
Pre- and Post-Transcriptional Control of HBV Gene Expression: The Road Traveled towards the New Paradigm of HBx, Its Isoforms, and Their Diverse Functions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Hepatitis B Virus and Its Replicative Cycle
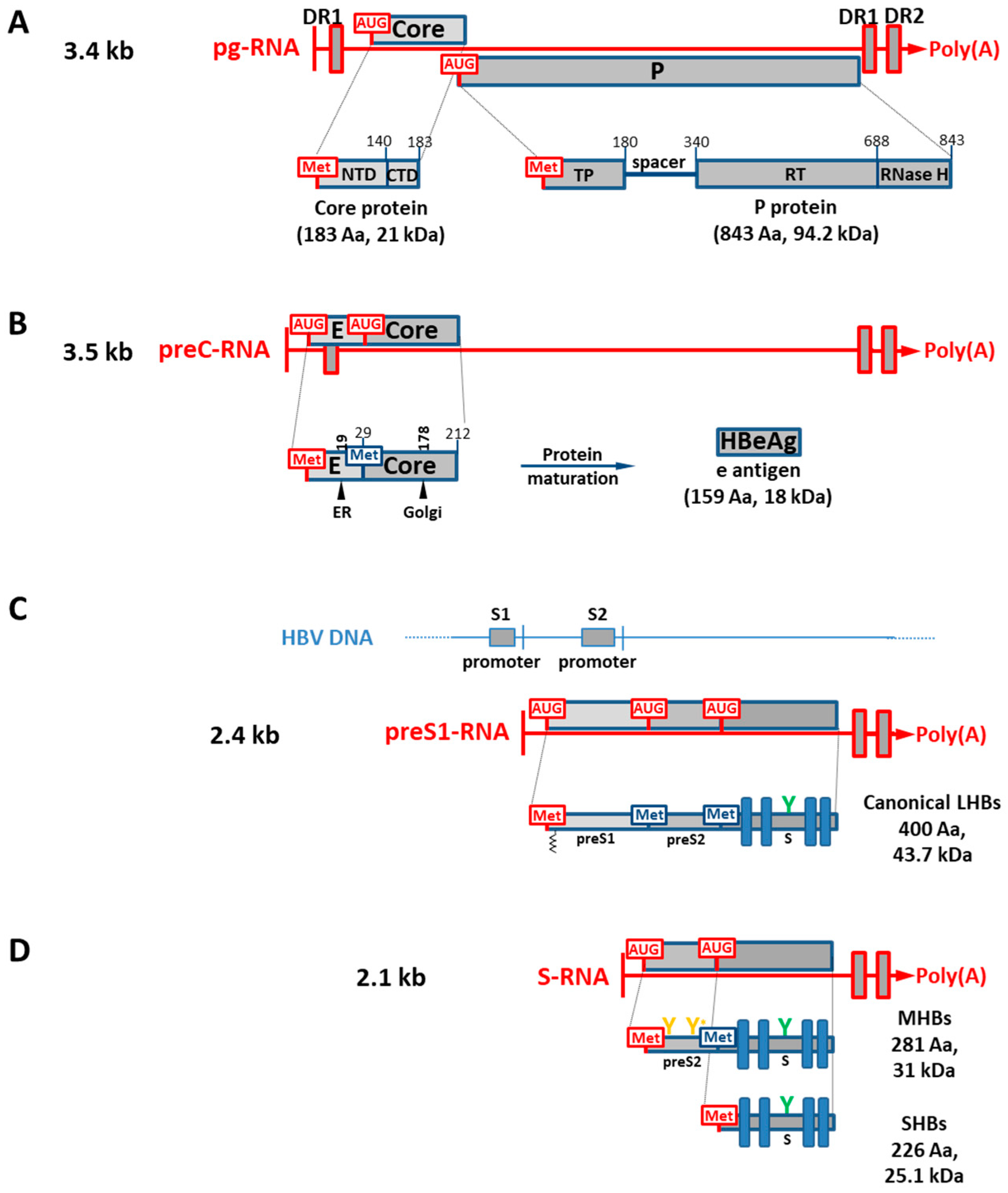
1.2. HBV Genes and Translation of Viral RNA Transcripts into Viral Isoform Proteins
1.3. HBV Viral Isoform Proteins Generated by RNA Splicing
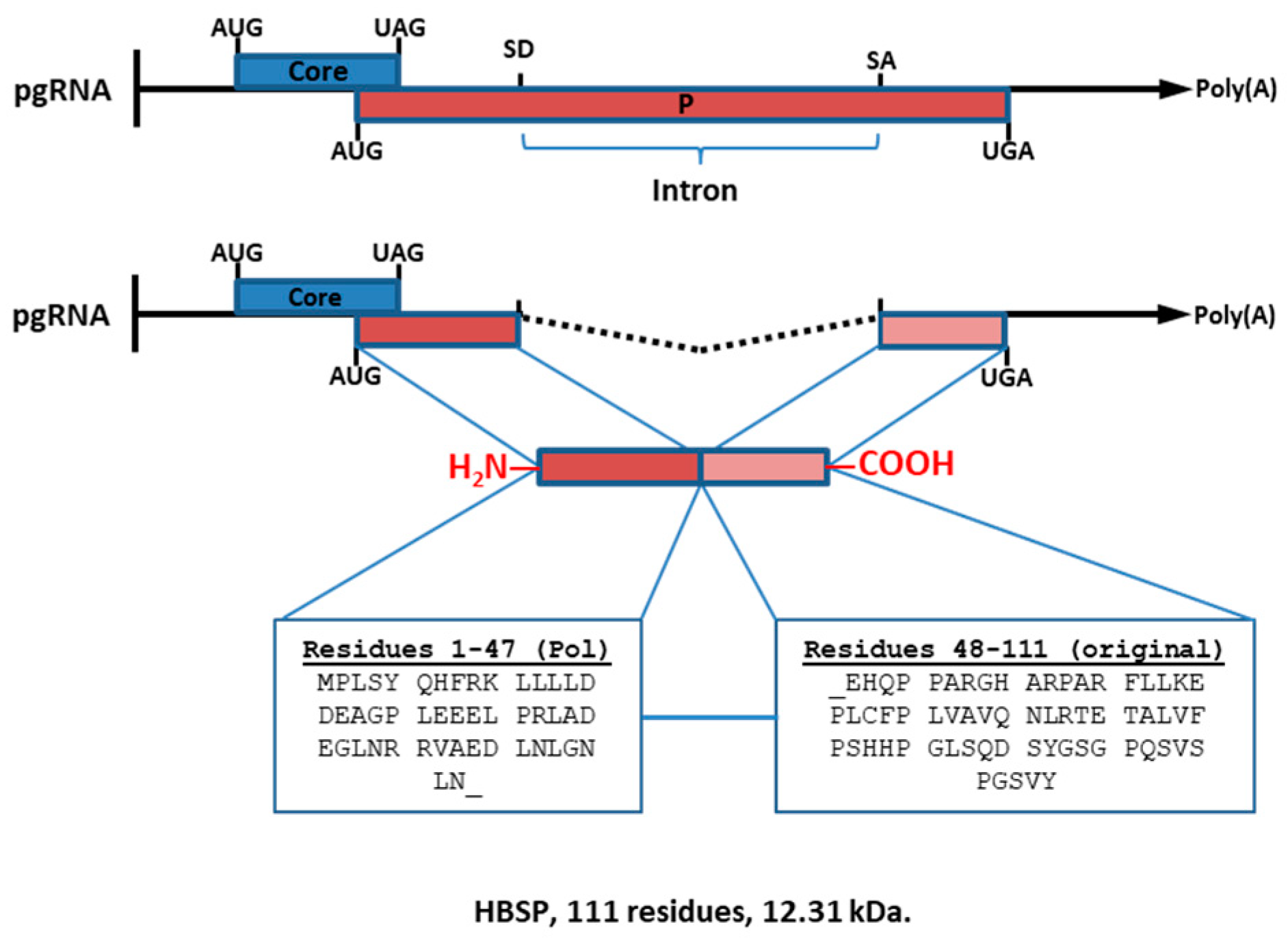
1.4. Hepatitis B-Spliced Protein (HBSP; 12.3 kDa)
1.5. HBV Polymerase-Surface Fusion Protein, HBV P-S Fusion ORF
1.6. Hepatitis B Double Splicing Protein, HBDSP
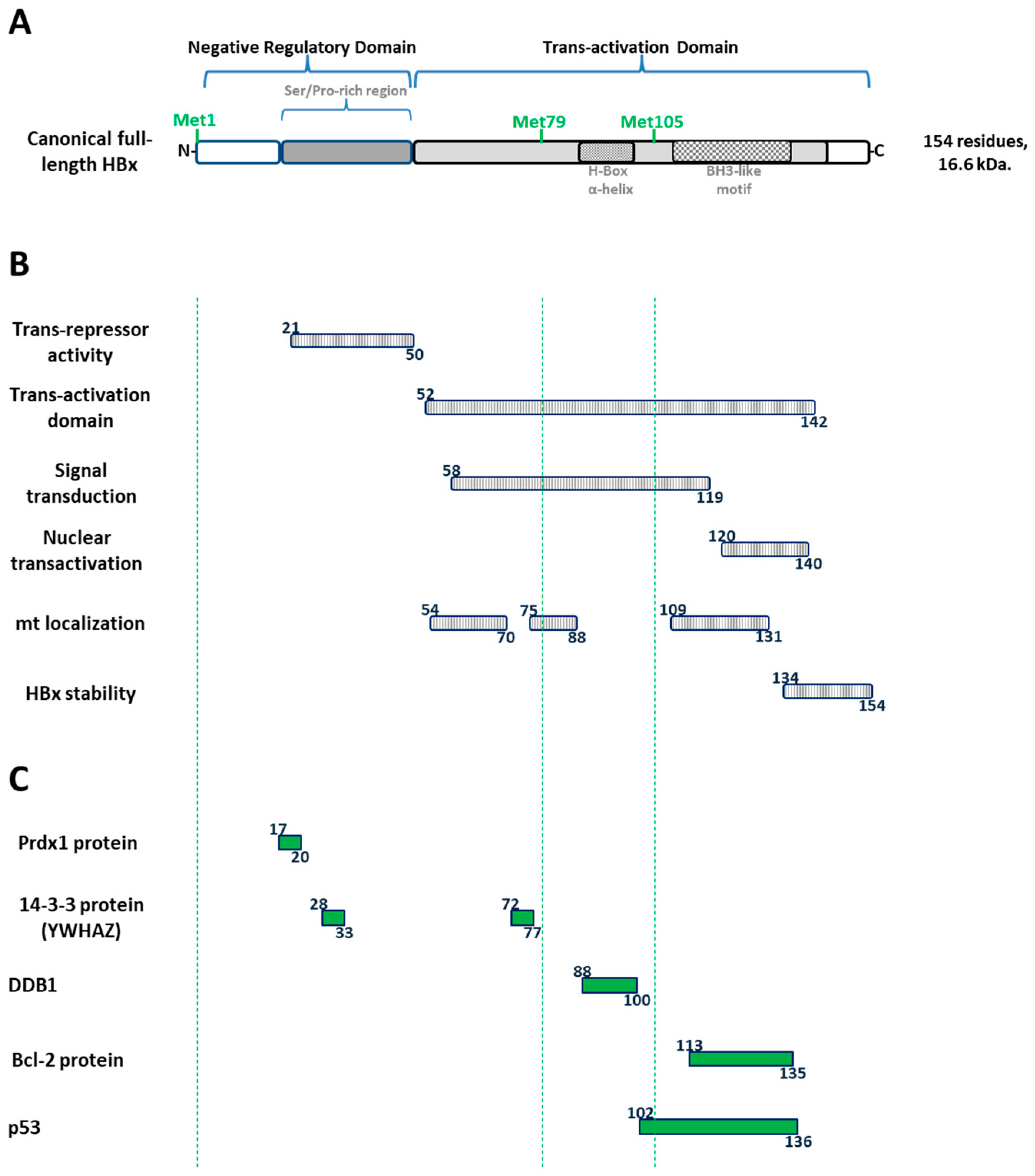
1.7. The Canonical HBx Is a Unique Multifunctional Regulatory HBV Protein
1.8. Regulation of the HBV HBx Gene Expression
1.9. HBV Canonical HBx Protein and Roles on the cccDNA Intermediate

1.10. An Unusual Protein Variant of HBV HBx: HBV Whole-X (HBVwx)
1.11. Prior Findings Revealing the True Paradigm of the Canonical HBx and Isoform Proteins
2. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seeger, C.; Mason, W. Molecular biology of hepatitis B virus infection. Virology 2015, 479, 672–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64, S4–S16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, S.; Venegas, M.; Brahm, J.; Villanueva, R. Full-genome sequence of a hepatitis B virus genotype F1b clone from a chronically infected Chilean patient. Genome Announc. 2014, 2, e01075-01014. [Google Scholar] [CrossRef] [Green Version]
- Hernández, S.; Jiménez, G.; Alarcón, V.; Prieto, C.; Muñoz, F.; Riquelme, C.; Venegas, M.; Brahm, J.; Loyola, A.; Villanueva, R. Replication of a chronic hepatitis B virus genotype F1b construct. Arch. Virol. 2016, 161, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.; Jaspe, R.; Loureiro, C.; Chemin, I. Hepatitis B virus American genotypes: Pathogenic variants? Clin. Res. Hepatol. Gastroenterol. 2020, 44, 825–835. [Google Scholar] [CrossRef]
- Livingston, S.; Simonetti, J.; McMahon, B.; Bulkow, L.; Hurlburt, K.; Homan, C.; Snowball, M.; Cagle, H.; Williams, J.; Chulanov, V. Hepatitis B virus genotypes in Alaska Native people with hepatocellular carcinoma: Preponderance of genotype F. J. Infect. Dis. 2007, 195, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Ching, L.; Gounder, P.; Bulkow, L.; Spradling, P.; Bruce, M.; Negus, S.; Snowball, M.; McMahon, B. Incidence of hepatocellular carcinoma according to hepatitis B virus genotype in Alaska Native people. Liver Int. 2016, 36, 1507–1515. [Google Scholar] [CrossRef] [Green Version]
- Kowalec, K.; Minuk, G.; Børresen, M.; Koch, A.; McMahon, B.; Simons, B.; Osiowy, C. Genetic diversity of hepatitis B virus genotypes B6, D and F among circumpolar indigenous individuals. J. Viral. Hepat. 2013, 20, 122–130. [Google Scholar] [CrossRef]
- Panduro, A.; Maldonado-Gonzalez, M.; Fierro, N.; Roman, S. Distribution of HBV genotypes F and H in Mexico and Central America. Antivir. Ther. 2013, 18, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Roman, S.; Panduro, A. HBV endemicity in Mexico is associated with HBV genotypes H and G. World J. Gastroenterol. 2013, 19, 5446–5453. [Google Scholar] [CrossRef]
- Roman, S.; Tanaka, Y.; Khan, A.; Kurbanov, F.; Kato, H.; Mizokami, M.; Panduro, A. Occult hepatitis B in the genotype H-infected Nahuas and Huichol native Mexican population. J. Med. Virol. 2010, 82, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Tachiquín, M.; Valdez-Salazar, H.; Juárez-Barreto, V.; Dehesa-Violante, M.; Torres, J.; Muñoz-Hernández, O.; Alvarez-Muñoz, M. Molecular analysis of hepatitis B virus “a” determinant in asymptomatic and symptomatic Mexican carriers. Virol. J. 2007, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobedo-Melendez, G.; Panduro, A.; Fierro, N.; Roman, S. High prevalence of occult hepatitis B virus genotype H infection among children with clinical hepatitis in west Mexico. Mem. Inst. Oswaldo Cruz 2014, 109, 728–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.; Lee, C. Epidemiology and prevention of hepatitis B virus infection. Korean J. Hepatol. 2011, 17, 87–95. [Google Scholar] [CrossRef]
- Liu, Y.; Maya, S.; Ploss, A. Animal Models of Hepatitis B Virus Infection—Success, Challenges, and Future Directions. Viruses 2021, 13, 777. [Google Scholar] [CrossRef]
- Rabe, B.; Delaleau, M.; Bischof, A.; Foss, M.; Sominskaya, I.; Pumpens, P.; Cazenave, C.; Castroviejo, M.; Kann, M. Nuclear entry of hepatitis B virus capsids involves disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog. 2009, 5, e1000563. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Kim, E.; Guo, H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: Implications for epigenetic therapy against chronic hepatitis B. Hepatology 2017, 66, 2066–2077. [Google Scholar] [CrossRef] [Green Version]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef]
- Stahl, M.; Beck, J.; Nassal, M. Chaperones activate hepadnavirus reverse transcriptase by transiently exposing a C-proximal region in the terminal protein domain that contributes to epsilon RNA binding. J. Virol. 2007, 81, 13354–13364. [Google Scholar] [CrossRef] [Green Version]
