1. Introduction
Sepsis, defined as “life-threatening organ dysfunction due to a dysregulated host response to infection”, is a common condition with high morbidity and mortality rates worldwide [
1]. In sepsis, dysregulated inflammation and oxidative stress (OS) can rapidly lead to multiorgan failure, primarily affecting the cardiovascular and respiratory systems [
2,
3,
4]. The lung is the most vulnerable organ in sepsis, and lung injury is initially characterized by vascular fluid leakage through paracellular pathways, clinically manifesting as acute respiratory distress syndrome (ARDS). Sepsis causes an imbalance between the levels of reactive oxygen species (ROS) and antioxidant agents in endothelial cells (ECs), which, in addition to mitochondrial dysfunction, exacerbates the dysfunction of ECs in multiple ways, such as glycocalyx breakdown, EC apoptosis, and junction protein remodeling [
5]. Since ECs serve as both a source and a target of oxidative stress during sepsis, severe vascular leakage caused by endothelial injury is currently regarded as one of the most important pathophysiological changes during septic lung injury [
6].
Under physiological and many pathophysiological conditions, mitochondria are the primary site of ROS production from aerobic respiration [
7]. Mitochondrial dysfunction is currently regarded as a significant factor in sepsis-induced organ failure, potentially impairing cellular energy supply and increasing OS [
8]. In sepsis, autophagy, a catabolic process that recycles cellular components and damaged organelles in response to various stressors such as oxidative stress, is commonly viewed as a protective adaptive mechanism that limits cellular damage and apoptosis [
9,
10,
11]. Mitophagy is the specific autophagic response that targets and removes cytotoxic mitochondria to avoid further damage [
8,
12]. Upon mitochondrial damage and reduced membrane potential (Δψm) caused by ROS overproduction, the mitochondrial kinase PINK1 and the E3 ubiquitin ligase Parkin cooperate to ubiquitinate outer membrane components, initiating autophagosome formation through the adaptor protein p62-mediated LC3 binding [
12,
13,
14]. Among the adaptor proteins, p62 plays a crucial role not only in mitophagy, but also in impeding the Keap1-mediated degradation of Nrf2 by directly engaging with Keap1 [
15,
16,
17,
18]. The activation of the antioxidant Keap1–Nrf2–ARE pathway is vital for maintaining mitochondrial structural and functional integrity under stress, as it regulates antioxidant response element (ARE)-dependent genes, including SQSTM1 (p62), and establishes a p62–Keap1–Nrf2 positive feedback axis for persistent Nrf2 activation and p62 replenishment [
19,
20].
Long noncoding RNAs (lncRNAs) play an important role in regulating vascular homeostasis, as previously demonstrated [
21]. The lncRNA, growth arrest-specific 5 (GAS5), has increasingly been recognized as a key modulator in sepsis-induced organ injury. In specific instances such as lipopolysaccharide (LPS)-induced myocardial cell damage, GAS5 has demonstrated potential in attenuating myocardial cell apoptosis, indicating a protective role [
22]. Nonetheless, the landscape of the available literature also includes a study positing that GAS5 may amplify myocardial injury and an inflammatory response under septic conditions [
23], which underscores the complexity of its role. In relation to septic kidney injury, emerging data suggest that GAS5 may serve as a protective mechanism, potentially through its ability to reduce cell pyroptosis [
24]. Additionally, GAS5 has been implicated in mitigating inflammatory response and apoptosis in both alveolar and bronchial epithelial cells [
25,
26]. This illustrates another dimension of GAS5’s role in organ-specific responses to sepsis. While the role of GAS5 in sepsis-induced organ injury has been increasingly acknowledged, the existing literature seems to slightly overlook the exploration of the upstream regulatory mechanisms that induce GAS5 expression. A specific area that requires additional attention is the impact of GAS5 on vascular endothelial cells under septic conditions. Considering the lung as a primary organ susceptible to sepsis, and vascular leakage being one of its principal injury mechanisms, this indeed highlights the necessity for our intended research in this specific area. Our study shows a significant upregulation of lncRNA GAS5 in the vascular endothelium of septic mice, but the exact mechanism remains unclear. Upon reviewing published studies, we discovered that the expression of GAS5 can be induced by oxidized low-density lipoprotein (Ox-LDL) and the autophagy agonist rapamycin [
27,
28,
29,
30,
31]. Rapamycin works as an autophagy agonist by inhibiting the function of mTOR complex 1 (mTORC1), which is composed of the mechanical target of rapamycin (mTOR) and multiple related proteins such as Raptor, PRAS40, and DEPTOR. Among them, mTOR is a member of the PI3K-related kinase family and functions as a redox sensor [
32,
33,
34]. When mTOR is dephosphorylated, the function of mTORC1 is inhibited, rendering mTORC1 incapable of preventing the nuclear localization of the transcription factor TFEB [
35], a member of the MiT–TFE family (TFEB, TFEC, TFE3, and MITF), which is an important regulator of lysosome biogenesis and autophagy [
36].
