3.1. Prediction of Protein Structure
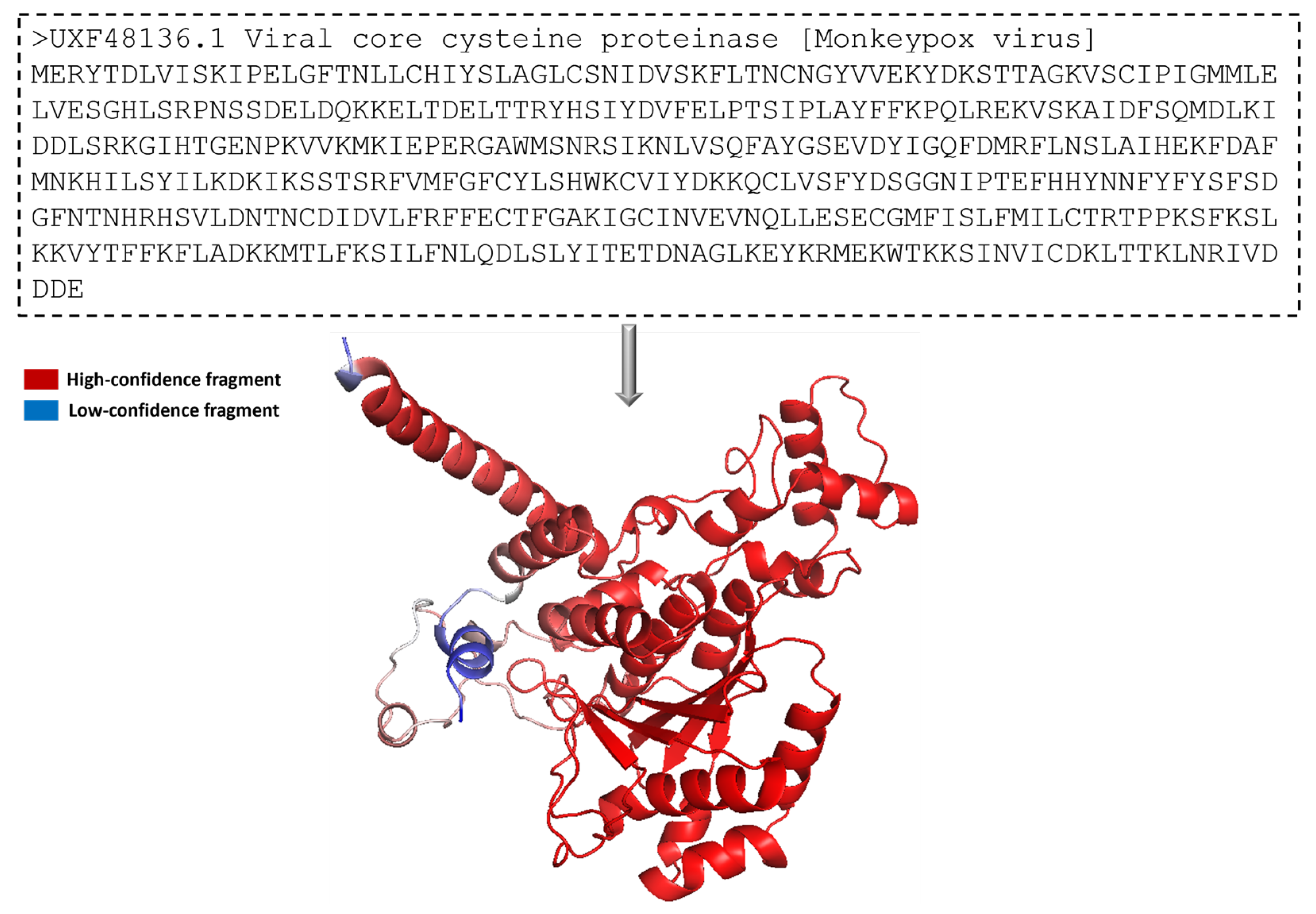
The protein 3-dimensional structure of the MPXV core cysteine proteinase was predicted using the AlphaFold [
25], as shown in
Figure 1. It also shows the FASTA sequence (GenBank ID: UXF48136.1) used to predict the 3D structure. The fragment exhibiting the highest level of confidence as predicted by AlphaFold is visually represented in red, whereas the fragment with the lowest degree of confidence is depicted in blue. The gradient from red to blue represents the confidence level of prediction for each fragment in the 0% to 100% range.
Figure 1 illustrates that most of the residues had a high confidence score. The overall prediction confidence score of the 3-dimensional structure of the MPXV core protein cysteine proteinase showed acceptable evidence to be used in this study. In this structure, a long loop is not appropriately modelled by AlphaFold, thus showing a low or medium confidence score. The model is prone to fluctuate in this region during and post-interaction with the ligand. However, the protein’s binding site was modelled accurately with a high confidence score. This provides high assurance for forming stable protein–ligand complexes in docking and molecular interaction during the molecular dynamic simulation. Overall, the protein structure is dominated by alpha helices. As shown in
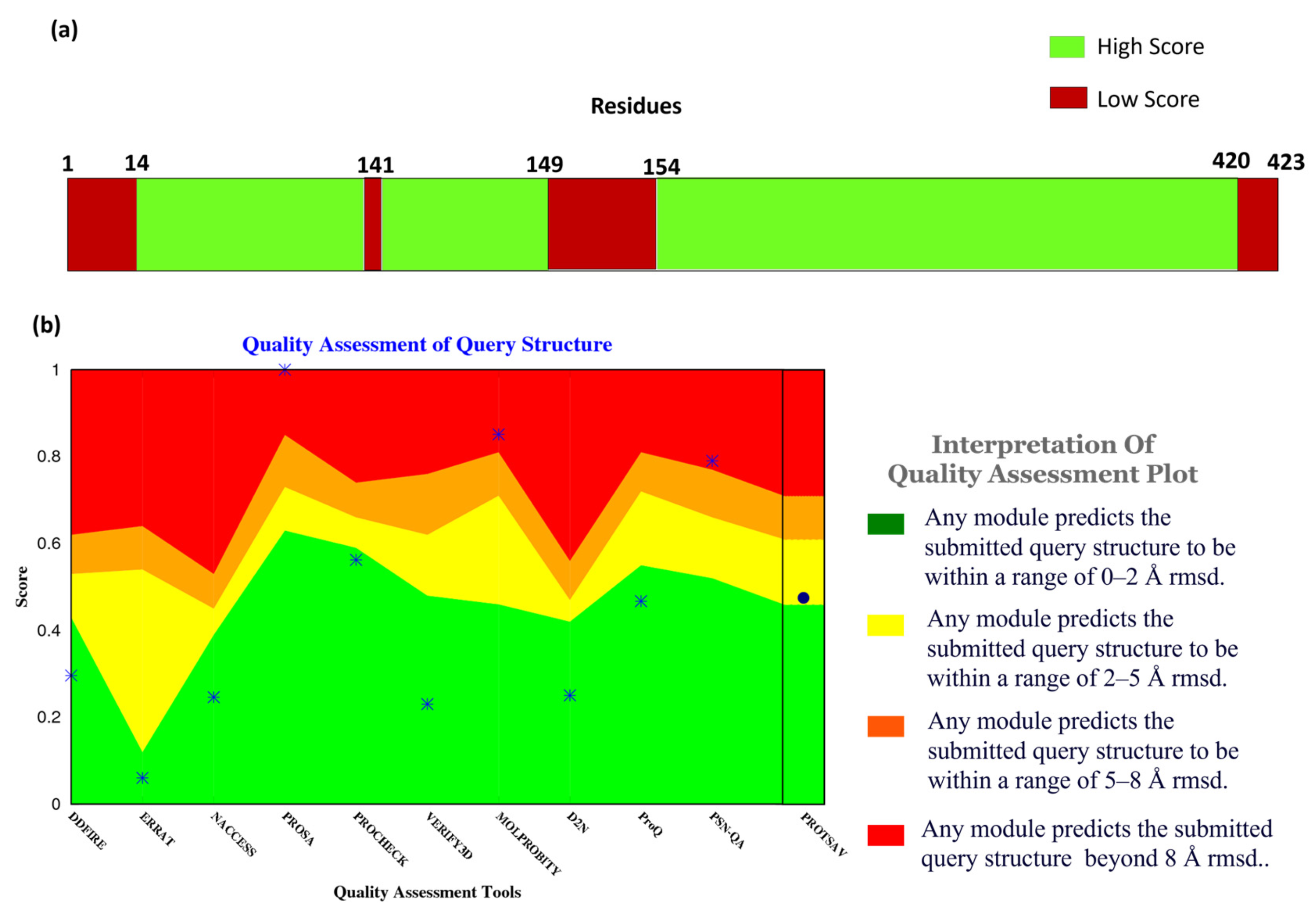
Figure 2a, the confidence score of the residues and their respective counts were plotted on the bar. It shows that most of the structural components of MPXV had high-quality scores given by Alpha Fold. Here, the confidence score ranging >70 indicates a high score, while <70 indicates a low score. Residues at the N- and C- terminals showed low confidence scores in prediction, which is widely accepted due to their high degree of freedom.
