanatomic Correlates of Longitudinal Clinical Decline in Atypical Alzheimer Disease. Neurology 2022, 98, e2436–e2445. [Google Scholar] [CrossRef]
- Johnson, J.K.; Head, E.; Kim, R.; Starr, A.; Cotman, C.W. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol. 1999, 56, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back-Madruga, C.; Boone, K.B.; Briere, J.; Cummings, J.; McPherson, S.; Fairbanks, L.; Thompson, E. Functional ability in executive variant Alzheimer’s disease and typical Alzheimer’s disease. Clin. Neuropsychol. 2002, 16, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Binetti, G.; Magni, E.; Padovani, A.; Cappa, S.F.; Bianchetti, A.; Trabucchi, M. Executive dysfunction in early Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1996, 60, 91–93. [Google Scholar] [CrossRef] [Green Version]
- Dickerson, B.C.; Wolk, D.A.; Alzheimer’s Disease Neuroimaging Initiative. Dysexecutive versus amnesic phenotypes of very mild Alzheimer’s disease are associated with distinct clinical, genetic and cortical thinning characteristics. J. Neurol. Neurosurg. Psychiatry 2011, 82, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mez, J.; Cosentino, S.; Brickman, A.M.; Huey, E.D.; Manly, J.J.; Mayeux, R. Dysexecutive versus amnestic Alzheimer disease subgroups: Analysis of demographic, genetic, and vascular factors. Alzheimer Dis. Assoc. Disord. 2013, 27, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Snowden, J.S.; Stopford, C.L.; Julien, C.L.; Thompson, J.C.; Davidson, Y.; Gibbons, L.; Pritchard, A.; Lendon, C.L.; Richardson, A.M.; Varma, A.; et al. Cognitive phenotypes in Alzheimer’s disease and genetic risk. Cortex 2007, 43, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Wolk, D.A.; Dickerson, B.C.; Alzheimer’s Disease Neuroimaging Initiative. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 10256–10261. [Google Scholar] [CrossRef]
- Balasa, M.; Gelpi, E.; Antonell, A.; Rey, M.J.; Sanchez-Valle, R.; Molinuevo, J.L.; Llado, A.; Neurological Tissue Bank/University of Barcelona/Hospital Clinic. Clinical features and APOE genotype of pathologically proven early-onset Alzheimer disease. Neurology 2011, 76, 1720–1725. [Google Scholar] [CrossRef]
- Blennerhassett, R.; Lillo, P.; Halliday, G.M.; Hodges, J.R.; Kril, J.J. Distribution of pathology in frontal variant Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 63–70. [Google Scholar] [CrossRef]
- Forman, M.S.; Farmer, J.; Johnson, J.K.; Clark, C.M.; Arnold, S.E.; Coslett, H.B.; Chatterjee, A.; Hurtig, H.I.; Karlawish, J.H.; Rosen, H.J.; et al. Frontotemporal dementia: Clinicopathological correlations. Ann. Neurol. 2006, 59, 952–962. [Google Scholar] [CrossRef] [Green Version]
- Mendez, M.F.; Joshi, A.; Tassniyom, K.; Teng, E.; Shapira, J.S. Clinicopathologic differences among patients with behavioral variant frontotemporal dementia. Neurology 2013, 80, 561–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larner, A.J. “Frontal variant Alzheimer’s disease”: A reappraisal. Clin. Neurol. Neurosurg. 2006, 108, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Woodward, M.; Jacova, C.; Black, S.E.; Kertesz, A.; Mackenzie, I.R.; Feldman, H.; ACCORD Investigator Group. Differentiating the frontal variant of Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2010, 25, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Habek, M.; Hajnsek, S.; Zarkovic, K.; Chudy, D.; Mubrin, Z. Frontal variant of Alzheimer’s disease: Clinico-CSF-pathological correlation. Can. J. Neurol. Sci. 2010, 37, 118–120. [Google Scholar] [CrossRef] [Green Version]