In this study, we found that GAS5 expression is regulated by the MiT–TFE family of transcription factors. Our examination of the effect of GAS5 revealed that it can inhibit oxidative stress and maintain mitochondrial homeostasis, ultimately leading to the attenuation of lung injury in sepsis. Through the inhibition of the MAPK and PKC signaling pathways, DGKE relieves the proinflammatory and prothrombotic state of ECs [
37,
38], and we demonstrated that the anti-inflammatory effect of GAS5 is achieved through the GAS5–miR-23–DGKE axis. Overall, GAS5, functioning as an antioxidant and anti-inflammatory lncRNA, may provide a promising target in sepsis.
2. Materials and Methods
2.1. Animals
All experiments were conducted on male C57BL/6J mice that were 8 to 10 weeks old and weighed between 24 and 27 g. The mice were obtained from the animal center of Chongqing Medical University. This research followed the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. The protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at The Second Affiliated Hospital of Chongqing Medical University. Prior to conducting the study, ethical approval was received.
2.2. In Vivo AAV–ENT-Mediated Endothelial-Specific Gene Overexpression
In vivo endothelial-specific gene expression was achieved through ENT serotype-associated adeno-associated viral (AAV) vectors. Recombinant AAV–ENT vectors carrying GAS5, MITF, DGKE, mmu–miR-23a, a GAS5–shRNA sequence, or an empty vector with a plasmid that had an intercellular adhesion molecule 2 (ICAM2p) promoter were manufactured by the Shanghai GeneChem Company Limited. The virus titer was 1012 vg/mL. AAV–ENT–ICAM2p–empty served as a negative control. AAV–ENT–ICAM2p–target gene/empty vector was delivered via tail vein injection (1 × 1011 vector genome/mice) combined with an intratracheal injection (5 × 1010 vector genome/mice) to achieve pulmonary and systemic vascular endothelium gene transduction. The C57BL/6J mice were placed in a gas anesthesia device and given oxygen and 2.5% isoflurane to induce anesthesia. After confirming that the effect of anesthesia was reliable, the skin of the anterior cervical region of the mice was incised to expose the trachea. Using an insulin syringe, 50 µL of the diluted virus was withdrawn, the needle was inserted into the trachea at a 45-degree angle, the virus was slowly injected, and then the needle was pulled out. Next, an insulin syringe was used to inject 100 µL of the diluted virus into the tail vein of the mice. During the whole operation, the anesthesia system was connected to the noses of the mice to maintain anesthesia. After the operation, the mice were placed in a warm environment while they recovered. Three weeks after adeno-associated virus infection, gene expression was detected, followed by sepsis modeling.
2.3. Cecal Ligation and Puncture (CLP) Sepsis Model
Before model induction, all mice were fasted for 8 h, but allowed to consume water ad libitum. The CLP model was constructed with modifications based on a previously published description. Briefly, mice were fully anesthetized by isoflurane inhalation, fixed on an aseptic operating table, and a 1 cm incision was made in the mid-abdomen to expose the cecum. The external two-thirds of the cecum were ligated and punctured once with a 19-gauge needle. After isolating a small amount of feces from the puncture site, the cecum was returned, and the abdominal incision was closed with sterile 6–0 silk sutures. Except for the ligation and perforation of the cecum, sham-operated animals were subjected to the same surgical procedure. All mice received subcutaneous fluid resuscitation with normal saline (50 mL/kg) at 37 °C after surgery.