- Prange, R. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 2012, 201, 449–461. [Google Scholar] [CrossRef]
- Hu, J.; Liu, K. Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousali, M.; Papatheodoridis, G.; Paraskevis, D.; Karamitros, T. Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma. Microorganisms 2021, 9, 1787. [Google Scholar] [CrossRef]
- Bill, C.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140. [Google Scholar] [CrossRef] [Green Version]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.; Yuen, M.; Chan, H.; Gish, R.; Locarnini, S.; Chavez, D.; Ferrari, C.; Given, B.; Hamilton, J.; Kanner, S.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaert, A.; Fernandez, E.; Gevaert, K. N-Terminal Proteoforms in Human Disease. Trends Biochem. Sci. 2020, 45, 308–320. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Smyth, J. Alternative mechanisms of translation initiation: An emerging dynamic regulator of the proteome in health and disease. Life Sci. 2018, 212, 138–144. [Google Scholar] [CrossRef]
- Kozak, M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 1986, 44, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. Cell 1987, 196, 947–950. [Google Scholar] [CrossRef]
- Kozak, M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Genes 2005, 361, 13–37. [Google Scholar] [CrossRef]
- Klerk, E.; Hoen, P. Alternative mRNA transcription, processing, and translation: Insights from RNA sequencing. Trends Genet. 2015, 31, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Sriram, A.; Bohlen, J.; Teleman, A. Translation acrobatics: How cancer cells exploit alternate modes of translational initiation. EMBO Rep. 2018, 19, e45947. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Ota, T.; Isogai, T. Prediction whether a human cDNA sequence contains initiation codon by combining statistical information and similarity with protein sequences. Bioinformatics 2000, 16, 960–967. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.; Sheu, S.; Lin, C.-G. Translational regulation of hepatitis B viral gene expression. J. Gastroenterol. Hepatol. 1993, 8, S45–S53. [Google Scholar] [CrossRef]
- Russnak, R.; Ganem, D. Sequences 5′ to the polyadenylation signal mediate differential poly(A) site use in hepatitis B viruses. Genes Dev. 1990, 4, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Slagle, B.; Bouchard, M. Hepatitis B Virus X and Regulation of Viral Gene Expression. Cold Spring Harb. Perspect. Med. 2016, 6, a021402. [Google Scholar] [CrossRef] [Green Version]
- Yuh, C.; Ting, L. The genome of hepatitis B virus contains a second enhancer: Cooperation of two elements within this enhancer is required for its function. J. Virol. 1990, 64, 4281–4287. [Google Scholar] [CrossRef] [Green Version]
- Turton, K.; Meier-Stephenson, V.; Badmalia, M.; Coffin, C.; Patel, T. Host Transcription Factors in Hepatitis B Virus RNA Synthesis. Viruses 2020, 12, 160. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Lo, S. Evidence for involvement of a ribosomal leaky scanning mechanism in the translation of the hepatitis B virus pol gene from the viral pregenome RNA. Virology 1992, 188, 342–352. [Google Scholar] [CrossRef]
- Fouillot, N.; Tlouzeau, S.; Rossignol, J.; Jean-Jean, O. Translation of the hepatitis B virus P gene by ribosomal scanning as an alternative to internal initiation. J. Virol. 1993, 67, 4886–4895. [Google Scholar] [CrossRef] [Green Version]
- Seitz, S.; Habjanič, J.; Schütz, A.; Bartenschlager, R. The Hepatitis B Virus Envelope Proteins: Molecular Gymnastics Throughout the Viral Life Cycle. Annu. Rev. Virol. 2020, 7, 263–288. [Google Scholar] [CrossRef]
- Diab, A.; Foca, A.; Zoulim, F.; Durantel, D.; Andrisani, O. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: Implications for the development of HBc-targeting antivirals. Antivir. Res. 2018, 149, 211–220. [Google Scholar] [CrossRef]
- Jones, S.; Hu, J. Hepatitis B virus reverse transcriptase: Diverse functions as classical and emerging targets for antiviral intervention. Emerg. Microbes Infect. 2013, 2, e56. [Google Scholar] [CrossRef]
- Grice, S.L. Human immunodeficiency virus reverse transcriptase: 25 years of research, drug discovery, and promise. J. Biol. Chem. 2012, 287, 40850–40857. [Google Scholar] [CrossRef] [Green Version]
- Ou, J.; Laub, O.; Rutter, W. Hepatitis B virus gene function: The precore region targets the core antigen to cellular membranes and causes the secretion of the e antigen. Proc. Natl. Acad. Sci. USA 1986, 83, 1578–1582. [Google Scholar] [CrossRef] [Green Version]
- Yaginuma, K.; Shirakata, Y.; Kobayashi, M.; Koike, K. Hepatitis B virus (HBV) particles are produced in a cell culture system by transient expression of transfected HBV DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 2678–2682. [Google Scholar] [CrossRef] [Green Version]