- Herrero-San Martin, A.; Villarejo-Galende, A.; Rabano-Gutierrez, A.; Guerrero-Marquez, C.; Porta-Etessam, J.; Bermejo-Pareja, F. Frontal variant of Alzheimer’s disease. Two pathologically confirmed cases and a literature review. Rev. Neurol. 2013, 57, 542–548. [Google Scholar]
- Taylor, K.I.; Probst, A.; Miserez, A.R.; Monsch, A.U.; Tolnay, M. Clinical course of neuropathologically confirmed frontal-variant Alzheimer’s disease. Nat. Clin. Pract. Neurol. 2008, 4, 226–232. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Pijnenburg, Y.A.; Perry, D.C.; Cohn-Sheehy, B.I.; Scheltens, N.M.; Vogel, J.W.; Kramer, J.H.; van der Vlies, A.E.; La Joie, R.; Rosen, H.J.; et al. The behavioural/dysexecutive variant of Alzheimer’s disease: Clinical, neuroimaging and pathological features. Brain 2015, 138, 2732–2749. [Google Scholar] [CrossRef] [Green Version]
- Bruen, P.D.; McGeown, W.J.; Shanks, M.F.; Venneri, A. Neuroanatomical correlates of neuropsychiatric symptoms in Alzheimer’s disease. Brain 2008, 131, 2455–2463. [Google Scholar] [CrossRef] [Green Version]
- Damasio, H.; Grabowski, T.; Frank, R.; Galaburda, A.M.; Damasio, A.R. The return of Phineas Gage: Clues about the brain from the skull of a famous patient. Science 1994, 264, 1102–1105. [Google Scholar] [CrossRef]
- Neufang, S.; Akhrif, A.; Riedl, V.; Forstl, H.; Kurz, A.; Zimmer, C.; Sorg, C.; Wohlschlager, A.M. Disconnection of frontal and parietal areas contributes to impaired attention in very early Alzheimer’s disease. J. Alzheimers Dis. 2011, 25, 309–321. [Google Scholar] [CrossRef]
- Pa, J.; Boxer, A.; Chao, L.L.; Gazzaley, A.; Freeman, K.; Kramer, J.; Miller, B.L.; Weiner, M.W.; Neuhaus, J.; Johnson, J.K. Clinical-neuroimaging characteristics of dysexecutive mild cognitive impairment. Ann. Neurol. 2009, 65, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, B.R.; Eberling, J.L.; Mungas, D.; Weiner, M.; Kramer, J.H.; Jagust, W.J. Effects of white matter lesions and lacunes on cortical function. Arch. Neurol. 2004, 61, 1545–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kang, S.J.; Kim, C.; Kim, G.H.; Jeon, S.; Lee, J.M.; Oh, S.J.; Kim, J.S.; Choe, Y.S.; Lee, K.H.; et al. The effects of small vessel disease and amyloid burden on neuropsychiatric symptoms: A study among patients with subcortical vascular cognitive impairments. Neurobiol. Aging 2013, 34, 1913–1920. [Google Scholar] [CrossRef]
- Sjobeck, M.; Elfgren, C.; Larsson, E.M.; Brockstedt, S.; Latt, J.; Englund, E.; Passant, U. Alzheimer’s disease (AD) and executive dysfunction. A case-control study on the significance of frontal white matter changes detected by diffusion tensor imaging (DTI). Arch. Gerontol. Geriatr. 2010, 50, 260–266. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Singleton, E.H.; Groot, C.; Dijkstra, A.A.; Eikelboom, W.S.; Seeley, W.W.; Miller, B.; Laforce, R.J.; Scheltens, P.; Papma, J.M.; et al. Research Criteria for the Behavioral Variant of Alzheimer Disease: A Systematic Review and Meta-analysis. JAMA Neurol. 2022, 79, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Alladi, S.; Xuereb, J.; Bak, T.; Nestor, P.; Knibb, J.; Patterson, K.; Hodges, J.R. Focal cortical presentations of Alzheimer’s disease. Brain 2007, 130, 2636–2645. [Google Scholar] [CrossRef] [Green Version]
- Griffin, W.S.; Sheng, J.G.; Royston, M.C.; Gentleman, S.M.; McKenzie, J.E.; Graham, D.I.; Roberts, G.W.; Mrak, R.E. Glial-neuronal interactions in Alzheimer’s disease: The potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998, 8, 65–72. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [Green Version]
- Leyns, C.E.G.; Holtzman, D.M. Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef] [Green Version]