2.4. Cell Lines and Cultures
Human umbilical vein endothelial cells (HUVECs), HEK-293T cells, and THP-1 cells were obtained from PROCELL Biotechnology (Wuhan, China). HUVECs were cultured in RPMI 1640 medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. HEK-293T cells were cultured in DMEM (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. THP-1 cells were cultured in RPMI 1640 medium with 12% fetal bovine serum, 1% penicillin-streptomycin, and 50 µmol/L β-mercaptoethanol. Throughout the experiments, the cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2 and in the exponential growth phase.
2.5. Cell Transfection
Small interfering RNA (siRNA) for GAS5, MITF, TFE3, TFEB, Raptor, DGKE, and its negative controls, as well as miR-23a mimics, miR-23b mimics, mimic NC, miR-23a inhibitor, miR-23b inhibitor, and inhibitor NC, were supplied by GenePharma (Shanghai, China). Overexpression plasmids for GAS5, MITF, TFE3, TFEB, and DGKE were supplied by GeneCreate Biological Engineering Co. (Wuhan, China), and the plasmid pcDNA-3.1 was used as a negative control. All transfections were carried out using Lipofectamine 3000 transfection reagent according to the manufacturer’s protocol (Thermo Fisher, Waltham, MA, USA, L3000075).
2.6. RNA Isolation and Quantitative Real-Time PCR
HUVECs were treated with TRIzol reagent (Thermo Fisher, Waltham, MA, USA, 15596026) to extract total RNA. Total RNA isolated from cells was subjected to reverse transcription using a PrimeScript RT Master Mix Kit (TaKaRa, Kyoto, Japan, RR036), and microRNA isolated from cells was subjected to reverse transcription using a Mir-X miRNA First-Strand Synthesis Kit (TaKaRa, Kyoto, Japan, 638315). Relative quantification was performed using 2xSYBR Green qPCR Master Mix (Bimake, Shanghai, China, B21203), and the primers were obtained from Sangon Biotech (Shanghai, China). Quantitative real-time PCR was used to determine the levels of mRNA expression using a Bio-Rad iCycler system (Bio-Rad, Hercules, CA, USA).
2.7. Western Blot
RIPA lysis buffer was used to lyse and extract total proteins from HUVECs (Beyotime, Shanghai, China, P0013B). Following quantification with a BCA Protein Assay Kit (Beyotime, Shanghai, China, P0010S), protein samples were separated using SDS-PAGE on a 10% or 12.5% acrylamide gel (EpiZyme, Shanghai, China). After that, the proteins were transferred to polyvinylidene fluoride (PVDF) membranes and blocked for 1 h with 5% BSA. Following overnight incubation with primary antibodies at 4 °C, the membranes were incubated for 1 h at room temperature with HRP-labeled secondary antibodies. The membrane was incubated with a substrate for enhanced chemiluminescence (ECL) (EpiZyme, Shanghai, China).
2.8. Cellular Immunofluorescence Staining
After 20 min of paraformaldehyde fixation, the samples were washed three times with PBS. The cells were then permeabilized for 10 min with 0.5% Triton X-100, washed three times with PBS, and blocked with goat serum for 1 h. Cellular samples were incubated with the indicated antibodies overnight at 4 °C, washed with PBS three times, and incubated with corresponding secondary antibodies for 1 h at 37 °C. Then, the samples were rinsed in PBS, counterstained with diamidino phenylindole (DAPI) (Beyotime, Shanghai, China, P0131), and mounted with anti-fading reagent (Solarbio, Shanghai, China). Representative images were visualized under a confocal microscope (Nikon A1R, Tokyo, Japan).
2.9. Immunofluorescence Staining of Paraffin Sections
Following quick fixation in 4% paraformaldehyde solution for 24 h, the samples were subjected to gradient dehydration (from 50% to 100% ethanol). After that, the samples were cleared with xylene, fixed in paraffin, and cut into 4 µm thick sections to mount onto slides. After deparaffinization and rehydration, the slides were warmed to 60 °C for 1 h in a dry oven to soften the paraffin. For ten minutes each, the slides were washed in xylene I, xylene II, 100% ethanol, 95% ethanol, 90% ethanol, and 80% ethanol. After washing the slides with deionized water for 5 min, they were heated in an oven at 95–100 °C for 20 min in an antigen retrieval solution. The sections were removed from the oven after antigen retrieval and allowed to cool at room temperature. After three washes with PBS, the tissue the sections were covered with goat serum and incubated at room temperature for 1 h. After blocking, the samples were incubated with the indicated antibodies overnight at 4 °C, washed with PBS three times, and incubated with corresponding secondary antibodies for 1 h at room temperature. After washing with PBS three times, the samples were counterstained with DAPI (Beyotime, Shanghai, P0131) and mounted with anti-fading reagent (Solarbio, China). Representative images were photographed using a confocal microscope (Nikon A1R, Tokyo, Japan).