- Standring, D.; Ou, J.; Masiarz, F.; Rutter, W. A signal peptide encoded within the precore region of hepatitis B virus directs the secretion of a heterogeneous population of e antigens in Xenopus oocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 8405–8409. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Kim, K.; Lok, A.; Tong, S. Characterization of genotype-specific carboxyl-terminal cleavage sites of hepatitis B virus e antigen precursor and identification of furin as the candidate enzyme. J. Virol. 2009, 83, 3507–3517. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Xie, X.; Tan, X.; Yu, H.; Tian, M.; Lv, H.; Qin, C.; Qi, J.; Zhu, Q. The Functions of Hepatitis B Virus Encoding Proteins: Viral Persistence and Liver Pathogenesis. Front. Immunol. 2021, 12, 691766. [Google Scholar] [CrossRef]
- Milich, D.; Liang, T. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology 2003, 38, 1075–1086. [Google Scholar] [CrossRef]
- Raney, A.; Milich, D.; Easton, A.; McLachlan, A. Differentiation-specific transcriptional regulation of the hepatitis B virus large surface antigen gene in human hepatoma cell lines. J. Virol. 1990, 64, 2360–2368. [Google Scholar] [CrossRef] [Green Version]
- Standring, D.; Ou, J.; Rutter, W. Assembly of viral particles in Xenopus oocytes: Pre-surface-antigens regulate secretion of the hepatitis B viral surface envelope particle. Proc. Natl. Acad. Sci. USA 1986, 83, 9338–9342. [Google Scholar] [CrossRef] [Green Version]
- Heermann, K.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W. Large surface proteins of hepatitis B virus containing the pre-s sequence. J. Virol. 1984, 52, 396–402. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, R.; Will, H.; Hernandez, N.; Schaller, H. Signals regulating hepatitis B surface antigen transcription. Nature 1983, 305, 336–338. [Google Scholar] [CrossRef]
- Standring, D.; Rutter, W.; Varmus, H.; Ganem, D. Transcription of the hepatitis B surface antigen gene in cultured murine cells initiates within the presurface region. J. Virol. 1984, 50, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Ou, J.; Rutter, W. Regulation of secretion of the hepatitis B virus major surface antigen by the preS-1 protein. J. Virol. 1987, 61, 782–786. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Rio, D. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.; Xu, Y.; Li, J.; Huang, L.; Lin, J.; Zhang, J.; Min, Q.; Yang, W.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Neves, G.; Zucker, J.; Daly, M.; Chess, A. Stochastic yet biased expression of multiple Dscam splice variants by individual cells. Nat Genet 2004, 36, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Ajiro, M.; Zheng, Z. Oncogenes and RNA splicing of human tumor viruses. Emerg. Microbes Infect. 2014, 3, e63. [Google Scholar] [CrossRef]
- Cross, S.; Michalski, D.; Miller, M.; Wilusz, J. RNA regulatory processes in RNA virus biology. Wiley Interdiscip. Rev. 2019, 10, e1536. [Google Scholar] [CrossRef] [Green Version]
- Emery, A.; Swanstrom, R. HIV-1: To Splice or Not to Splice, That Is the Question. Viruses 2021, 13, 181. [Google Scholar] [CrossRef]
- Zheng, Z. Viral oncogenes, noncoding RNAs, and RNA splicing in human tumor viruses. Int. J. Biol. Sci. 2010, 6, 730–755. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Kajitani, N.; Schwartz, S. Splicing and Polyadenylation of Human Papillomavirus Type 16 mRNAs. Int. J. Mol. Sci. 2017, 18, 366. [Google Scholar] [CrossRef] [Green Version]
- Sommer, G.; Heise, T. Posttranscriptional control of HBV gene expression. Front. Biosci. 2008, 13, 5533–5547. [Google Scholar] [CrossRef] [Green Version]
- Hantz, O.; Baginski, I.; Fourel, I.; Chemin, I.; Trepo, C. Viral spliced RNA are produced, encapsidated and reverse transcribed during in vivo woodchuck hepatitis virus infection. Virology 1992, 190, 193–200. [Google Scholar] [CrossRef]
- Obert, S.; Zachmann-Brand, B.; Deindl, E.; Tucker, W.; Bartenschlager, R.; Schaller, H. A splice hepadnavirus RNA that is essential for virus replication. EMBO J. 1996, 15, 2565–2574. [Google Scholar] [CrossRef]
- Lim, C.; Sozzi, V.; Littlejohn, M.; Yuen, L.; Warner, N.; Betz-Stablein, B.; Luciani, F.; Revill, P.; Brown, C. Quantitative analysis of the splice variants expressed by the major hepatitis B virus genotypes. Microb. Genom. 2021, 7, mgen000492. [Google Scholar] [CrossRef]
- Rosmorduc, O.; Petit, M.; Pol, S.; Capel, F.; Bortolotti, F.; Berthelot, P.; Brechot, C.; Kremsdorf, D. In vivo and in vitro expression of defective hepatitis B virus particles generated by spliced hepatitis B virus RNA. Hepatology 1995, 22, 10–19. [Google Scholar]
- Günther, S.; Sommer, G.; Iwanska, A.; Will, H. Heterogeneity and common features of defective hepatitis B virus genomes derived from spliced pregenomic RNA. Virology 1997, 238, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Soussan, P.; Garreau, F.; Zylberberg, H.; Ferray, C.; Brechot, C.; Kremsdorf, D. In vivo expression of a new hepatitis B virus protein encoded by a spliced RNA. J. Clin. Investig. 2000, 105, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Soussan, P.; Tuveri, R.; Nalpas, B.; Garreau, F.; Zavala, F.; Masson, A.; Pol, S.; Brechot, C.; Kremsdorf, D. The expression of hepatitis B spliced protein (HBSP) encoded by a spliced hepatitis B virus RNA is associated with viral replication and liver fibrosis. J. Hepatol. 2003, 38, 343–348. [Google Scholar] [CrossRef]