- Beach, T.G.; Monsell, S.E.; Phillips, L.E.; Kukull, W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J. Neuropathol. Exp. Neurol. 2012, 71, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Ossenkoppele, R.; Prins, N.D.; Pijnenburg, Y.A.; Lemstra, A.W.; van der Flier, W.M.; Adriaanse, S.F.; Windhorst, A.D.; Handels, R.L.; Wolfs, C.A.; Aalten, P.; et al. Impact of molecular imaging on the diagnostic process in a memory clinic. Alzheimers Dement. 2013, 9, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Rabinovici, G.D.; Rosen, H.J.; Alkalay, A.; Kornak, J.; Furst, A.J.; Agarwal, N.; Mormino, E.C.; O’Neil, J.P.; Janabi, M.; Karydas, A.; et al. Amyloid vs. FDG-PET in the differential diagnosis of AD and FTLD. Neurology 2011, 77, 2034–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varma, A.R.; Snowden, J.S.; Lloyd, J.J.; Talbot, P.R.; Mann, D.M.; Neary, D. Evaluation of the NINCDS-ADRDA criteria in the differentiation of Alzheimer’s disease and frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 1999, 66, 184–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.K.; Diehl, J.; Mendez, M.F.; Neuhaus, J.; Shapira, J.S.; Forman, M.; Chute, D.J.; Roberson, E.D.; Pace-Savitsky, C.; Neumann, M.; et al. Frontotemporal lobar degeneration: Demographic characteristics of 353 patients. Arch. Neurol. 2005, 62, 925–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattsson, N.; Groot, C.; Jansen, W.J.; Landau, S.M.; Villemagne, V.L.; Engelborghs, S.; Mintun, M.M.; Lleo, A.; Molinuevo, J.L.; Jagust, W.J.; et al. Prevalence of the apolipoprotein E epsilon4 allele in amyloid beta positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 913–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, M.; Ricci, M.; Gerace, C.; Blundo, C. Late-onset obsessive-compulsive disorder as the initial manifestation of possible behavioural variant Alzheimer’s disease. Cogn. Neuropsychiatry 2022, 27, 11–19. [Google Scholar] [CrossRef]
- Lagana, V.; Bruno, F.; Altomari, N.; Bruni, G.; Smirne, N.; Curcio, S.; Mirabelli, M.; Colao, R.; Puccio, G.; Frangipane, F.; et al. Neuropsychiatric or Behavioral and Psychological Symptoms of Dementia (BPSD): Focus on Prevalence and Natural History in Alzheimer’s Disease and Frontotemporal Dementia. Front. Neurol. 2022, 13, 832199. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.C.; Brown, J.A.; Possin, K.L.; Datta, S.; Trujillo, A.; Radke, A.; Karydas, A.; Kornak, J.; Sias, A.C.; Rabinovici, G.D.; et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain 2017, 140, 3329–3345. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.S.; Da Re, F.; Dratch, L.; Xie, S.X.; Irwin, D.J.; McMillan, C.T.; Vaishnavi, S.N.; Ferrarese, C.; Lee, E.B.; Shaw, L.M.; et al. Neocortical origin and progression of gray matter atrophy in nonamnestic Alzheimer’s disease. Neurobiol. Aging 2018, 63, 75–87. [Google Scholar] [CrossRef]
- Phillips, J.S.; Da Re, F.; Irwin, D.J.; McMillan, C.T.; Vaishnavi, S.N.; Xie, S.X.; Lee, E.B.; Cook, P.A.; Gee, J.C.; Shaw, L.M.; et al. Longitudinal progression of grey matter atrophy in non-amnestic Alzheimer’s disease. Brain 2019, 142, 1701–1722. [Google Scholar] [CrossRef]
- Therriault, J.; Pascoal, T.A.; Savard, M.; Benedet, A.L.; Chamoun, M.; Tissot, C.; Lussier, F.; Kang, M.S.; Thomas, E.; Terada, T.; et al. Topographic Distribution of Amyloid-beta, Tau, and Atrophy in Patients With Behavioral/Dysexecutive Alzheimer Disease. Neurology 2021, 96, e81–e92. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, D.; Beauregard, J.M.; Soucy, J.P.; Verret, L.; Poulin, S.; Matias-Guiu, J.A.; Cabrera-Martin, M.N.; Bouchard, R.W.; Laforce, R. Posterior Cingulate Cortex Hypometabolism in Non-Amnestic Variants of Alzheimer’s Disease. J. Alzheimers Dis. 2020, 77, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Calvo, B.F.; de Lucena, V.M. Frontal variant of Alzheimer’s disease and typical Alzheimer’s disease: A comparative study. Ann. Psychol. 2013, 29, 293–300. [Google Scholar]