2.10. Tissue Reactive Oxygen Species Detection
Frozen sections were thawed at room temperature, and a histochemical pen was used to draw a circle around the tissue. An autofluorescence quencher was added to the area for 5 min, and then the tissue was rinsed with running water for 10 min. ROS staining solution (SIGMA, Darmstadt, Germany, D7008 1:500) was added dropwise to the circular tissue area and incubated in a dark incubator at 37 °C for 30 min. The nuclei were counterstained with DAPI, and the slides were washed three times in PBS (pH 7.4) while shaking on a destaining shaker for 5 min each time. After the sections were slightly dried, DAPI staining solution was added dropwise to the circular area and incubated at room temperature for 10 min in the dark. The slides were washed three times in PBS (pH 7.4) while shaking on a destaining shaker for 5 min each time. After drying, the sections were mounted with anti-fluorescence quenchers.
2.11. Immunochemistry (IHC)
Paraffin sections of the largest cross-sections of lung tissue were deparaffinized, hydrated and incubated with 3% H2O2 for 5–10 min at room temperature to eliminate endogenous peroxidase activity. After washing the sections with PBS, antigen retrieval was performed, and then the specimens were blocked with 5–10% goat serum (diluted in PBS). After 10 min of incubation at room temperature, the serum was removed, and the working concentrations of anti-NRF2, p62, Ly6g, ICAM, and VCAM primary antibody solutions were added dropwise. The sections were incubated with the primary antibodies overnight at 4 °C. After washing with PBS, an appropriate amount of biotin-labeled secondary antibody working solution (Servicebio, Wuhan, China) was added dropwise and incubated at 37 °C for 30 min. The slides were rinsed with PBS again, an appropriate amount of a horseradish peroxidase-labeled streptavidin working solution was added dropwise, and the slides were incubated at 37 °C for 10–30 min. After rinsing with PBS, the color developing agent (DAB) (Servicebio, Wuhan, China) was added and the sections were incubated 3–15 min for color development. Then, they were fully rinsed with tap water, counterstained with hematoxylin, routinely dehydrated, cleared, dried, and mounted. Finally, representative images were photographed under an Olympus 600 microscope (Olympus, Tokyo, Japan).
2.12. Histological Analysis
After CLP modelling, mouse lung tissue was fixed in DEPC-treated water-containing 4% paraformaldehyde for 24 h. After paraffin embedding, the largest cross-section of the lung was taken for further tissue sectioning. After sectioning, H&E staining was performed, and the sections were photographed for analysis.
2.13. Fluorescence In Situ Hybridization (FISH) and Colocalization
HUVECs were used for the FISH and colocalization assay. Shanghai GenePharma Biotechnology Co. Ltd. (Shanghai, China) designed and synthesized Cy3-labeled miR-23a-3p and miR-23b-3p probes, as well as fluorescein amidite (FAM)-labeled human-GAS5, MITF, and DGKE probes. The probe signals were detected using the Fluorescent In Situ Hybridization Kit (GenePharma Biotechnology, Shanghai, China), in accordance with the manufacturer’s instructions. During the procedure, miR-23a-3p and miR-23b-3p red fluorescent probes were incubated with GAS5, MITF, and DGKE green fluorescent probes. Paraffin sections of mouse lung tissue were used for the tissue FISH assay. The Cy3-labeled mouse GAS5 probe was designed and synthesized by Shanghai GenePharma Biotechnology Co., Ltd. (Shanghai, China). The probe signal was detected using a paraffin section fluorescence in situ hybridization kit (GenePharma Biotechnology, Shanghai, China), according to the manufacturer’s instructions. All photos were taken with a Nikon A1R Laser Scanning confocal microscope (Nikon A1R, Tokyo, Japan).