In addition to the confidence score given by AlphaFold, the structure quality assessment was performed using ProTSAV (Protein Structure Analysis and Validation) [
26].
Figure 2b demonstrates the quality assessment of the modelled structure using ProTSAV, which utilizes various tools (such as Procheck, ProSA-web, ERRAT, Verify3D, dDFire, Naccess, MolProbity, D2N, ProQ, and PSN-QA) and metrics to evaluate protein structure quality. Except for PSN-QA, MolProbity, and ProSA-web, all the other tools showed high quality for the modelled protein structure. Notably, various quality assessment instruments employ distinct criteria and metrics to assess protein structure, and they may exhibit varying degrees of efficacy and limitations. PSN-QA uses a network-based approach to evaluate the quality of a protein’s 3D structure. The MolProbity tool analyses the overall geometry and stereochemistry of a protein structure.
In contrast, the ProSA web tool uses a potential statistical approach to predict the quality of a protein structurally. These tools may be more sensitive to certain types of discrepancy detected in the structure, while others may be less sensitive. This might be due to insufficient sampling during the modelling process. However, the other tools suggested that the quality of the protein structure was high (predicted RMSD < 2 Å), including Procheck, which calculates a set of stereochemical parameters, including the bond lengths, the bond angles, and the torsion angles, and compares them to traditional values to identify potential errors or complications within the structure, ERRAT quantifies the quality of a protein 3-dimensional structure by analyzing the agreement between the structure and the atomic environment around each residue. The high score in Procheck indicates the appropriate geometry of the model structure. However, it does not assure native confirmation. Verify3D uses a statistical approach to assess whether or not the structure of the protein is compatible with the sequence of its amino acids, dDFire uses a machine learning approach to investigate the quality of a protein structure based on structural features, including solvent accessibility, secondary structure, and packing density, whereas Naccess estimates the solvent accessibility of each residue in the protein structure, which can provide insight into the protein’s interactions with its environment.
Moreover, D2N (distance to native) predicts the modelled structure’s RMSD (root-mean-square deviation), which measures the structural similarity with the native contacts. ProQ uses machine learning and potential statistical approaches to predict a protein structure’s quality. The results indicated that most of these tools favored the quality of the protein structure, validating the modelled structure of MPXV core cysteine proteinase and, thus, progressing with further calculations.
A modelled protein structure of cysteine protease was used in structure-based drug design to screen phytochemicals against the MPXV [
53]. This study discussed the application of molecular docking that identified N-(2-Allylcarbamoyl-4-chloro-phenyl)-3,4-dimethoxy-benzamide, which exhibited promising molecular binding results. A recent research study employed the structural model of cysteine protease as an approach to screening FDA-approved compounds [
54].
3.2. MD (Molecular Dynamics) Simulation and Clustering of Protein Structure
Further, the protein structure of MPXV cysteine proteinase generated by AlphaFold was used in a molecular dynamics (MD) simulation, followed by clustering analysis to identify the most probable conformations. MD simulation has been widely used to refine the modeled protein structure [
55]. Using water solvent molecules during the protein structure simulation has been recommended to refine the modelled conformation [
56]. The criterion for selection stipulated that a cluster’s population must exceed 10% of the total conformations. Therefore, three clusters (Cluster-1, Cluster-2, and Cluster-3) were identified, accounting for most of the simulation trajectory. Cluster-1 had the highest number of conformations, with 65% of the total population; cluster-2 had a population of 16% of the total conformations; and cluster-3 had 11% of the members. These clusters collectively had more than 90% of the population, thus covering the major conformations of the protein structural change. This justified the selection of the top 3 representative structures through clustering. Each middle structure of these three clusters was selected for further calculation to cover the conformational variability of the binding site. They were docked individually using molecular docking to detect potential hit compounds. Here, the binding pocket was predicted using these three structures.
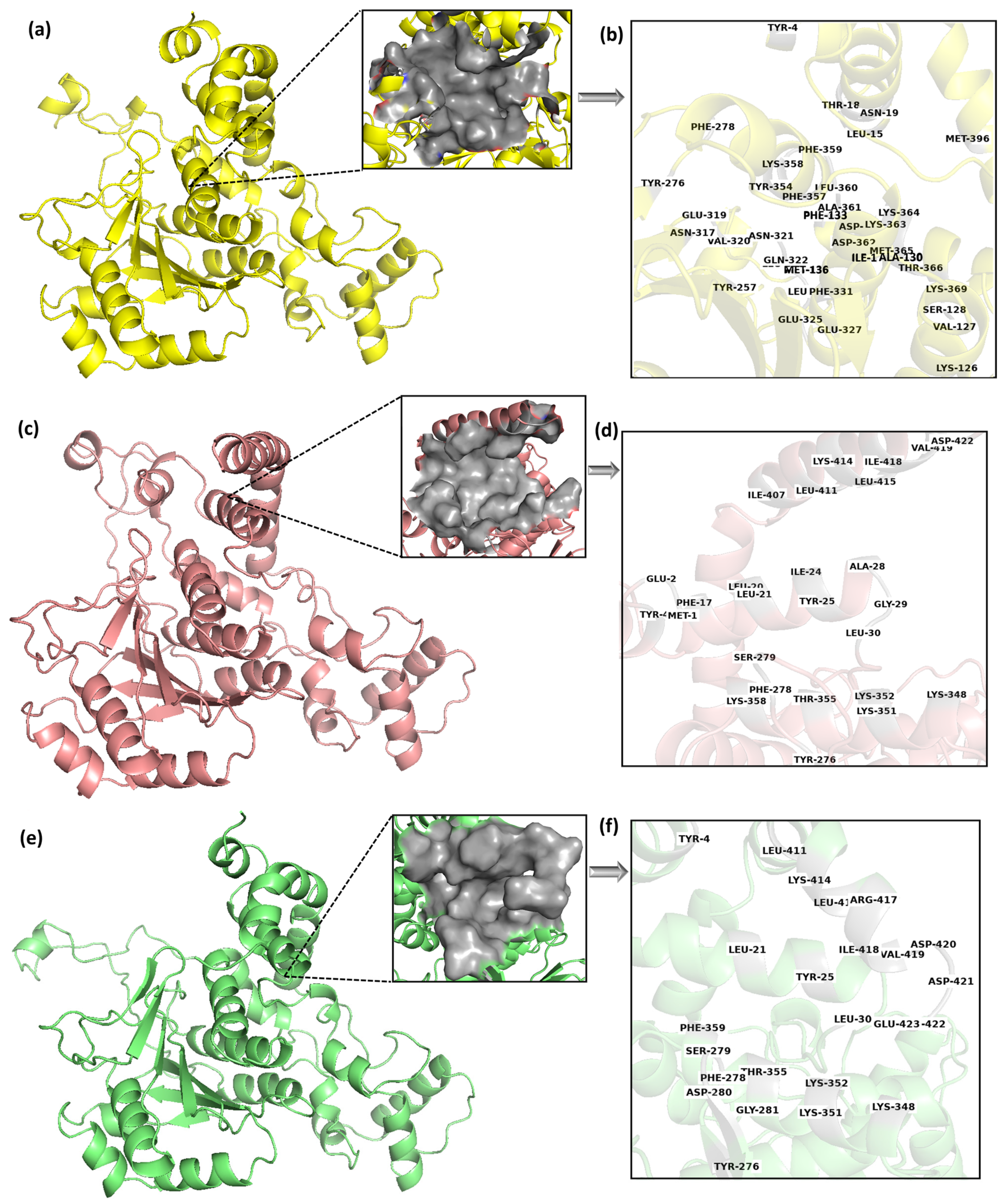
Figure 3a,c,e shows the 3D representation of the middle structures of these selected clusters, Cluster-1, Cluster-2, and Cluster-3, respectively. Protein conformation largely changes with time, which can cause the movement of binding site residues. The multi-docking approach can address this challenge by considering different poses of the binding site [
57]. However, enhanced sampling methods are recommended for more flexible proteins. This approach allowed us to cover the different states of the protein structure during docking.
3.3. Binding Pocket Prediction
The binding pockets of the selected middle structures (Cluster-1, Cluster-2, and Cluster-3) were predicted in the CASTp web server [
36]. Here, the CASTp predicted different binding pockets for the three representative structures of MPXV cysteine proteinase generated during clustering (Cluster-1, Cluster-2, and Cluster-3), as shown in
Figure 3.
Table 1 lists the binding site residues of these binding pockets. The reason for the different binding pocket residues reflects the conformational changes in the protein structures; they are significant enough to cause changes in the geometrical characteristics of binding sites. It is also possible that the changes in conformation can affect the electrostatic properties and accessibility of the binding site, which can further contribute to differences in the predicted binding poses.