- Singleton, E.H.; Pijnenburg, Y.A.L.; Sudre, C.H.; Groot, C.; Kochova, E.; Barkhof, F.; La Joie, R.; Rosen, H.J.; Seeley, W.W.; Miller, B.; et al. Investigating the clinico-anatomical dissociation in the behavioral variant of Alzheimer disease. Alzheimers Res. Ther. 2020, 12, 148. [Google Scholar] [CrossRef] [PubMed]
- Seeley, W.W.; Menon, V.; Schatzberg, A.F.; Keller, J.; Glover, G.H.; Kenna, H.; Reiss, A.L.; Greicius, M.D. Dissociable intrinsic connectivity networks for salience processing and executive control. J. Neurosci. 2007, 27, 2349–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corriveau-Lecavalier, N.; Machulda, M.M.; Botha, H.; Graff-Radford, J.; Knopman, D.S.; Lowe, V.J.; Fields, J.A.; Stricker, N.H.; Boeve, B.F.; Jack, C.R., Jr.; et al. Phenotypic subtypes of progressive dysexecutive syndrome due to Alzheimer’s disease: A series of clinical cases. J. Neurol. 2022, 269, 4110–4128. [Google Scholar] [CrossRef]
- Rivera-Escalera, F.; Matousek, S.B.; Ghosh, S.; Olschowka, J.A.; O’Banion, M.K. Interleukin-1beta mediated amyloid plaque clearance is independent of CCR2 signaling in the APP/PS1 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2014, 69, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Adolphs, R. Conceptual challenges and directions for social neuroscience. Neuron 2010, 65, 752–767. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarty, P.; Tianbai, L.; Herring, A.; Ceballos-Diaz, C.; Das, P.; Golde, T.E. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol. Neurodegener. 2012, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [Green Version]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, O. Biomarkers for neurodegenerative diseases. Nat. Med. 2021, 27, 954–963. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, Z.; Zhang, N.; Cai, L.; Li, Y.; Yang, H.; Yao, S.; Xing, X.; Ji, Y.; Gao, S. Spatial Patterns of Hypometabolism and Amyloid Deposition in Variants of Alzheimer’s Disease Corresponding to Brain Networks: A Prospective Cohort Study. Mol. Imaging Biol. 2019, 21, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, D.; Sellami, L.; Poulin, S.; Verret, L.; Bouchard, R.W.; Laforce, R., Jr. The Behavioral/Dysexecutive Variant of Alzheimer’s Disease: A Case Series with Clinical, Neuropsychological, and FDG-PET Characterization. Dement. Geriatr. Cogn. Disord. 2020, 49, 518–525. [Google Scholar] [CrossRef]
- Lehingue, E.; Gueniat, J.; Jourdaa, S.; Hardouin, J.B.; Pallardy, A.; Courtemanche, H.; Rocher, L.; Etcharry-Bouyx, F.; Auriacombe, S.; Mollion, H.; et al. Improving the Diagnosis of the Frontal Variant of Alzheimer’s Disease with the DAPHNE Scale. J. Alzheimers Dis. 2021, 79, 1735–1745. [Google Scholar] [CrossRef]
- Sala, A.; Caprioglio, C.; Santangelo, R.; Vanoli, E.G.; Iannaccone, S.; Magnani, G.; Perani, D. Brain metabolic signatures across the Alzheimer’s disease spectrum. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 256–269. [Google Scholar] [CrossRef]
- Woodward, M.C.; Rowe, C.C.; Jones, G.; Villemagne, V.L.; Varos, T.A. Differentiating the frontal presentation of Alzheimer’s disease with FDG-PET. J. Alzheimers Dis. 2015, 44, 233–242. [Google Scholar] [CrossRef]