- Wu, S.; Chen, W.; Jing, Z.; Liu, W.; Lin, X.; Lin, X. Hepatitis B Spliced Protein (HBSP) Suppresses Fas-Mediated Hepatocyte Apoptosis via Activation of PI3K/Akt Signaling. J. Virol. 2018, 92, e01273-18. [Google Scholar] [CrossRef] [Green Version]
- Park, G.; Kim, H.; Shin, H.; Park, S.; Shin, H.; Kim, K. Modulation of hepatitis B virus replication by expression of polymerase-surface fusion protein through splicing: Implications for viral persistence. Virus Res. 2008, 136, 166–174. [Google Scholar] [CrossRef]
- Huang, H.; Jeng, K.; Hu, C.; Tsai, C.; Lo, S.; Chang, C. Identification and characterization of a structural protein of hepatitis B virus: A polymerase and surface fusion protein encoded by a spliced RNA. Virology 2000, 275, 398–410. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Lin, X.; Wang, Y.; Tian, X.; Xie, Y.; Wen, Y. A double-spliced defective hepatitis B virus genome derived from hepatocellular carcinoma tissue enhanced replication of full-length virus. J. Med. Virol. 2009, 81, 230–237. [Google Scholar] [CrossRef]
- Chen, W.; Chen, J.; Lin, W.; Lin, J.; Lin, X. Hepatitis B doubly spliced protein, generated by a 2.2 kb doubly spliced hepatitis B virus RNA.; is a pleiotropic activator protein mediating its effects via activator protein-1- and CCAAT/enhancer-binding protein-binding sites. J. Gen. Virol. 2010, 91, 2592–2600. [Google Scholar] [CrossRef]
- Blum, H.E.; Zhang, Z.; Galun, E.; Weizsäcker, F.; Garner, B.; Liang, T.; Wands, J. Hepatitis B virus X protein is not central to the viral life cycle in vitro. J. Virol. 1992, 66, 1223–1227. [Google Scholar] [CrossRef] [Green Version]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Melegari, M.; Scaglioni, P.; Wands, J. Cloning and characterization of a novel hepatitis B virus × binding protein that inhibits viral replication. J. Virol. 1998, 72, 1737–1743. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Cheong, J.; Kaneko, S. Human hepatitis B virus X gene encodes a regulatory domain which represses transactivation of X protein. J. Biol. Chem. 1994, 269, 15118–15123. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular functions and biological roles of hepatitis B virus × protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef]
- Kumar, V.; Sarkar, D. Hepatitis B Virus X Protein: Structure–Function Relationships and Role in Viral Pathogenesis. HEP 2004, 166, 377–407. [Google Scholar]
- Misra, K.; Mukherji, A.; Kumar, V. The conserved amino-terminal region (amino acids 1–20) of the hepatitis B virus X protein shows a transrepression function. Virus Res. 2004, 105, 157–165. [Google Scholar] [CrossRef]
- Gottlob, K.; Pagano, S.; Levrero, M.; Graessmann, A. Hepatitis B virus X protein transcription activation domains are neither required nor sufficient for cell transformation. Cancer Res. 1998, 58, 3566–3570. [Google Scholar] [PubMed]
- Kumar, V.; Jayasuryan, N.; Kumar, R. A truncated mutant (residues 58–140) of the hepatitis B virus X protein retains transactivation function. Proc. Natl. Acad. Sci. USA 1996, 93, 5647–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arii, M.; Takada, S.; Koike, K. Identification of three essential regions of hepatitis B virus X protein for trans-activation function. Oncogene 1992, 7, 397–403. [Google Scholar]
- Nijhara, R.; Jana, S.; Goswami, S.; Kumar, V.; Sarkar, D. An internal segment (residues 58–119) of the hepatitis B virus X protein is sufficient to activate MAP kinase pathways in mouse liver. FEBS Lett. 2001, 504, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Reddi, H.; Kumar, R.; Jain, S.; Kumar, V. A carboxy-terminal region of the hepatitis B virus X protein promotes DNA interaction of CREB and mimics the native protein for transactivation function. Virus Genes 2003, 26, 227–238. [Google Scholar] [CrossRef]
- Lizzano, R.; Yang, B.; Clippinger, A.; Bouchard, M. The C-terminal region of the hepatitis B virus X protein is essential for its stability and function. Virus Res. 2011, 155, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Huh, K.; Siddiqui, A. Characterization of the mitochondrial association of hepatitis B virus X protein, HBx. Mitochondrion 2002, 1, 349–359. [Google Scholar] [CrossRef]