2.14. Mitochondrial ROS Detection
According to the manufacturer’s protocol, mitochondrial ROS levels were detected using a MitoSOX™ Red Mitochondrial Superoxide Indicator (Thermo Fisher, Waltham, MA, USA, M36008) and observed under a confocal microscope (Nikon A1R, Tokyo, Japan).
2.15. Dual Luciferase Reporter Gene Assays
PCR was used to amplify the wild-type (Wt) and mutant (Mut) fragments of the MITF, DGKE 3’UTR, and GAS5 sequences containing the predicted miR-23a and miR-23b binding sites. To construct wild-type luciferase reporter gene vectors, the miR-23 target site was PCR-amplified from human genomic DNA, and the DNA fragment was cloned into the XhoI and NotI sites on the 3’ end of the luc2 gene in the REPORT vector. To construct mutant-type luciferase reporter gene vectors, the miR-23 target site was changed by PCR mutagenesis. The Wt-REPORT vector and Mut-REPORT vector were cotransfected into HEK-293T cells along with miR-23a mimics, miR-23b mimics, and negative control mimics using Lipofectamine 3000 reagent (Thermo Fisher, Waltham, MA, USA, L3000075) according to the manufacturer’s protocol. Following 48 h of transfection, the dual-luciferase reporter assay system (Promega, Durham, NC, USA) was used according to the manufacturer’s protocol to determine the luciferase activity of the samples compared with that of the Renilla luciferase positive control.
2.16. Chromatin Immunoprecipitation (ChIP)
ChIP was performed using the ChIP assay kit (Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. DNA and protein crosslinking was achieved by incubating the cells for 20 min at 37 °C in 1% formaldehyde solution. After sonication, chromatin was immunoprecipitated overnight with 6 μg of anti-MITF, TFEB, Nrf2, Flag antibodies, or 2 μg of a normal IgG antibody (CST, USA). The genomic regions of GAS5, SQSTM1, MITF, TFE3, and NFE2L2 containing the Nrf2-, MITF-, TFEB-, and TFE3-binding sites were amplified by RT-PCR in 20 μL volumes for 30–35 cycles to determine the appropriate conditions for the PCR products of each region. Primer sequences and antibodies are described below.
2.17. Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were performed using an EMSA kit (Viagene, Shanghai, China) according to the manufacturer’s instructions. The oligonucleotide probes containing the MITF-binding sequence from the GAS5, SQSTM1, and NFE2L2 promoters were synthesized and labeled with biotin at the 5′ end. The probes were incubated with the nuclear extract at room temperature for 30 min. The entire reaction mixture was run on a nondenaturing 0.5 × TBE 6% polyacrylamide gel for 1 h at 60 V at 4 °C and then transferred onto Biodyne® B nylon membranes (Pall Corporation). Signals were visualized with ChemiDoc XRS (Bio-Rad, Hercules, CA, USA).
2.18. Evaluation of Autophagic Flux
mRFP-GFP-LC3 adenovirus transfection was used to monitor autophagic flux by marking and tracking LC3. The mRFP–GFP–LC3 adenovirus construct was obtained from Hanbio Inc. HUVECs were transfected with mRFP–GFP–LC3 adenovirus for 24 h following the manufacturer’s instructions and then transfected with siRNA or plasmid for 48 h. The cells were then transferred to a confocal dish and treated with H2O2 after the cells adhered. Finally, a confocal microscope was used to observe the cells and acquire images (Nikon A1R, Tokyo, Japan).
2.19. Evaluation of Mitophagy
A mitochondrial green fluorescent probe (Beyotime, Shanghai, C1049) and lysosomal red fluorescent probe (Beyotime, Shanghai, C1046) were used to identify and track mitochondria and lysosomes. Following the manufacturer’s instructions, HUVECs were transfected with siRNA or plasmids for 48 h. HUVECs were labeled with probes and then treated with H2O2. Finally, the cells were observed and images were acquired using a confocal microscope (Nikon A1R, Tokyo, Japan).
2.20. Monocyte Adhesion Assays
HUVECs were transfected with siRNA or plasmids in a 24-well plate, and a confluent monolayer of HUVECs was formed after 48 h. Then, HUVECs were treated with TNF-α for 24 h to express adhesion factors. THP-1 cells were labeled with Calcein AM (Beyotime, Shanghai, C2012) following the manufacturer’s instructions, and then 1 × 105 THP-1 monocytes were added to a 24-well plate and incubated with HUVECs. After 30 min, the cells were washed twice with warm PBS to gently remove nonadherent cells, and then the number of adherent cells was counted using an inverted microscope.