The binding pocket of Cluster-1 had an area of 515.961 Å
2 and a volume of 417.746 Å
3, Cluster-2 had an area of 537.845 Å
2 and volume of 797.627 Å
3 while Cluster-3 had an area of 411.089 Å
2 and volume of 439.660 Å
3. It was observed that Cluster-1 had the highest number of binding site residues compared to Cluster-2 and Cluster-3. However, Cluster-2 had the largest surface area and surface volume compared to Cluster-1 and Cluster-3. It was found that Tyr
4, Tyr
276 and Phe
278 were common in all the binding site pockets of the clusters. This indicates that these pockets were distinct but not distant.
Figure 3a,c,e showed the binding pocket of Cluster-1, Cluster-2, and Cluster-3, respectively, while
Figure 3b,d,f showed the binding site residues of the Cluster-1, Cluster-2, and Cluster-3, respectively. A total of 39 residues were detected as the prime site in the cluster 1 binding pocket. However, out of the total, merely seven residues were found to be exclusively hydrophobic, while the majority of the residues were polar or charged in nature. Comparable patterns were noted in the remaining two clusters, albeit with a smaller number of residues. This indicates a high possibility of charged/polar ligands interacting with the target protein.
Generating ensembles of protein structures using MD simulation and detecting their binding sites accounts for the protein’s flexibility. This approach has been deployed earlier for drug discovery and resulted in a successful hit compound [
58].
The grid box was built for each selected structure guided by its binding pocket (Cluster-1, Cluster-2, and Cluster-3) covering all their binding site residues. The binding sites were encompassed by grid boxes, where the dimensions of the grid box for Cluster-1 were 30 Å, 30 Å, and 40 Å in the x, y, and z axes, respectively, with the center of the box located at −8.102 Å, 8.790 Å, and −1.358 Å. Similarly, the dimensions of the grid box for Cluster-2 were 30 Å, 30 Å, and 32 Å in the x, y, and z axes, respectively, with the center of the box located at -1.823 Å, 10.733 Å, and 18.198 Å. Lastly, the grid box for Cluster-3 had dimensions of 30 Å, 30 Å, and 30 Å in the x, y, and z axes, respectively, with the center of the box located at 3.273 Å, 9.693 Å, and 17.796 Å, encompassing the binding sites. These grid boxes were used for docking with the anti-dengue compounds. As can be observed, the grid box for the Cluster-1 structure had the maximum dimension with an 8–10 Å edge in the Z-direction.
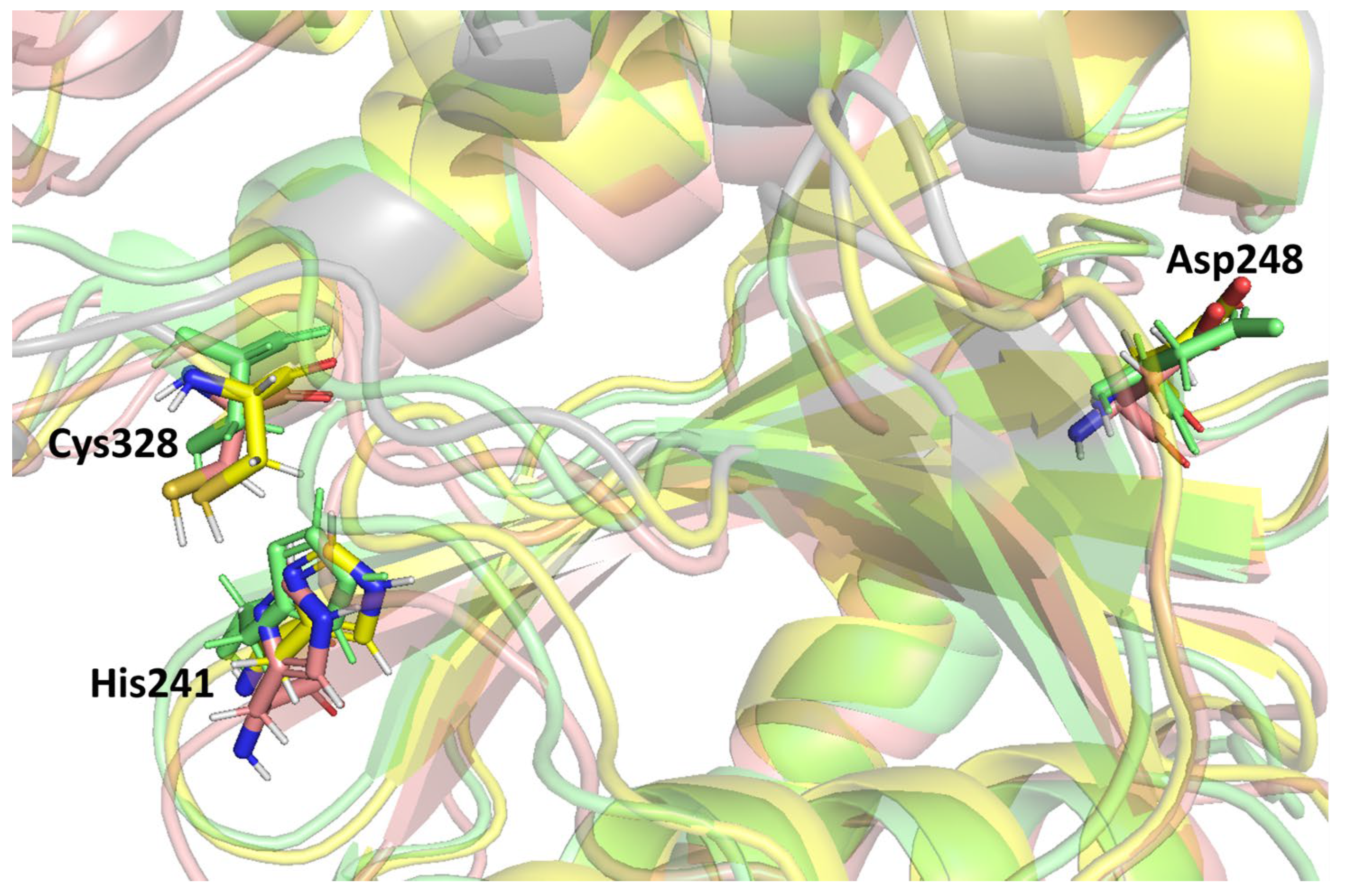
Previous studies stated that the cysteine proteinase enzyme consists of the putative catalytic triad residues, histidine (His) and cysteine (Cys), with the additional residue aspartate (Asp) embedded in a conserved region of the protein structure [
59]. A catalytic triad is a group of three amino acids present at the active site of cysteine proteases that are essential for catalytic activity. The catalytic triad of MPXV core cysteine proteinases His
241, Cys
328, and Asp
248 is shown in
Figure 4, as are all three clusters. These catalytic residues are shared among the cysteine proteinases in this family. Moreover, these were detected in the binding grid box used for molecule docking in this study.
Figure 4 shows the orientation and alignment differences among all of the cluster structures.
3.4. Ensemble Docking
In this study, the efficacy of anti-dengue compounds was evaluated against three distinct clusters of MPXV cysteine proteinase using the ensemble docking approach. The clusters were screened separately, and for each cluster, the middle structure was prepared and then docked with 1627 unique anti-dengue compounds. However, some compounds’ docking was unsuccessful due to conformational errors, resulting in only 1467 docking results.
Table S1 lists the binding scores of the 1467 compounds and ranks them accordingly. The top hit was selected from docking for a given cluster, and their best pose was used for further interaction analysis.
Table 2 lists the binding energies of the top hit from each cluster given by Autodock in kcal/mol. Cluster-3 docked with the compound CHEMBL4861364 had the best binding energy of −10.9 kcal/mol, compared to the other two with their respective clusters. Here, the compound CHEMBL4549312 had a minimum binding energy of −10.5 kcal/mol for Cluster-1, while CHEMBL32926 had a binding energy of −10.7 kcal/mol for Cluster-2. It was observed that the protein–ligand complex of Cluster-3 had the highest negative binding score, Cluster-2 was the second best, and Cluster-1 had the lowest negative binding score. All of the binding scores were <−10 kcal, showing a high affinity of these compounds with the target protein and, therefore, being good candidates for further development and investigation. TTP-6171 (control) was also docked with the same protocol with the three clusters as shown in
Supplementary Table S2. Cluster-2 showed the highest negative binding score of −8.9 kcal/mol; thus, it was selected for further investigation. A similar study was performed with natural products, and the top-hit compound showed −10.56 kcal/mol of binding energy [
60]. Based on these findings, it seems reasonable to recommend conducting additional research into whether these compounds could be useful in combating MPXV cysteine proteinase. The best hits from repurposing the FDA drugs showed binding scores between −9.0 and −7.0 kcal/mol [
18]. Here, from a previous study, the known inhibitor TTP-6171 had a binding score of −9.3 kcal/mol [
18]. This indicates that all three hits captured in this study have a higher binding potential.