- McClain, S.; Clippinger, A.; Lizzano, R.; Bouchard, M. Hepatitis B virus replication is associated with an HBx-dependent mitrochondrion-regulated increase in cytosolic calcium levels. J. Virol. 2007, 81, 12061–12065. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Cha, E.; Lim, J.; Kwon, S.; Kim, D.; Cho, H.; Han, K. Structural characterization of an intrinsically unfolded mini-HBX protein from hepatitis B virus. Mol. Cells 2012, 34, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Moura, P.; Rui, E.; Gonçalves, K.D.A.; Kobarg, J. The cysteine residues of the hepatitis B virus onco-protein HBx are not required for its interaction with RNA or with human p53. Virus Res. 2005, 108, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Damme, E.V.; Vanhove, J.; Severyn, B.; Verschueren, L.; Pauwels, F. The Hepatitis B Virus Interactome: A Comprehensive Overview. Front. Microbiol. 2021, 12, 724877. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Gan, X.; Ito, M.; Chen, M.; Aly, H.; Matsui, C.; Abe, T.; Watashi, K.; Wakita, T.; Suzuki, T.; et al. Peroxiredoxin 1, a Novel HBx-Interacting Protein, Interacts with Exosome Component 5 and Negatively Regulates Hepatitis B Virus (HBV) Propagation through Degradation of HBV RNA. J. Virol. 2019, 93, e02203–e02218. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zhang, Y.; Wang, C.; Sun, Z.; Li, L.; Dong, J.; Zhou, W. 14-3-3ζ binds to hepatitis B virus protein X and maintains its protein stability in hepatocellular carcinoma cells. Cancer Med. 2018, 7, 5543–5553. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Robert, E.; Breugel, P.; Strubin, M.; Zheng, N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Chen, H.; Cao, J.; Xiong, H.; Mo, X.; Li, T.; Kang, X.; Zhao, J.; Yin, B.; Zhao, X.; et al. Structural and functional analyses of hepatitis B virus X protein BH3-like domain and Bcl-xL interaction. Nat. Commun. 2019, 10, 3192. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Nomura, T.; Yamashita, T.; Dorjsuren, D.; Tang, H.; Murakami, S. The transactivation and p53-interacting functions of hepatitis B virus X protein are mutually interfering but distinct. Cancer Res. 1997, 57, 5137–5142. [Google Scholar] [PubMed]
- Dandri, M.; Schirmache, P.; Rogler, C. Woodchuck hepatitis virus X protein is present in chronically infected woodchuck liver and woodchuck hepatocellular carcinomas which are permissive for viral replication. J. Virol. 1996, 70, 5246–5254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirma, H.; Giannini, C.; Poussin, K.; Paterlini, P.; Kremsdorf, D.; Bréchot, C. Hepatitis B virus X mutants, present in hepatocellular carcinoma tissue abrogate both the antiproliferative and transactivation effects of HBx. Oncogene 1999, 18, 4848–4859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoare, J.; Henkler, F.; Dowling, J.; Errington, W.; Goldin, R.; Fish, D.; McGarvey, M. Subcellular localisation of the X protein in HBV infected hepatocytes. J. Med. Virol. 2001, 64, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Weil, R.; Sirma, H.; Giannini, C.; Kremsdorf, D.; Bessia, C.; Dargemont, C.; Bréchot, C.; Israël, A. Direct association and nuclear import of the hepatitis B virus X protein with the NF-kappaB inhibitor IkappaBalpha. Mol. Cell. Biol. 1999, 19, 6345–6354. [Google Scholar] [CrossRef] [Green Version]
- Kornyeyev, D.; Ramakrishnan, D.; Voitenleitner, C.; Livingston, C.; Xing, W.; Hung, M.; Kwon, H.; Fletcher, S.; Beran, R. Spatiotemporal Analysis of Hepatitis B Virus X Protein in Primary Human Hepatocytes. J. Virol. 2019, 93, e00248-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkler, F.; Hoare, J.; Waseem, N.; Goldin, R.; McGarvey, M.; Koshy, R.; King, I. Intracellular localization of the hepatitis B virus HBx protein. J. Gen. Virol. 2001, 82, 871–882. [Google Scholar] [CrossRef]
- Cha, M.; Ryu, D.; Jung, H.; Chang, H.; Ryu, W. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J. Gen. Virol. 2009, 90, 978–986. [Google Scholar] [CrossRef]
- Ma, J.; Sun, T.; Park, S.; Shen, G.; Liu, J. The role of hepatitis B virus X protein is related to its differential intracellular localization. Acta Biochim. Biophys. Sin. 2011, 43, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Prieto, C.; Montecinos, J.; Jiménez, G.; Riquelme, C.; Garrido, D.; Hernández, S.; Loyola, A.; Villanueva, R. Phosphorylation of Phylogenetically Conserved Amino Acid Residues Confines HBx within Different Cell Compartments of Human Hepatocarcinoma Cells. Molecules 2021, 26, 1254. [Google Scholar] [CrossRef]
- Bouchard, M.; Wang, L.; Schneider, R. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef] [PubMed]
- Benn, J.; Su, F.; Doria, M.; Schneider, R. Hepatitis B virus HBx protein induces transcription factor AP-1 by activation of extracellular signal-regulated and c-Jun N-terminal mitogen-activated protein kinases. J. Virol. 1996, 70, 4978–4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Yun, Y. HBx protein of hepatitis B virus activates Jak1-STAT signaling. J. Biol. Chem. 1998, 273, 25510–25515. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, M.; Wang, L.; Schneider, R. Activation of focal adhesion kinase by hepatitis B virus HBx protein: Multiple functions in viral replication. J. Virol. 2006, 80, 4406–4414. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Khine, A.; Sarangi, F.; Hsu, E.; Iorio, C.; Tibbles, L.; Woodgett, J.; Penninger, J.; Richardson, C. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J. Biol. Chem. 2001, 276, 8328–8340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, M.; Kim, C.; Park, Y.; Ryu, W. Hepatitis B virus X protein is essential for the activation of Wnt/beta-catenin signaling in hepatoma cells. Hepatology 2004, 39, 1683–1693. [Google Scholar] [CrossRef]