- Song, W.M.; Joshita, S.; Zhou, Y.; Ulland, T.K.; Gilfillan, S.; Colonna, M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 2018, 215, 745–760. [Google Scholar] [CrossRef]
- Singleton, E.; Hansson, O.; Pijnenburg, Y.A.L.; La Joie, R.; Mantyh, W.G.; Tideman, P.; Stomrud, E.; Leuzy, A.; Johansson, M.; Strandberg, O.; et al. Heterogeneous distribution of tau pathology in the behavioural variant of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2021, 92, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Dominguez Perez, S.; Phillips, J.S.; Norise, C.; Kinney, N.G.; Vaddi, P.; Halpin, A.; Rascovsky, K.; Irwin, D.J.; McMillan, C.T.; Xie, L.; et al. Neuropsychological and Neuroanatomical Features of Patients with Behavioral/Dysexecutive Variant Alzheimer’s Disease (AD): A Comparison to Behavioral Variant Frontotemporal Dementia and Amnestic AD Groups. J. Alzheimers Dis. 2022, 89, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.G.; Kuiper, W.; Ahmed, R.M.; Halliday, G.M.; Burrell, J.R.; Hodges, J.R.; Guastella, A.J.; Piguet, O.; Kumfor, F. Plasma Oxytocin Is Not Associated with Social Cognition or Behavior in Frontotemporal Dementia and Alzheimer’s Disease Syndromes. Dement. Geriatr. Cogn. Disord. 2022, 51, 241–248. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Argyrophilic grain disease: Frequency of occurrence in different age categories and neuropathological diagnostic criteria. J. Neural Transm. 1998, 105, 801–819. [Google Scholar] [CrossRef]
- Hamilton, R.L. Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000, 10, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Salmon, D.; Galasko, D.; Masliah, E.; Katzman, R.; DeTeresa, R.; Thal, L.; Pay, M.M.; Hofstetter, R.; Klauber, M.; et al. The Lewy body variant of Alzheimer’s disease: A clinical and pathologic entity. Neurology 1990, 40, 1–8. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Attems, J. Sporadic cerebral amyloid angiopathy: Pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol. 2005, 110, 345–359. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Ballard, C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis. Assoc. Disord. 1999, 13 (Suppl. S3), S115–S123. [Google Scholar] [CrossRef]
- Munoz, D.G.; Woulfe, J.; Kertesz, A. Argyrophilic thorny astrocyte clusters in association with Alzheimer’s disease pathology in possible primary progressive aphasia. Acta Neuropathol. 2007, 114, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F.; Maraganore, D.M.; Parisi, J.E.; Ahlskog, J.E.; Graff-Radford, N.; Caselli, R.J.; Dickson, D.W.; Kokmen, E.; Petersen, R.C. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999, 53, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Cordato, N.J.; Halliday, G.M.; McCann, H.; Davies, L.; Williamson, P.; Fulham, M.; Morris, J.G. Corticobasal syndrome with tau pathology. Mov. Disord. 2001, 16, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F.; Lang, A.E.; Litvan, I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann. Neurol. 2003, 54 (Suppl. S5), S15–S19. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Josephs, K.A.; Aiba, I.; Yoshida, M.; Dickson, D.W. Neuropathology and emerging biomarkers in corticobasal syndrome. J. Neurol. Neurosurg. Psychiatry 2022, 93, 919–929. [Google Scholar] [CrossRef]