3.5. Interactions Analysis
The top three hits’ best-docked poses of the protein–ligand complex were investigated for interaction analysis. The binding interactions were predicted using the PoseView component of the ProteinsPlus server [
44].
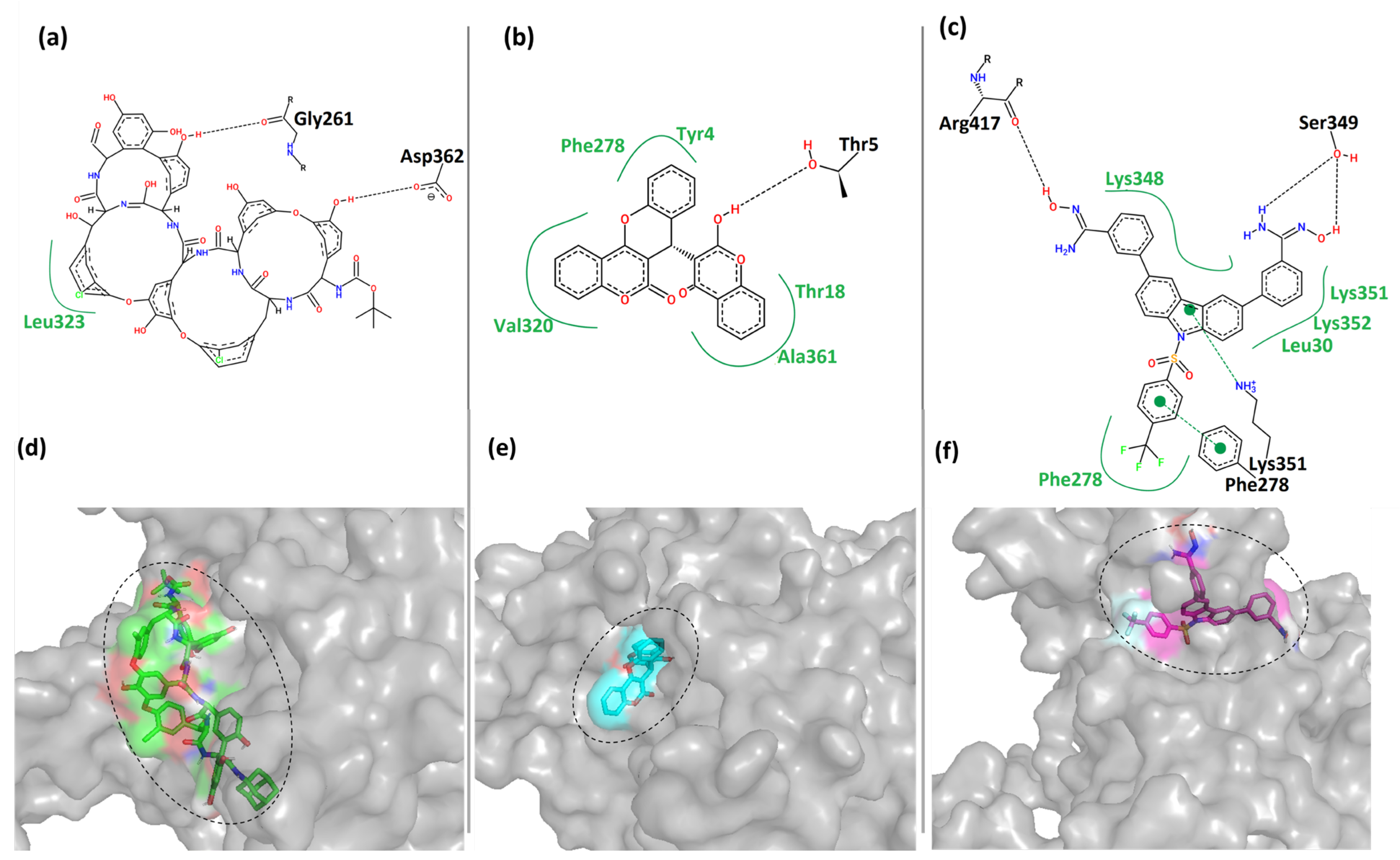
Figure 5 shows the 2- and 3-dimensional representations of the binding interactions between the protein MPXV cysteine proteinase and the hit compounds. Here, it was observed that the compound CHEMBL4549312 had two hydrogen bonds with the residues Gly
261 and Asp
362 of Cluster-1 of MPXV cysteine proteinase. It also had hydrophobic contact with residue Leu
323 of the protein, as shown in
Figure 5a. This was also reflected by its relatively low binding score in
Table 2. CHEMBL4549312 is a peculiar structure that has a lower degree of freedom. This prevents it from covering the larger sample space and forming more interactions.
Moreover, the compound CHEMBL32926 had multiple hydrophobic contacts with its corresponding binding pocket residues Tyr
4, Thr
18, Phe
278, Val
320, and Ala
361, and a single hydrogen bond interaction with residue Thr
5 (
Figure 5b). This shows that CHEMBL4549312 had more hydrogen bonds than CHEMBL32926, but the overall interaction was lesser with the pocket-lining residues.
Figure 5c showed four hydrogen bonds between CHEMBL4861364 and Cluster-3 protein with residues Phe
278, Lys
351, Ser
349, and Arg
417. It also showed multiple hydrophobic contacts with residues Leu
30, Phe
278, Lys
348, Lys
351, and Lys
352.
Figure 5d–f showed that the compounds were bound to each cluster’s different binding locations of the MPXV protein. The compound CHEMBL4549312 was relatively bulkier as compared to the other two hits. The compound CHEMBL32926 bound to Cluster-2 was compact in nature compared to the other two compounds, while CHEMBL4861364 bound to Cluster-3 was of moderate size with a high degree of freedom. Here, it was observed that CHEMBL32926 and CHEMBL4861364 had greater interactions (polar and non-polar) with the MPXV cysteine proteinase than the large compound CHEMBL4549312.
Additionally, the control TTP-6171 was used for interaction analysis as shown in the
Supplementary Figure S1. Two hydrogen bonds were formed between the ligand and the protein with Thr
5 and Arg
3. Multiple hydrophobic contacts were formed with the residues Val
8, Thr
18, Phe
133, Ala
361 and Leu
360.
3.7. Molecular Dynamics Simulation
Molecular dynamics (MD) simulation was used in this investigation to ascertain the protein–ligand binding interaction strength and to draw conclusions. The root mean square deviation (RMSD) and root mean square fluctuation (RMSF) for the MD trajectory provide essential information on the system’s degree of adaptability in protein–ligand complexes. In the last stage of this process, the binding site residues were broken down into their parts and examined individually for how they interacted with the top three hit compounds.
3.7.1. Root Mean Square Deviation (RMSD)
The highest-ranking ligands identified from molecular docking were evaluated for their binding stability to the MPXV core cysteine proteinase. The degree of conformational change induced by the protein–ligand interaction was measured using the root mean square deviation (RMSD) calculation.
Figure S2 shows the RMSD of the protein Cα-atoms and the ligand for the protein–ligand complex of Cluster-1 and CHEMBL4549312. The RMSD of the protein Cα-atoms was stable at the initial conformation until 0.15 ns and increased to 0.45 ns, where it fluctuated between the ranges of 0.4 nm to 0.5 nm. However, the RMSD of the ligand (calculated concerning protein) CHEMBL4549312 showed high fluctuations in the latter half of the 100 ns MD simulation. It was observed that, for the first 50 ns, the RMSD of the ligand was <0.5 nm; however, it increased with a jump to 1 nm and heavily fluctuated between 0.4 nm and 1 nm for the rest of the simulation. The analysis showed that the ligand CHEMBL4549312 exhibited an unstable binding towards the protein MPXV core cysteine proteinase and, hence, it was excluded from further experimental investigation. The structural conformation was impeded by its voluminous dimensions, thereby hindering the attainment of its optimal stability and leading to a limited duration of persistence.