- Bouchard, M.; Schneider, R. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [Green Version]
- Keasler, V.; Hodgson, A.; Madden, C.; Slagle, B. Hepatitis B virus HBx protein localized to the nucleus restores HBx-deficient virus replication in HepG2 cells and in vivo in hydrodynamically-injected mice. Virology 2009, 390, 122–129. [Google Scholar] [CrossRef] [Green Version]
- Rossner, M. Review: Hepatitis B virus X-gene product: A promiscuous transcriptional activator. J. Med. Virol. 1992, 36, 101–117. [Google Scholar] [CrossRef]
- Qadri, I.; Ferrari, M.; Siddiqui, A. The hepatitis B virus transactivator protein, HBx, interacts with single-stranded DNA (ssDNA). Biochemical characterizations of the HBx-ssDNA interactions. J. Biol. Chem. 1996, 271, 15443–15450. [Google Scholar] [CrossRef] [Green Version]
- Qadri, I.; Maguire, H.; Siddiqui, A. Hepatitis B virus transactivator protein X interacts with the TATA-binding protein. Proc. Natl. Acad. Sci. USA 1995, 92, 1003–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cougot, D.; Wu, Y.; Cairo, S.; Caramel, J.; Renard, C.; Lévy, L.; Buendia, M.; Neuveut, C. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J. Biol. Chem. 2007, 282, 4277–4287. [Google Scholar] [CrossRef] [Green Version]
- Slagle, B.; Bouchard, M. Role of HBx in hepatitis B virus persistence and its therapeutic implications. Curr. Opin. Virol. 2018, 30, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Raney, A.; McLachlan, A. Characterization of the hepatitis B virus X- and nucleocapsid gene transcriptional regulatory elements. Virology 1992, 191, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Treinin, M.; Laub, O. Identification of a promoter element located upstream from the hepatitis B virus X gene. Mol. Cell Biol. 1987, 7, 545–548. [Google Scholar] [PubMed] [Green Version]
- Landry, J.; Mager, D.; Wilhelm, B. Complex controls: The role of alternative promoters in mammalian genomes. Trends Genet. 2003, 19, 640–648. [Google Scholar] [CrossRef]
- Forrest, A.; Kawaji, H.; Rehli, M.; Baillie, J.; Hoon, M.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar]
- Davuluri, R.; Suzuki, Y.; Sugano, S.; Plass, C.; Huang, T. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008, 24, 167–177. [Google Scholar] [CrossRef]
- Lenhard, B.; Sandelin, A.; Carninci, P. Metazoan promoters: Emerging characteristics and insights into transcriptional regulation. Nat. Rev. Genet. 2012, 13, 233–245. [Google Scholar] [CrossRef]
- Kimura, K.; Wakamatsu, A.; Suzuki, Y.; Ota, T.; Nishikawa, T.; Yamashita, R.; Yamamoto, J.; Sekine, M.; Tsuritani, K.; Wakaguri, H.; et al. Diversification of transcriptional modulation: Large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2006, 16, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Riegler, J.; Wu, J.; Yen, T. Novel short transcripts of hepatitis B virus X gene derived from intragenic promoter. J. Biol. Chem. 1994, 269, 22593–22598. [Google Scholar] [CrossRef]
- Altinel, K.; Hashimoto, K.; Wei, Y.; Neuveut, C.; Gupta, I.; Suzuki, A.; Santos, A.D.; Moreau, P.; Xia, T.; Kojima, S.; et al. Single-Nucleotide Resolution Mapping of Hepatitis B Virus Promoters in Infected Human Livers and Hepatocellular Carcinoma. J. Virol. 2016, 90, 10811–10822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmayer, B.; Diederichs, A.; Chapus, F.; Rivoire, M.; Neveu, G.; Alam, A.; Fraisse, L.; Carter, K.; Testoni, B.; Zoulim, F. Full-length 5′RACE identifies all major HBV transcripts in HBV-infected hepatocytes and patient serum. J. Hepatol. 2020, 73, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Schranz, I.; Schroder, C.; Zentgraf, H. Hepatitis B Virus Genome Is Organized into Nucleosomes in the Nucleus of the Infected Cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Newbold, J.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J. The Covalently Closed Duplex Form of the Hepadnavirus Genome Exists in situ as a Heterogeneous Population of Viral Minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, C.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef]
- Belloni, L.; Pollicino, T.; Nicola, F.D.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Wu, C.; Xu, Z.; Teng, Y.; Zhao, L.; Zhao, K.; Wang, J.; Wang, W.; Zhan, Q.; Zhu, C.; et al. Hepatitis B Virus Core Protein Is Not Required for Covalently Closed Circular DNA Transcriptional Regulation. J. Virol. 2022, 96, e0136222. [Google Scholar] [CrossRef]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef]
- Alarcon, V.; Hernández, S.; Rubio, L.; Alvarez, F.; Flores, Y.; Varas-Godoy, M.; Ferrari, G.D.; Kann, M.; Villanueva, R.; Loyola, A. The enzymes LSD1 and Set1A cooperate with the viral protein HBx to establish an active hepatitis B viral chromatin state. Sci. Rep. 2016, 6, 25901. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Astudillo, F.; Garrido, D.; Varas-Godoy, M.; Gutiérrez, J.; Villanueva, R.; Loyola, A. The histone variant H3.3 regulates the transcription of the hepatitis B virus. Ann. Hepatol. 2021, 21, 100261. [Google Scholar] [CrossRef] [PubMed]