2.21. Flow Cytometry
Apoptosis: The cells were collected and incubated in binding buffer, followed by staining with Annexin V-FITC and PI solution using an Annexin V-FITC Apoptosis Detection kit (Beyotime, Shanghai, C1062S). The apoptotic cells were measured by a flow cytometer, and the apoptotic rate of HUVECs at early and late apoptosis was presented as the percentage of cells with positive Annexin V-FITC staining and negative or positive PI staining.
ROS: Following the treatment, the cells were incubated with DCFH-DA for 20 min at 37 °C and then measured via flow cytometry at 488 nm excitation and 525 nm emission wavelengths.
2.22. Transmission Electron Microscopy Assay
HUVECs were transfected with siRNA or plasmids in a 6-well plate for 48 h. Then, HUVECs were treated with H2O2, and the cells were collected in a 1.5 mL microcentrifuge tube for each sample. After, the cell sample was fixed with 3% glutaraldehyde overnight at 4 °C and washed twice with PBS. Then, all specimens were washed in 4% (w/v) sucrose solution before being transferred to 1% osmium tetroxide solution. All samples were dehydrated in a series of gradients of ethanol and acetone and then embedded in epoxy resin. All samples were then cut into thin slices, stained with uranylacetate and lead citrate, and visualized with a transmission electron microscope (FEI inspect, Corvallis, OR, USA).
2.23. Transendothelial Resistance (TER) Measurement
HUVECs were seeded in a Transwell chamber and then measured with a transmembrane resistance meter (EVOM2, World Precision Instruments, Sarasota, FL, USA) after the cell monolayer was fully confluent. The electrodes were inserted vertically into the Transwell, with the long electrode in the lower chamber and the short electrode in the upper chamber; the value was recorded after stabilization, the measurement was repeated three times, and the average value was taken. The TER value = (measured resistance value − blank resistance value) * transwell effective membrane area.
2.24. Statistical Analysis
Data are presented as the mean ± standard deviation. Independent sample t-tests were performed to compare the differences using GraphPad Prism 8.0 software (GraphPad, San Diego, CA, USA). p-values < 0.05 indicated statistical significance.
4. Discussion
Based on the known biological mechanisms of Ox-LDL, H2O2, and rapamycin, we hypothesized that autophagy-associated transcription factors activated under oxidative stress contribute to GAS5 transcription. While existing knowledge highlights the role of the redox–sensor mTORC1 complex in regulating TFEB—a member of the MiT-TFE family—in response to ROS stimulation [
36], our investigations suggested that TFEB silencing had minimal influence on GAS5 expression in endothelial cells (ECs). Analysis from sequencing data showed that expression levels of TFEB and TFEC were comparatively lower in ECs than MITF and TFE3, suggesting that the latter were the primary contributors to GAS5 transcription.
During sepsis, mitochondria, as the main source of intracellular ROS, are particularly vulnerable to active molecules released by activated immune cells [
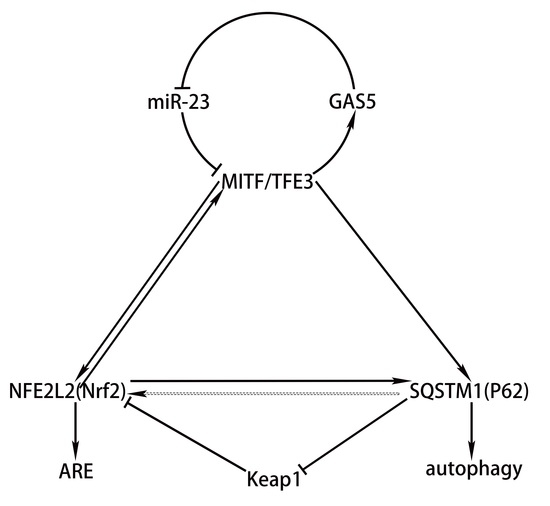
39]. We observed that GAS5 upregulated autophagy adapter protein and ECs with enhanced GAS5 expression typically maintain healthier mitochondrial populations. This sparked a pertinent inquiry: Does GAS5 regulate mitochondrial quality control (MQC) via mitophagy? However, our subsequent data seemed to counter this hypothesis, indicating that GAS5 predominantly suppressed mitophagy. The manifestation of mitophagy after GAS5 silencing seemed to be tied to the severity of oxidative stress. Under mildly oxidative conditions, GAS5 knockdown prompted mitochondria to transform into myelin-like figures, coupled with amplified mitophagy, suggesting that GAS5 silencing exacerbates oxidative damage but leaves the mitophagy mechanism intact. As oxidative stress escalated, the absence of GAS5 triggered irreversible mitochondrial swelling, alongside impaired mitophagy, culminating in cell death. In contrast, ECs that overexpressed GAS5 neither display mitochondrial swelling nor myelin-like figures, with only typical autolysosomes observed. We proposed that GAS5’s function represented a synergistic effect arising from MITF’s transcription of multiple genes. Notably, mitophagy was not the primary choice, as MITF additionally mediated Nrf2 transcription, which in turn inhibited mitochondrial oxidative damage via the MITF–Nrf2–ARE pathway. Concurrently, the normal functioning of mitophagy necessitated the antioxidant mechanism, providing a relatively stable intracellular environment.