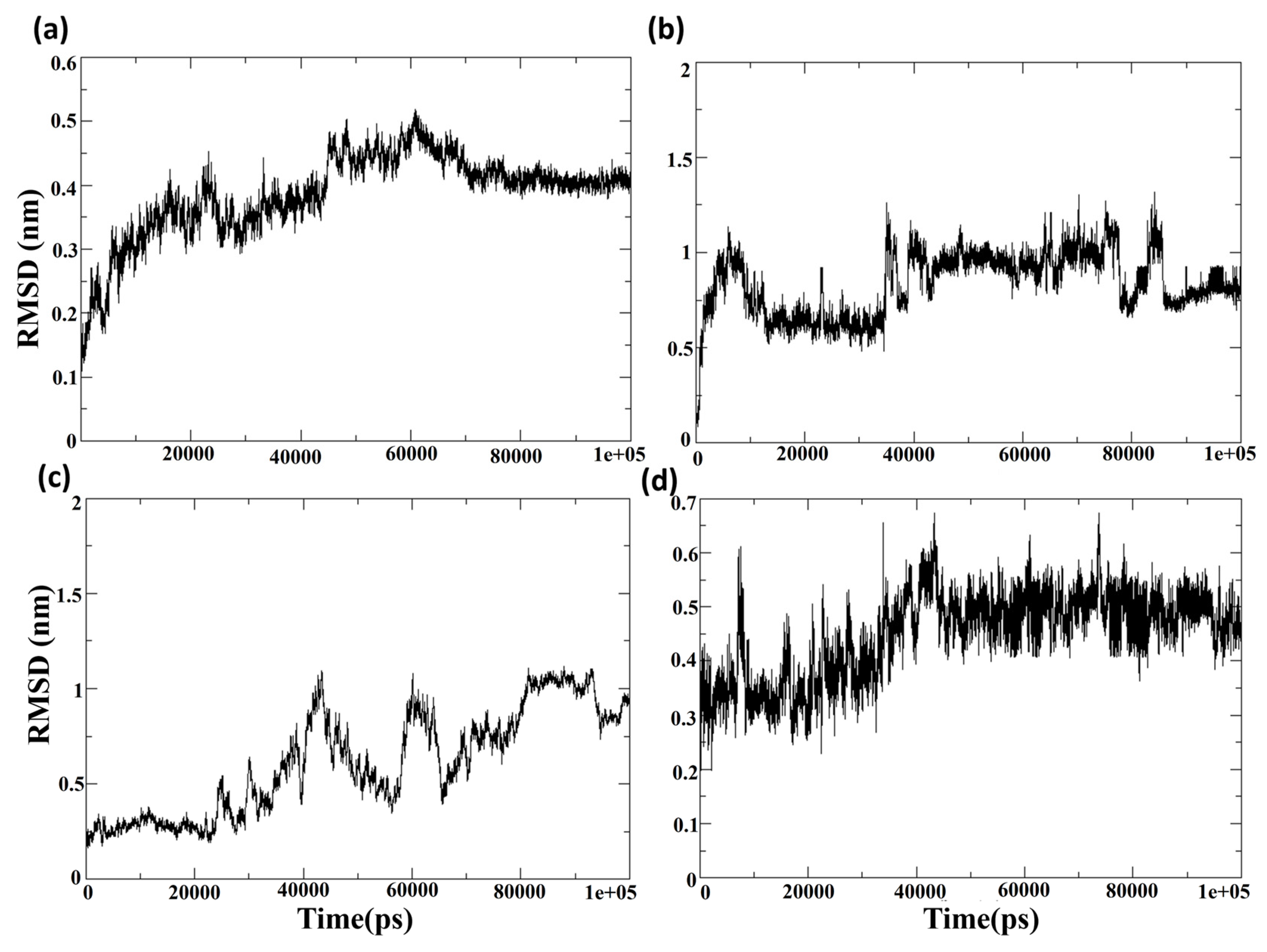
Figure 6a,c shows the root mean square deviation of the protein Cα atoms when the protein is in the bound state with the compounds CHEMBL32926 and CHEMBL4861364. After ligand binding, the simulation demonstrated cluster-2 and CHEMBL32926 complex protein structure changes. It was observed in
Figure 6a that the RMSD of the protein Cα-atoms was 0.15 nm at the initial conformation, while it increased to 0.4 nm to 0.5 nm by the 60 ns simulation timeframe. However, it became stable for the last 30 ns of the simulation, with an RMSD of 0.4 nm. The ligand’s binding can change the protein’s conformation if the ligand binding site is located in a flexible protein region. Moreover, it is noted that the protein structure is the modelled structure and thus could be more prone to conformational changes than the experimental crystal structure. The RMSD of the protein Cα-atoms for the compound CHEMBL4861364 had an RMSD of 0.3 for the first 20 ns, increased to 1 nm at 40 ns, and fluctuated between 0.5 nm and 1 nm for the rest of the simulation, as shown in
Figure 6c. This showed more fluctuation than the Cluster-2 complex, which indicates a higher effect of the binding of the ligand at the given binding site of the protein. Additionally, the RMSD of the protein and ligand was calculated for the control TTP-6171, as shown in the
Supplementary Figure S3. It was observed that the protein had stable RMSD for the majority of the simulation as shown in
Supplementary Figure S3a. Initially, the RMSD of the protein was 0.2 nm which gradually increased to 0.4 nm and with dip decrease to 0.2 nm at 40 ns. After 40 ns, the RMSD of the protein was consistent with 0.4 nm for the rest of the simulation. The RMSD of the ligand showed high fluctuation for the first 60 ns simulation as shown in
Figure S3b. It increased from 0.5 nm to 1 nm and eventually to 1.5 nm till 60 ns; however, post 60 it decreased to 1 nm and stayed stable and consistent with no major fluctuations for the rest of the simulation. This indicated that the ligand TTP-6171 (control) changed its conformation; however, it had a stable binding for the last 20 ns simulation.
Later, the RMSD of the ligand showed that the compounds CHEMBL32926 and CHEMBL4861364 were stable compared to the compound CHEMBL4549312.
Figure 6b showed a high increase in the RMSD at the 5 ns, 40 ns and 85 ns simulation with RMSD of >1 nm; however, the RMSD was consistent for maximum frames of the 100 ns simulation. This indicated that the compound CHEMBL32926 bound to the protein deviated from its initial docked conformation at the 5 ns, 40 ns and 85 ns simulation, as shown in
Figure 6b. However, it stabilized in patches during the simulation. The RMSD of the ligand CHEMBL4861364 had a relatively high stability and consistency in RMSD for the 100 ns simulation, as shown in
Figure 6d. It was observed that the RMSD was 0.3 nm to 0.4 nm for the first 25 ns, where it increased gradually to 0.6 nm, which then stabilized at 0.5 nm for the remainder of the simulation. However, the compound CHEMBL4861364 showed a high jump in RMSD at the 10 ns, 45 ns and 70 ns, where it deviated from its initial conformation, as shown in
Figure 6d. Similarly, the control was analyzed at the 60 ns, 70 ns, 80 ns, and 100 ns to validate the change in conformation of the ligand to stabilize its binding with the protein.
3.7.2. Interaction Analysis (MD)
The RMSD plots showed some deviations shown by the ligands CHEMBL32926 and CHEMBL32926 during the 100 ns simulation. The conformations at the 5 ns, 40 ns, and 85 ns timeframes of the simulation showed high deviation from the initial state for the compound CHEMBL32926 bound to the protein, while the 10 ns, 45 ns, and 70 ns timeframes also showed deviation for the compound CHEMBL4861364 bound to the protein. Thus, these conformations were extracted and compared to the initial states of the protein–ligand complex of Cluster-2 and CHEMBL32926 and Cluster-3 and CHEMBL4861364, respectively.
Figure 7 and
Figure 8 show the 3D conformations and the interactions of the complexes at the deviated state, along with the initial states of the complexes for CHEMBL32926 and CHEMBL32926, respectively.