- Shelley, B.P.; Hodges, J.R.; Kipps, C.M.; Xuereb, J.H.; Bak, T.H. Is the pathology of corticobasal syndrome predictable in life? Mov. Disord. 2009, 24, 1593–1599. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.T.; Rippon, G.W.; Boeve, B.F.; Knopman, D.S.; Petersen, R.C.; Parisi, J.E.; Josephs, K.A. Alzheimer’s disease and corticobasal degeneration presenting as corticobasal syndrome. Mov. Disord. 2009, 24, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.E.; Rabinovici, G.D.; Mayo, M.C.; Wilson, S.M.; Seeley, W.W.; DeArmond, S.J.; Huang, E.J.; Trojanowski, J.Q.; Growdon, M.E.; Jang, J.Y.; et al. Clinicopathological correlations in corticobasal degeneration. Ann. Neurol. 2011, 70, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Sakae, N.; Josephs, K.A.; Litvan, I.; Murray, M.E.; Duara, R.; Uitti, R.J.; Wszolek, Z.K.; van Gerpen, J.; Graff-Radford, N.R.; Dickson, D.W. Clinicopathologic subtype of Alzheimer’s disease presenting as corticobasal syndrome. Alzheimers Dement. 2019, 15, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Brettschneider, J.; Grossman, M.; Arnold, S.E.; Hu, W.T.; Xie, S.X.; Lee, V.M.; Shaw, L.M.; Trojanowski, J.Q. CSF biomarkers cutoffs: The importance of coincident neuropathological diseases. Acta Neuropathol. 2012, 124, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossenkoppele, R.; Jansen, W.J.; Rabinovici, G.D.; Knol, D.L.; van der Flier, W.M.; van Berckel, B.N.; Scheltens, P.; Visser, P.J.; Amyloid, P.E.T.S.G.; Verfaillie, S.C.; et al. Prevalence of amyloid PET positivity in dementia syndromes: A meta-analysis. JAMA 2015, 313, 1939–1949. [Google Scholar] [CrossRef] [Green Version]
- Palleis, C.; Brendel, M.; Finze, A.; Weidinger, E.; Botzel, K.; Danek, A.; Beyer, L.; Nitschmann, A.; Kern, M.; Biechele, G.; et al. Cortical [(18)F]PI-2620 Binding Differentiates Corticobasal Syndrome Subtypes. Mov. Disord. 2021, 36, 2104–2115. [Google Scholar] [CrossRef]
- Tagai, K.; Ono, M.; Kubota, M.; Kitamura, S.; Takahata, K.; Seki, C.; Takado, Y.; Shinotoh, H.; Sano, Y.; Yamamoto, Y.; et al. High-Contrast In Vivo Imaging of Tau Pathologies in Alzheimer’s and Non-Alzheimer’s Disease Tauopathies. Neuron 2021, 109, 42–58.e8. [Google Scholar] [CrossRef]
- Ali, F.; Whitwell, J.L.; Martin, P.R.; Senjem, M.L.; Knopman, D.S.; Jack, C.R.; Lowe, V.J.; Petersen, R.C.; Boeve, B.F.; Josephs, K.A. [(18)F]AV-1451 uptake in corticobasal syndrome: The influence of beta-amyloid and clinical presentation. J. Neurol. 2018, 265, 1079–1088. [Google Scholar] [CrossRef]
- Ono, M.; Sahara, N.; Kumata, K.; Ji, B.; Ni, R.; Koga, S.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M.; Yoshida, M.; et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 2017, 140, 764–780. [Google Scholar] [CrossRef] [Green Version]
- Marquie, M.; Verwer, E.E.; Meltzer, A.C.; Kim, S.J.W.; Aguero, C.; Gonzalez, J.; Makaretz, S.J.; Siao Tick Chong, M.; Ramanan, P.; Amaral, A.C.; et al. Lessons learned about [F-18]-AV-1451 off-target binding from an autopsy-confirmed Parkinson’s case. Acta Neuropathol. Commun. 2017, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Soleimani-Meigooni, D.N.; Iaccarino, L.; La Joie, R.; Baker, S.; Bourakova, V.; Boxer, A.L.; Edwards, L.; Eser, R.; Gorno-Tempini, M.L.; Jagust, W.J.; et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer’s disease and other neurodegenerative diseases. Brain 2020, 143, 3477–3494. [Google Scholar] [CrossRef] [PubMed]
- Kroth, H.; Oden, F.; Molette, J.; Schieferstein, H.; Capotosti, F.; Mueller, A.; Berndt, M.; Schmitt-Willich, H.; Darmency, V.; Gabellieri, E.; et al. Discovery and preclinical characterization of [(18)F]PI-2620, a next-generation tau PET tracer for the assessment of tau pathology in Alzheimer’s disease and other tauopathies. Eur. J. Nucl. Med. Mol. Imaging. 2019, 46, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abate, F.; Dati, G.; Ginevrino, M.; Valente, E.M.; Barone, P.; Picillo, M. APP-Related Corticobasal Syndrome: Expanding the List of Corticobasal Degeneration Look Alikes. Mov. Disord. Clin. Pract. 2020, 7, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.; Khan, A.; Keith, J.; Rogaeva, E.; Bilbao, J.; St George-Hyslop, P.; Ghani, M.; Freedman, M.; Stuss, D.T.; Chow, T.; et al. Characterizing familial corticobasal syndrome due to Alzheimer’s disease pathology and PSEN1 mutations. Alzheimers Dement. 2017, 13, 520–530. [Google Scholar] [CrossRef]