In the context of sepsis-induced oxidative damage, what is the functional significance of the MITF–GAS5–miR-23 loop-activated p62 expression? Current research has revealed a transient increase in autophagy during sepsis, followed by a long-term decline in autophagic flux, ultimately resulting in organ dysfunction [
40,
41,
42]. To date, the mechanisms leading to the decline in autophagic flux in sepsis remain to be studied. One potential explanation is that overactive autophagy in the initial stage depletes autophagy-related proteins, rendering them unable to be effectively replenished later [
43]. Our experimental findings indicated that under oxidative stress, the MITF–GAS5–miR-23 loop persistently elevated levels of autophagy-related proteins, while the high expression of p62 did not yield a transient increase in autophagy or mitophagy levels. On the contrary, autophagy levels were dynamically regulated in response to the intensity of oxidative stress. The high expression of MITF or GAS5 correlated with relatively lower autophagy intensity, attributable to the antioxidant effect of MITF-transcribed Nrf2, which in turn contributes to the maintenance of autophagic flux during sepsis.
Despite the GAS5 inhibition of mitophagy, numerous autolysosomes were still present in the cytoplasm. This suggests the possibility of other types of autophagy that may exert antioxidant effects, potentially superior to mitophagy. As previously reported, p62 can bind to Keap1, inhibiting Keap1-mediated polyubiquitinylation, and the subsequent proteasomal degradation of Nrf2 [
16]. Our observations further revealed that p62-mediated autophagy resulted in more pronounced Keap1 degradation in GAS5-high-expressing ECs, explaining the asynchronous levels of mitophagy and autophagy and showed that the MITF–GAS5–miR-23 loop enhanced Nrf2 expression via P62-mediated post-transcriptional mechanisms. Besides MITF, Nrf2 also mediated the transcription of p62, thus forming a positive feedback loop of p62–Keap1–Nrf2 [
17]. We further propose that MITF, p62, and Nrf2 formed a triangular regulatory system (hereinafter referred to as the “MITF–Nrf2–p62 triangle”). Upon confirming the transcriptional effects of Nrf2 on MITF and TFE3, we observed that the MITF–Nrf2–p62 triangle components exhibited a pairwise mutual regulation pattern, thus enriching the regulatory theory of the MITF–Nrf2–p62 triangle. In summary, the protective effect of GAS5 in sepsis is mainly the result of the synergistic effect of the target gene transcribed by MITF. The triangular regulatory system comprising MITF, Nrf2, and p62 integrates antioxidant and autophagy mechanisms, increasing autophagy adaptor protein reserves while preventing transient autophagy surges caused by oxidative stress, thus contributing to the maintenance of autophagic flux in sepsis (
Figure 8 A model for the anti-inflammatory and anti-oxidative mechanisms of the MITF–GAS5–miR-23 loop in sepsis vascular endothelium). The selective regulation of autophagy by GAS5 under oxidative stress has developed the theory of sepsis-related autophagy regulation, and the biological role of GAS5 in anti-oxidation and anti-inflammation also suggests that GAS5 is expected to become a new target for the treatment of sepsis.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