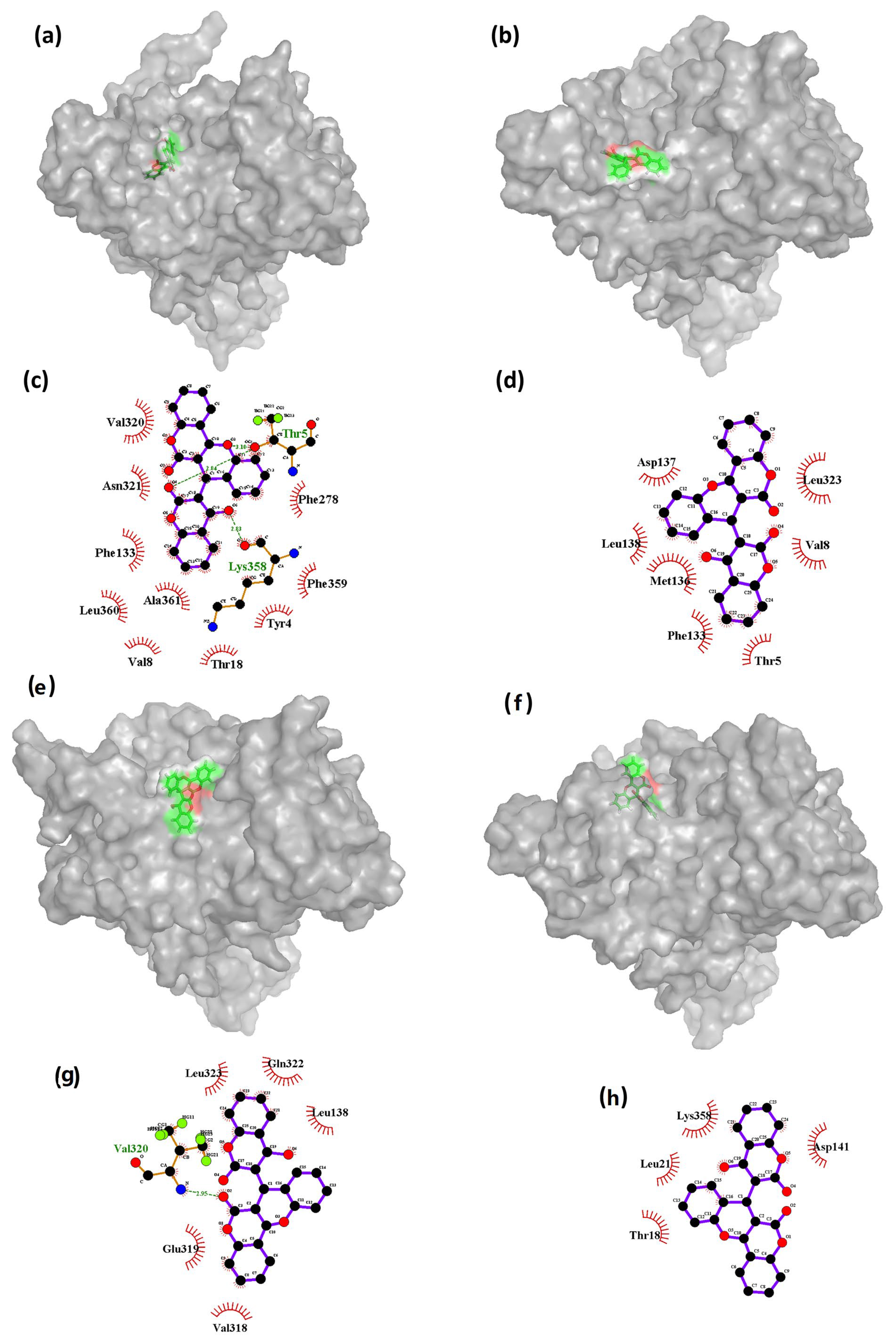
The results presented in
Figure 7 suggest that the binding interactions between the ligand CHEMBL32926 and Cluster-2 vary throughout the molecular dynamics (MD) simulation. Specifically, it was observed that the initial phase of the simulation had the highest number of bonds formed between the ligand and the protein, which gradually decreased at the 5 ns timeframe and then increased again at the 40 ns timeframe. This behavior is likely due to the changing conformations of both the protein and the ligand throughout the entirety of the simulation.
Figure 7c shows that the initial state had two hydrogen bond interactions with residues Thr
5 and Lys
358. However, as the conformation of the ligand changed (
Figure 7b), the ligand interactions cut down at the 5 ns timeframe, and there were no hydrogen bond interactions with either of these residues at 5 ns or 85 ns (as shown in
Figure 7d,h). At 40 ns, only one hydrogen bond interaction was observed with residue Val320 (
Figure 7g). These findings suggest that the binding interactions between the ligand and the protein are highly dependent on the conformational changes that occur in both the protein and the ligand throughout the MD simulation. The decrease in hydrogen bond interactions at 5 ns and 85 ns may indicate that the ligand has adopted less favorable conformations for binding to the protein. However, the increase in interactions at 40 ns suggests that the ligand may have adopted a more favorable conformation that is conducive to binding.
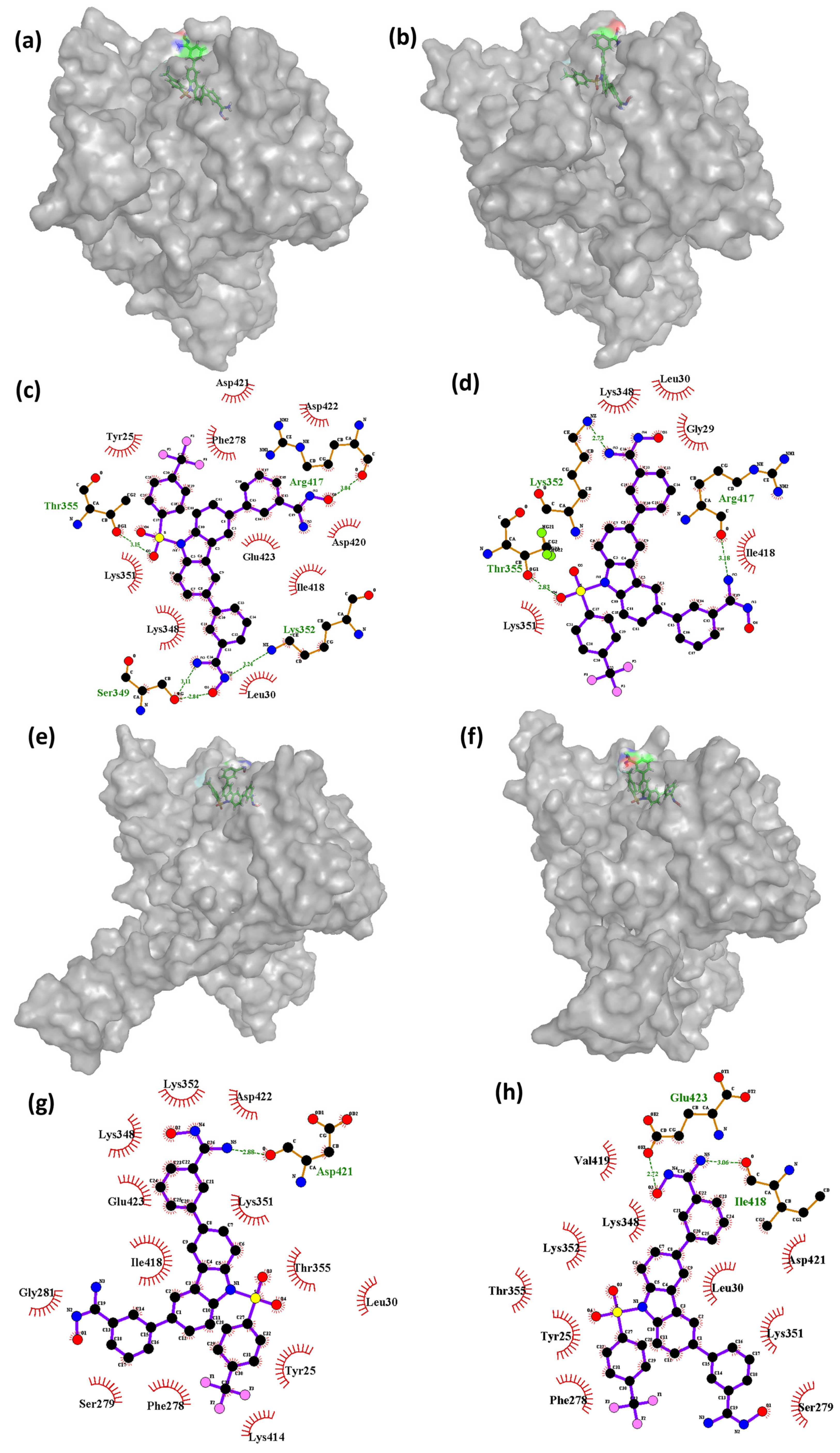
Further analysis may be necessary to determine the specific factors contributing to these observations. The findings indicate that, during the simulation, the complex of Cluster-3 and CHEMBL4861364 had a variable number of hydrogen bond interactions with the protein residues. At 0 ns, the complex formed four hydrogen bonds with residues Ser349, Thr355, Lys352, and Arg417. However, as the simulation progressed, the number of hydrogen bond interactions decreased, with only one hydrogen bond interaction observed at the 45 ns timeframe with residue Asp421. At the 70 ns timeframe, the compound showed two hydrogen bonds with the protein residues Glu423 and Ile418. The decrease in the number of hydrogen bond interactions was found to be correlated with changes in the conformation of the protein. Interestingly, the compound CHEMBL4861364 showed greater hydrogen bond interactions than the compound CHEMBL32926, which may explain why CHEMBL4861364 showed better performance overall, as observed in the RMSD fluctuation and molecular docking results. These findings suggest that the specific chemical structure of CHEMBL4861364 may be more favorable for binding to the protein and forming hydrogen bonds with key residues, resulting in a more stable and effective complex formation during the simulation.
Supplementary Figure S4 shows the conformations of the ligand TTP-6171 (control) bound to the protein at different timeframes during the MD simulation. This also includes the interactions formed between the protein and the ligand during the MD simulation. As shown in the
Figure S4a, the control at the 60 ns timeframe was bound at the cleavage region of the protein; however, over the course for the next 10 ns, the conformation of the ligand changed. At 70 ns, TTP-6171 shifted to a different region of the protein, still bound to it as shown in the
Figure S4c. Post-70 ns, the ligand moved to the inner cavity of the protein and became stable for the rest of the simulation as shown at the 80 ns, 90 ns, and 100 ns (
Figure S4e,g,i). Similarly, the interaction analysis showed that the ligand bound to the protein during the 60 ns and 70 ns had no interaction however, at the 80 ns and 90 ns timeframe, a single hydrogen bond was formed between the ligand and the protein, with the residue Glu14 indicating stable binding with the protein.