- Lopez-Garcia, S.; Jimenez-Bonilla, J.; Lopez Delgado, A.; Orizaola Balaguer, P.; Infante Ceberio, J.; Banzo Marraco, I.; Rodriguez Rodriguez, E.; Sanchez-Juan, P. A Rare PSEN1 (Leu85Pro) Mutation Causing Alzheimer’s Disease in a 29-Year-Old Woman Presenting as Corticobasal Syndrome. J. Alzheimers Dis. 2019, 70, 655–658. [Google Scholar] [CrossRef]
- Navarro, E.; De Andres, C.; Guerrero, C.; Gimenez-Roldan, S. Corticobasal Syndrome in a Family with Early-Onset Alzheimer’s Disease Linked to a Presenilin-1 Gene Mutation. Mov. Disord. Clin. Pract. 2015, 2, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, S.; Ascari, R.; Maderna, E.; Catania, M.; Ghetti, B.; Tagliavini, F.; Giaccone, G.; Di Fede, G. Microglial Heterogeneity and Its Potential Role in Driving Phenotypic Diversity of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2780. [Google Scholar] [CrossRef]
- Neff, R.A.; Wang, M.; Vatansever, S.; Guo, L.; Ming, C.; Wang, Q.; Wang, E.; Horgusluoglu-Moloch, E.; Song, W.M.; Li, A.; et al. Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci. Adv. 2021, 7, abb5398. [Google Scholar] [CrossRef]
- Tijms, B.M.; Gobom, J.; Teunissen, C.; Dobricic, V.; Tsolaki, M.; Verhey, F.; Popp, J.; Martinez-Lage, P.; Vandenberghe, R.; Lleo, A.; et al. CSF Proteomic Alzheimer’s Disease-Predictive Subtypes in Cognitively Intact Amyloid Negative Individuals. Proteomes 2021, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Feng, Z.; Gao, F.; Zang, W.; Liu, J. Mitoepigenetics: An intriguing regulatory layer in aging and metabolic-related diseases. Free Radic. Biol. Med. 2021, 177, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, G.C.; Magalhaes, L.; Ribeiro-Dos-Santos, A.; Vidal, A.F. Mitochondrial Epigenetics: Non-Coding RNAs as a Novel Layer of Complexity. Int. J. Mol. Sci. 2020, 21, 1838. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; London, A. Neuroimmunity: A New Science That Will Revolutionize How We Keep Our Brains Healthy and Young, 1st ed.; Yale University Press: New Haven, CT, USA, 2015. [Google Scholar]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139 (Suppl. S2), 237–252. [Google Scholar] [CrossRef] [PubMed]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Prokop, S.; Miller, K.R.; Labra, S.R.; Pitkin, R.M.; Hoxha, K.; Narasimhan, S.; Changolkar, L.; Rosenbloom, A.; Lee, V.M.; Trojanowski, J.Q. Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer’s disease patient brain samples. Acta Neuropathol. 2019, 138, 613–630. [Google Scholar] [CrossRef]
- Scapagnini, U. Psychoneuroendocrinoimmunology: The basis for a novel therapeutic approach in aging. Psychoneuroendocrinology 1992, 17, 411–420. [Google Scholar] [CrossRef]
- Russell, W.L.; Russell, L.B.; Kelly, E.M. Radiation dose rate and mutation frequency. Science 1958, 128, 1546–1550. [Google Scholar] [CrossRef]