- Hernández, S.; Álvarez-Astudillo, F.; Garrido, D.; Prieto, C.; Loyola, A.; Villanueva, R. Canonical and Divergent N-Terminal HBx Isoform Proteins Unveiled: Characteristics and Roles during HBV Replication. Biomedicines 2021, 9, 1701. [Google Scholar] [CrossRef] [PubMed]
- Rivière, L.; Gerossier, L.; Ducroux, A.; Dion, S.; Deng, Q.; Michel, M.; Buendia, M.-A.; Hantz, O.; Neuveut, C. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J. Hepatol. 2015, 63, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Escobar, T.; Loyola, A.; Reinberg, D. Parental nucleosome segregation and the inheritance of cellular identity. Nat. Rev. Genet. 2021, 22, 379–392. [Google Scholar] [CrossRef]
- Benhenda, S.; Ducroux, A.; Rivière, L.; Sobhian, B.; Ward, M.; Dion, S.; Hantz, O.; Protzer, U.; Michel, M.; Benkirane, M.; et al. Methyltransferase PRMT1 is a binding partner of HBx and a negative regulator of hepatitis B virus transcription. J. Virol. 2013, 87, 4360–4371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Zhao, H.; Wu, Y.; Peng, B.; Gao, Z.; Sun, Y.; Duan, J.; Qi, Y.; Li, Y.; Zhou, Z.; et al. Transcriptionally inactive hepatitis B virus episome DNA preferentially resides in the vicinity of chromosome 19 in 3D host genome upon infection. Cell. Rep. 2021, 35, 109288. [Google Scholar] [CrossRef]
- Sozzi, V.; Shen, F.; Chen, J.; Colledge, D.; Jackson, K.; Locarnini, S.; Yuan, Z.; Revill, P. In vitro studies identify a low replication phenotype for hepatitis B virus genotype H generally associated with occult HBV and less severe liver disease. Virology 2018, 519, 190–196. [Google Scholar] [CrossRef]
- Loncarević, I.; Schranz, P.; Zentgraf, H.; Liang, X.; Herrmann, G.; Tang, Z.; Schröder, C. Replication of hepatitis B virus in a hepatocellular carcinoma. Virology 1990, 174, 158–168. [Google Scholar] [CrossRef]
- Loncarevic, I.; Zentgraf, H.; Schröder, C. Sequence of a replication competent hepatitis B virus genome with a preX open reading frame. Nucleic Acids Res. 1990, 18, 4940. [Google Scholar] [CrossRef] [Green Version]
- Faure, E. Alternative peptide-fusion proteins generated by out-of-frame mutations, just upstream ORFs or elongations in mutants of human hepatitis B viruses. Virus Res. 2006, 117, 185–201. [Google Scholar] [CrossRef]
- Yang, Q.; Cheng, J.; Dong, J.; Zhang, J.; Zhang, S. Molecular epidemiological study on pre-X region of hepatitis B virus and identification of hepatocyte proteins interacting with whole-X protein by yeast two-hybrid. World J. Gastroenterol. 2005, 11, 3473–3478. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Liu, H.; He, Y.; Yi, R.; Niu, Y.; Chen, T.; Yang, Q.; Zhao, Y. Comparative study of the different activities of hepatitis B virus whole-X protein and HBx in hepatocarcinogenesis by proteomics and bioinformatics analysis. Arch. Virol. 2015, 160, 1645–1656. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, H.; Yi, R.; Yan, T.; He, Y.; Zhao, Y.; Liu, J. Hepatitis B virus whole-X and X protein play distinct roles in HBV-related hepatocellular carcinoma progression. J. Exp. Clin. Cancer Res. 2016, 35, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsano, C.; Avantaggiati, M.; Natoli, G.; Marzio, E.D.; Will, H.; Perricaudet, M.; Levrero, M. Full-length and truncated versions of the hepatitis B virus (HBV) X protein (pX) transactivate the cmyc protooncogene at the transcriptional level. Biochem. Biophys. Res. Commun. 1991, 176, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Kwee, L.; Lucito, R.; Aufiero, B.; Schneider, R. Alternate translation initiation on hepatitis B virus X mRNA produces multiple polypeptides that differentially transactivate class II and III promoters. J. Virol. 1992, 66, 4382–4389. [Google Scholar] [CrossRef] [Green Version]
- Nakatake, H.; Chisaka, O.; Yamamoto, S.; Matsubara, K.; Koshy, R. Effect of X protein on transactivation of hepatitis B virus promoters and on viral replication. Virology 1993, 195, 305–314. [Google Scholar] [CrossRef]
- Leach, J.; Qiao, L.; Fang, Y.; Han, S.; Gilfor, D.; Fisher, P.; Grant, S.; Hylemon, P.; Peterson, D.; Dent, P. Regulation of p21 and p27 expression by the hepatitis B virus X protein and the alternate initiation site X proteins, AUG2 and AUG3. J. Gastroenterol. Hepatol. 2003, 18, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–E5724. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Liao, G.; Zhang, M.; Zhu, Y.; Wang, K.; Xiao, W.; Jia, C.; Dong, M.; Sun, N.; Walch, A.; et al. Translatomic profiling reveals novel self-restricting virus-host interactions during HBV infection. J. Hepatol. 2021, 21, 00109-4. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Li, G.; Hu, H.; Yang, C.; Zhang, X.; Leng, Q.; Xie, Y.; Yu, D.; Zhang, X.; Gao, Y.; et al. Recombinant covalently closed circular hepatitis B virus DNA induces prolonged viral persistence in immunocompetent mice. J. Virol. 2014, 88, 8045–8056. [Google Scholar] [CrossRef] [Green Version]