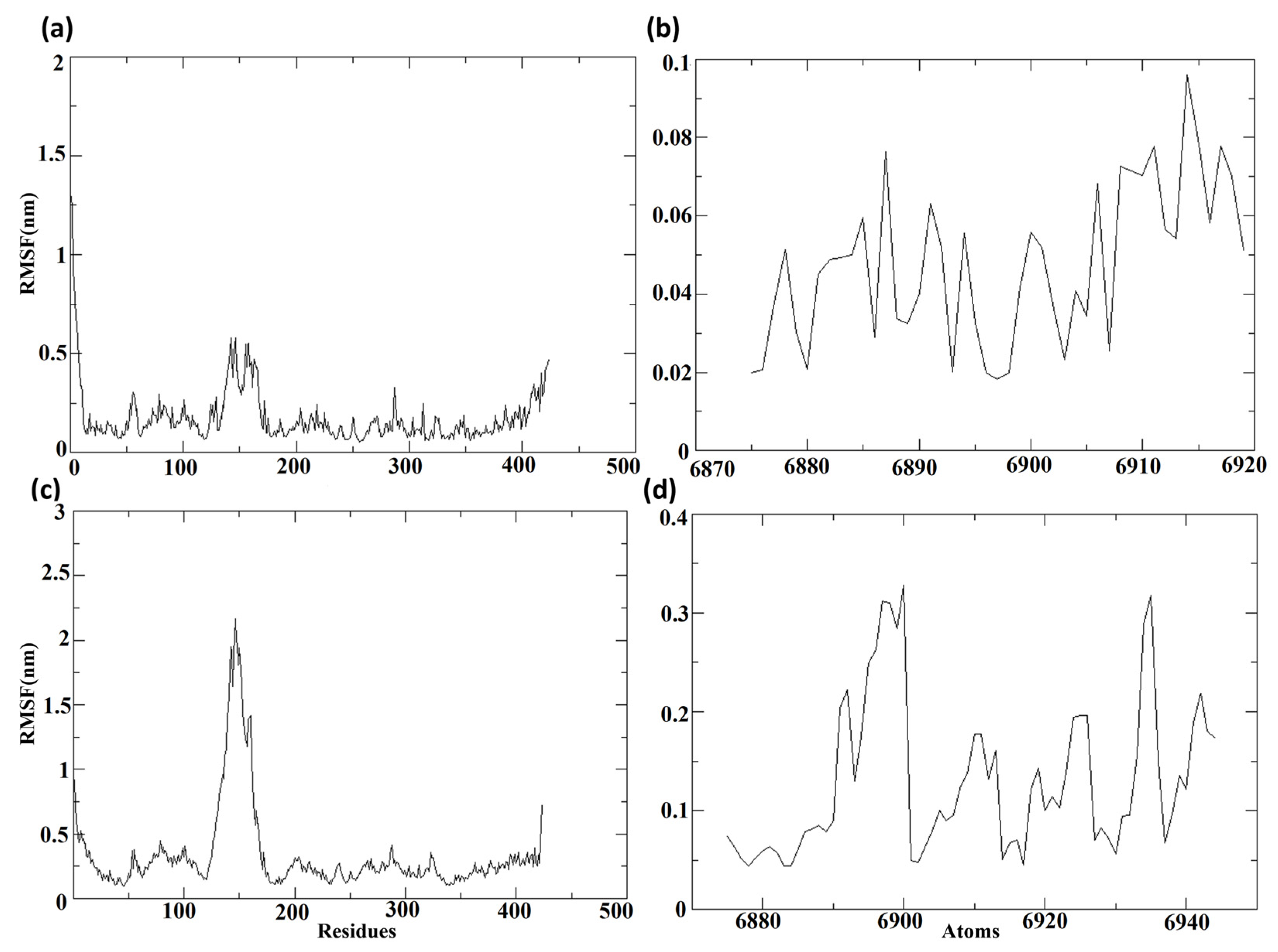
3.7.3. Root Mean Square Fluctuation (RMSF)
The root mean square fluctuations, or RMSF values, were computed regarding the protein and the ligand molecules following their binding, to determine the individual fluctuations of each residue or atom in the bound state.
Figure 9 depicts the RMSF of the protein and ligand for complexes composed of Cluster-2 and CHEMBL32926, and Cluster-3 and CHEMBL4861364, respectively.
Figure 9a,c displays the RMSF of the protein MPXV cysteine proteinase from the protein–ligand complexes of the selected compounds. Throughout the molecular dynamics (MD) simulation, the RMSF of proteins displayed comparable lower fluctuations, with RMSF values of less than 0.3 nm in both complexes. Nonetheless, a major peak was observed in the protein bound to CHEMBL32926, with an RMSF of 0.5 nm and 51 residues showing RMSF values of over 0.3 nm. Additionally, the protein bound to the compound CHEMBL4861364 exhibited greater RMSF fluctuations.
Figure 9b,d shows the RMSF of the ligands, the chosen compounds CHEMBL32926 and CHEMBL4861364. The protein–ligand complex of CHEMBL32926 had no atoms with an RMSF of over 0.3 nm, while the compound CHEMBL4861364 had four residues with an RMSF of over 0.3 nm. Both compounds exhibited lower fluctuations, indicating the potential for stable binding to the MPXV protein.
Supplementary Figure S5 showed the RMSF of the protein and ligand for TTP-6171 (control). It was observed that the protein had 40 residues showing RMSF values over 0.3 nm among them five residues had RMSF over 0.8 nm. The RMSF of the ligand was less than 0.4 nm during the MD simulation. Here, the RMSF of the control was comparable with the compounds with a high peak for the protein, as shown in
Figure S5a.
3.8. MM/GBSA Analysis
Protein–ligand complexes for compounds CHEMBL32926 and CHEMBL4861364 showed deviation at a certain timeframe of the simulations (as discussed in RMSD). These conformations were extracted, and their MMGBSA binding energies were determined to evaluate the binding of the extreme poses. The total free binding energy for the top two hits, which are listed in
Table 4, was computed with the help of the tool known as the gmx MMPBSA test [
49]. It was observed that the compound CHEMBL4861364 had a stronger total binding free energy than the compound CHEMBL32926. The initial docked complex of Cluster-2 with CHEMBL32926 and Cluster-3 with CHEMBL4861364 had positive total binding free energy. However, the other conformations at 5 ns, 40 ns, and 85 ns were 19.25 kcal/mol, 13.57 kcal/mol, and −9.65 kcal/mol, respectively, for CHEMBL32926. It was observed that the conformations at 10 ns, 45 ns, and 70 ns showed better total binding free energy for the complex Cluster-3 and CHEMBL4861364. The conformation at 45 ns had the best total binding free energy of −43.24 kcal/mol, while the complex at the 70 ns timeframe had −41.66 kcal/mol, and the 10 ns complex had −31.47 kcal/mol. This indicated that the compound CHEMBL4861364 had relatively better binding to the protein MPXV cysteine proteinase, compared to the compound CHEMBL32926. The magnitude of MMGBSA energy for CHEMBL4861364 was 3-fold compared to CHEMBL32926. This was demonstrated by more hydrogen bonds between the ligand and the protein during the simulation of the protein–ligand complex. These findings suggest that CHEMBL4861364 has a stronger binding affinity and inhibitory potential against MPXV cysteine proteinase and could be a promising candidate for experimental testing. Additionally, the control had total free binding energy of −32.30 kcal/mol indicating that the compound CHEMBL4861364 had better binding than the control, TTP-6171.
In a previous investigation, the natural ligand TTP-6171 was docked with the protein and its complex was simulated for 100 ns. This study reported ΔGTotal = −53.86 kcal/mol, which is very compatible with the binding free energy showed by CHEMBL486136453 [
60].
3.9. IC50 Prediction and Analogue Search
Molecular docking and dynamic simulation techniques were employed to identify two potential hit compounds against a target protein. Subsequently, DeepPurpose, a machine learning-based activity prediction algorithm, was used to validate the binding scores of these compounds. Pretrained models were utilized in this analysis, and five distinct encoding method pairs were used for the protein and ligand. The corresponding encoding methods for these models are listed in
Table 5. The models use the protein sequence and SMILES to predict their binding affinities. Six encoding techniques were employed: CNN, Morgan, AAC, MPNN, Daylight, and AAC. It is worth noting that Morgan and Daylight methods were used specifically for the ligand compound, while AAC was used exclusively for the protein sequence.
Five different models were employed to predict the binding affinity of the cysteine proteinase amino acid sequence and SMILES of CHEMBL32926 and CHEMBL4861364. These models utilized the IC
50 concentration required to inhibit the protein’s function by 50% as an indicator of binding affinity, where a lower binding score (IC
50) indicates a higher affinity.