- Zhao, P.; Xu, Y.; Jiang, L.L.; Fan, X.; Ku, Z.; Li, L.; Liu, X.; Deng, M.; Arase, H.; Zhu, J.J.; et al. LILRB2-mediated TREM2 signaling inhibition suppresses microglia functions. Mol. Neurodegener. 2022, 17, 44. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Gordon, M.N. Diversity of transcriptomic microglial phenotypes in aging and Alzheimer’s disease. Alzheimers Dement. 2022, 18, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Walker, D. Defining activation states of microglia in human brain tissue: An unresolved issue for Alzheimer’s disease. Neuroimmunol. Neuroinflam. 2020, 7, 194–214. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Innate immunity, local inflammation, and degenerative disease. Sci. Aging Knowl. Environ. 2002, 2002, re3. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Ceballos-Diaz, C.; Beccard, A.; Janus, C.; Dickson, D.; Golde, T.E.; Das, P. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J. Immunol. 2010, 184, 5333–5343. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarty, P.; Jansen-West, K.; Beccard, A.; Ceballos-Diaz, C.; Levites, Y.; Verbeeck, C.; Zubair, A.C.; Dickson, D.; Golde, T.E.; Das, P. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: Evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010, 24, 548–559. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [Green Version]
- Deleidi, M.; Jaggle, M.; Rubino, G. Immune aging, dysmetabolism, and inflammation in neurological diseases. Front. Neurosci. 2015, 9, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahidehpour, R.K.; Higdon, R.E.; Crawford, N.G.; Neltner, J.H.; Ighodaro, E.T.; Patel, E.; Price, D.; Nelson, P.T.; Bachstetter, A.D. Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol. Aging 2021, 99, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.E.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Bisht, K.; Sharma, K.; Lacoste, B.; Tremblay, M.E. Dark microglia: Why are they dark? Commun. Integr. Biol. 2016, 9, e1230575. [Google Scholar] [CrossRef] [Green Version]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sanchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gomez-Nicola, D.; Luheshi, G.; Vallieres, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- El Hajj, H.; Savage, J.C.; Bisht, K.; Parent, M.; Vallieres, L.; Rivest, S.; Tremblay, M.E. Ultrastructural evidence of microglial heterogeneity in Alzheimer’s disease amyloid pathology. J. Neuroinflamm. 2019, 16, 87. [Google Scholar] [CrossRef] [Green Version]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Arauzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proc. Natl. Acad. Sci. USA. 2013, 110, 19802–19807. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.L.; Salmen, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Jordao, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar] [CrossRef] [PubMed]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.N.; Ceballos, C.; Rosario, A.; Ladd, T.; Moore, B.; Golde, G.; Wang, X.; Allen, M.; Ertekin-Taner, N.; Funk, C.C.; et al. Microglia show differential transcriptomic response to Abeta peptide aggregates ex vivo and in vivo. Life Sci. Alliance 2021, 4, e202101108. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donato, L.; Mordà, D.; Scimone, C.; Alibrandi, S.; D’Angelo, R.; Sidoti, A. How Many Alzheimer–Perusini’s Atypical Forms Do We Still Have to Discover? Biomedicines 2023, 11, 2035. https://doi.org/10.3390/biomedicines11072035
Donato L, Mordà D, Scimone C, Alibrandi S, D’Angelo R, Sidoti A. How Many Alzheimer–Perusini’s Atypical Forms Do We Still Have to Discover? Biomedicines. 2023; 11(7):2035. https://doi.org/10.3390/biomedicines11072035
Chicago/Turabian StyleDonato, Luigi, Domenico Mordà, Concetta Scimone, Simona Alibrandi, Rosalia D’Angelo, and Antonina Sidoti. 2023. "How Many Alzheimer–Perusini’s Atypical Forms Do We Still Have to Discover?" Biomedicines 11, no. 7: 2035. https://doi.org/10.3390/biomedicines11072035