- Babu, M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [Green Version]
- Buljan, M.; Chalancon, G.; Dunker, A.; Bateman, A.; Balaji, S.; Fuxreiter, M.; Babu, M. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr. Opin. Struct. Biol. 2013, 23, 443–450. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, S.; Dunker, A. Intrinsically Disordered Proteins Link Alternative Splicing and Post-translational Modifications to Complex Cell Signaling and Regulation. J. Mol. Biol. 2018, 430, 2342–2359. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Romero, P.; Rani, M.; Dunker, A.; Obradovic, Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. 1999, 10, 30–40. [Google Scholar]
- Romero, P.; Obradovic, Z.; Dunker, A. Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Inform. 1997, 8, 110–124. [Google Scholar]
- McNaughton, A.; Revill, P.; Littlejohn, M.; Matthews, P.; Ansari, M. Analysis of genomic-length HBV sequences to determine genotype and subgenotype reference sequences. J. Gen. Virol. 2020, 101, 271–283. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, M.; Wu, J.; Shi, Y. Structural and biochemical analysis of Bcl-2 interaction with the hepatitis B virus protein HBx. Proc. Natl. Acad. Sci. USA 2016, 113, 2074–2079. [Google Scholar] [CrossRef] [Green Version]
- Bohrer, A.; Yoshimoto, N.; Sekiguchi, A.; Rykulski, N.; Saito, K.; Takahashi, H. Alternative translational initiation of ATP sulfurylase underlying dual localization of sulfate assimilation pathways in plastids and cytosol in Arabidopsis thaliana. Front. Plant Sci. 2015, 5, 750. [Google Scholar] [CrossRef] [Green Version]
- Outten, C.; Culotta, V. Alternative start sites in the Saccharomyces cerevisiae GLR1 gene are responsible for mitochondrial and cytosolic isoforms of glutathione reductase. J. Biol. Chem. 2004, 279, 7785–7791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brubaker, S.; Gauthier, A.; Mills, E.; Ingolia, N.; Kagan, J. A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell 2014, 156, 800–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, P.; Anderson, P. Alternative translation initiation in immunity: MAVS learns new tricks. Trends Immunol. 2014, 35, 188–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyama, S.; Schibler, J.; Mulloy, J. Alternative translation initiation generates the N-terminal truncated form of RUNX1 that retains hematopoietic activity. Exp. Hematol. 2019, 72, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.; Bourdon, J. The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2010, 2, a000927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logette, E.; Wotawa, A.; Solier, S.; Desoche, L.; Solary, E.; Corcos, L. The human caspase-2 gene: Alternative promoters, pre-mRNA splicing and AUG usage direct isoform-specific expression. Oncogene 2003, 22, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Trulley, P.; Snieckute, G.; Bekker-Jensen, D.; Menon, M.; Freund, R.; Kotlyarov, A.; Olsen, J.; Diaz-Muñoz, M.; Turner, M.; Bekker-Jensen, S.; et al. Alternative Translation Initiation Generates a Functionally Distinct Isoform of the Stress-Activated Protein Kinase MK2. Cell. Rep. 2019, 27, 2859–2870. [Google Scholar] [CrossRef] [Green Version]
- Kadmiel, M.; Cidlowski, J. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Chen, X.; Yin, Q.; Ruan, D.; Zhao, X.; Zhang, C.; McNutt, M.; Yin, Y. PTENβ is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat. Commun. 2017, 8, 14771. [Google Scholar] [CrossRef] [Green Version]
- Benassayag, C.; Montero, L.; Colombié, N.; Gallant, P.; Cribbs, D.; Morello, D. Human c-Myc isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol. Cell. Biol. 2005, 25, 9897–9909. [Google Scholar] [CrossRef] [Green Version]
- Abbas, Z.; Afzal, R. Life cycle and pathogenesis of hepatitis D virus: A review. World J. Hepatol. 2013, 5, 666–675. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Müller, M. Post-Translational Modifications of Protein Backbones: Unique Functions, Mechanisms, and Challenges. Biochemistry 2008, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villanueva, R.A.; Loyola, A. Pre- and Post-Transcriptional Control of HBV Gene Expression: The Road Traveled towards the New Paradigm of HBx, Its Isoforms, and Their Diverse Functions. Biomedicines 2023, 11, 1674. https://doi.org/10.3390/biomedicines11061674
Villanueva RA, Loyola A. Pre- and Post-Transcriptional Control of HBV Gene Expression: The Road Traveled towards the New Paradigm of HBx, Its Isoforms, and Their Diverse Functions. Biomedicines. 2023; 11(6):1674. https://doi.org/10.3390/biomedicines11061674
Chicago/Turabian StyleVillanueva, Rodrigo A., and Alejandra Loyola. 2023. "Pre- and Post-Transcriptional Control of HBV Gene Expression: The Road Traveled towards the New Paradigm of HBx, Its Isoforms, and Their Diverse Functions" Biomedicines 11, no. 6: 1674. https://doi.org/10.3390/biomedicines11061674