Table 6 shows the binding scores that each model predicted for the compounds CHEMBL32926 and CHEMBL4861364. Subsequently, all individual binding scores were aggregated, and an average score was calculated, which is referred to as the final binding score (shown in the last row of
Table 6).
Table 6 indicates that both compounds exhibit comparable predicted IC
50 values, which strongly implies their potential binding affinity for the target protein due to the low IC
50 observed.
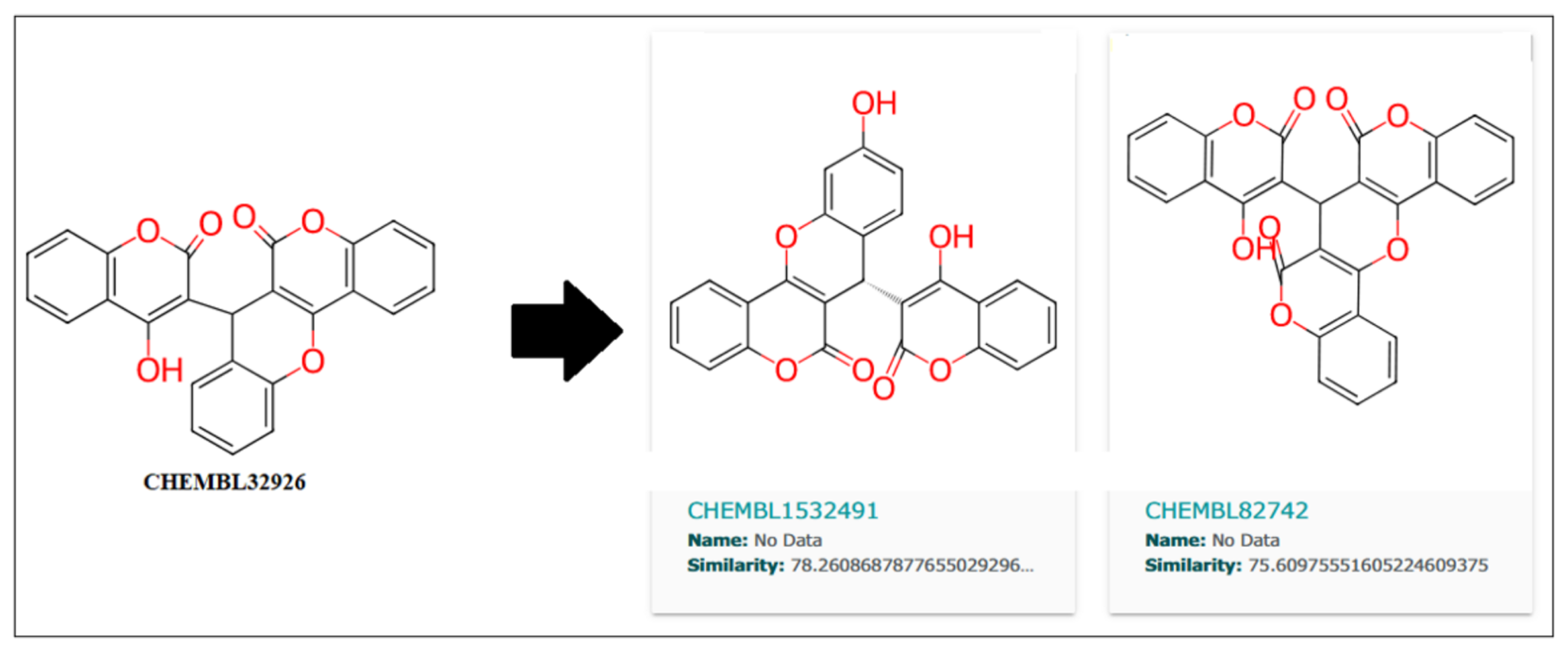
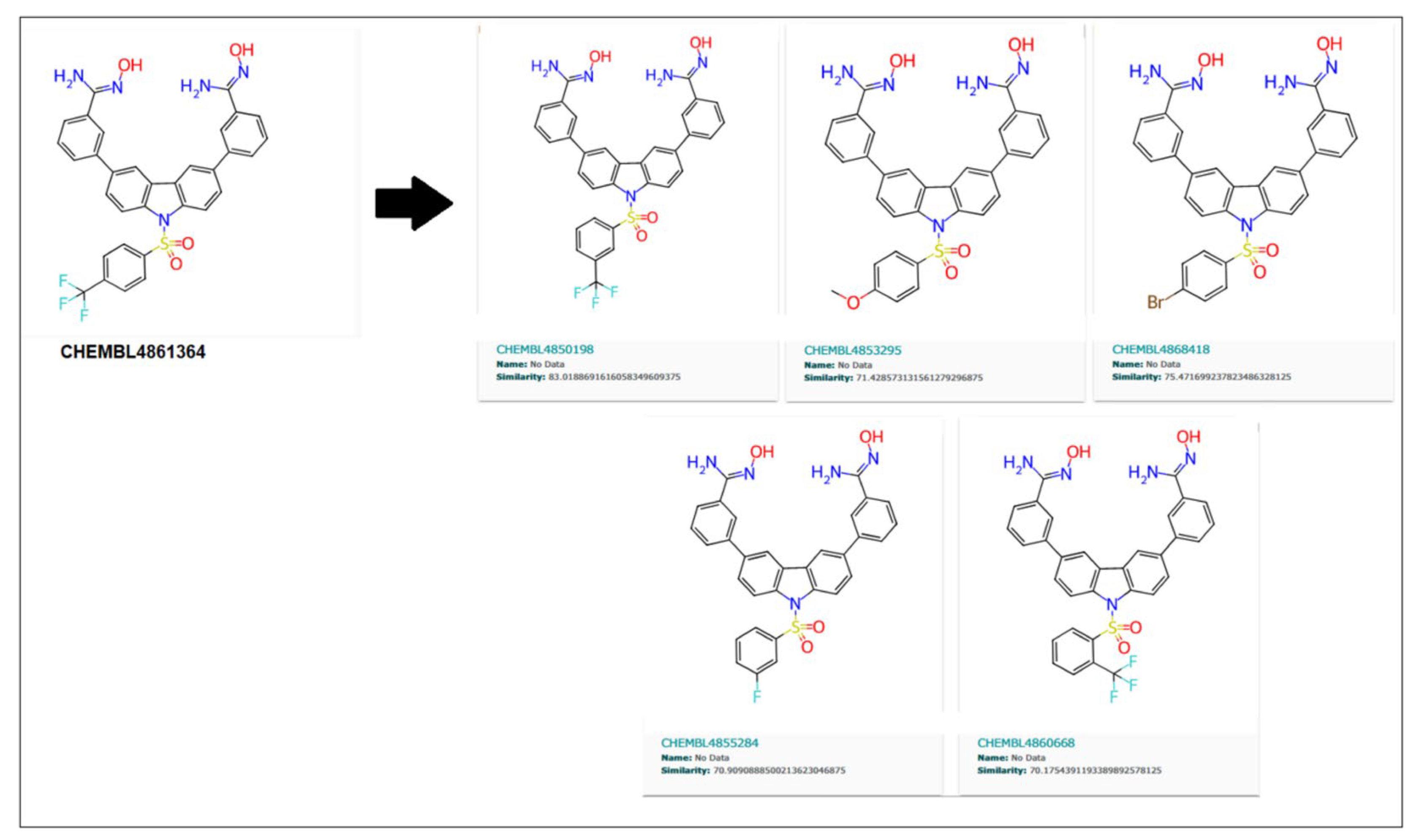
Further, the close analogs for these compounds were searched in the ChemBL database with a 70% similarity threshold. These analogs are shown in
Figure 10 and
Figure 11 for compounds CHEMBL32926 and CHEMBL4861364. Two similar analogs for CHEMBL32926 were identified with 78.26% and 75.60% similarity, as shown in
Figure 10.
Similarly, 70% of similar structures were searched for the CHEMBL4861364 compound, resulting in five analogs, as shown in
Figure 11. These are 83.01%, 71.42%, 75.47%, 70.9%, and 70.17%, similar to the parent compound.
Later, these similar compounds were tested using the five DeepPurpose ML models to predict their IC
50.
Table 7 shows the average IC
50 of the two analogues of CHEMBL32926 and the five analogues of CHEMBL4861364.
As shown in
Table 7, CHEMBL1532491 showed a better IC
50 of 5.22 nM than its parent compound (CHEMBL32926), while CHEMBL4850198 was the best analogue detected for CHEMBL4861364, with an IC
50 of 5.84 nM. Moreover, the other two analogs, CHEMBL82742 and CHEMBL4868418, also showed high binding with IC
50s of 5.97 nM and 5.99 nM for the target protein, as detected by the ML models. Analog findings added four more compounds with similar structures and binding to the target protein.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}