
Simultaneous Inhibition of Mcl-1 and Bcl-2 Induces Synergistic Cell Death in Hepatocellular Carcinoma
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cells
2.3. Cell Death Assays
2.4. Luciferase Activity Assays
2.5. Immunofluorescence
2.6. Western Blot
2.7. Graphics and Calculations
3. Results
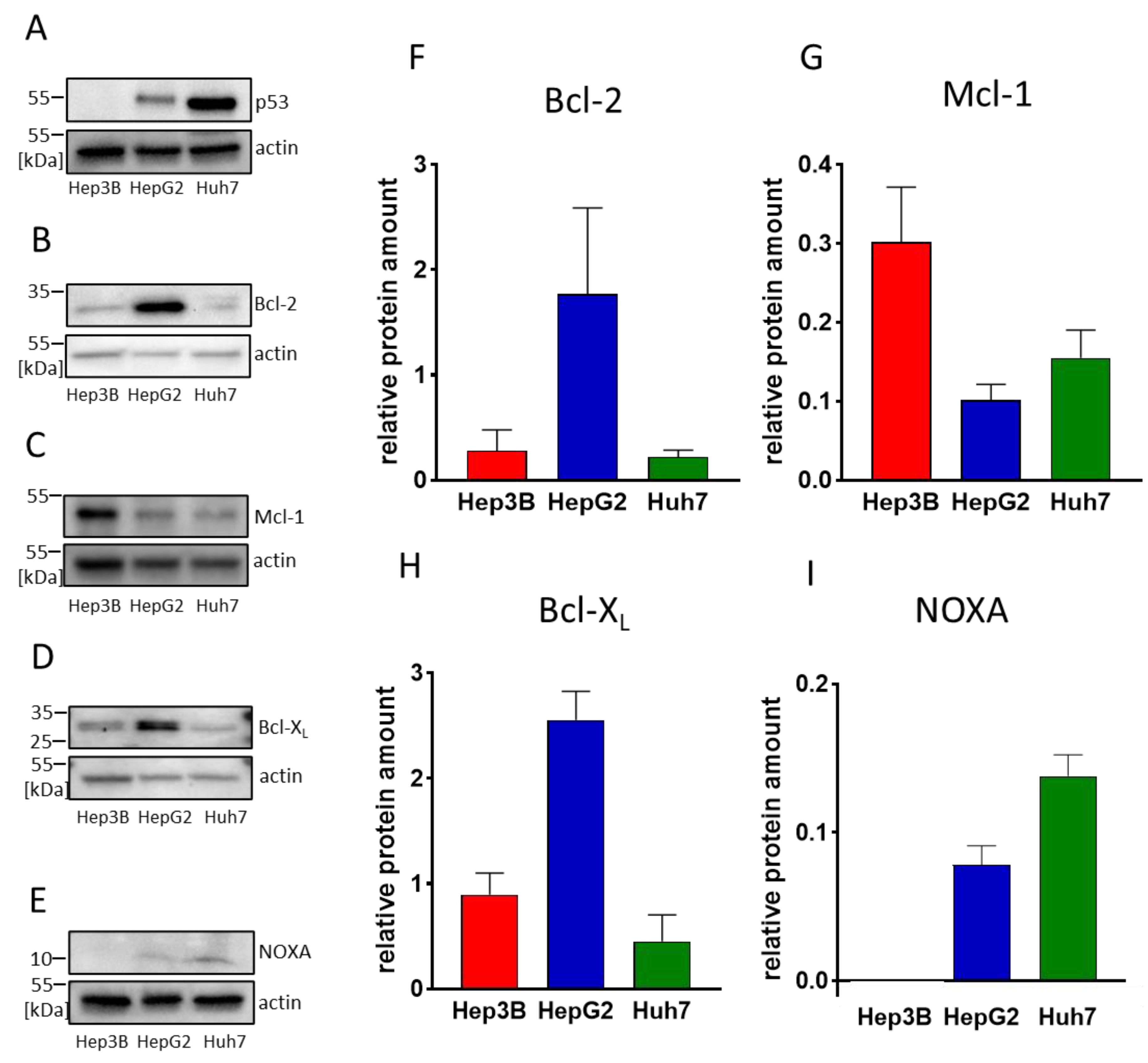
3.1. HCC Cell Lines Display Different Expression Levels of Bcl-2 and Mcl-1
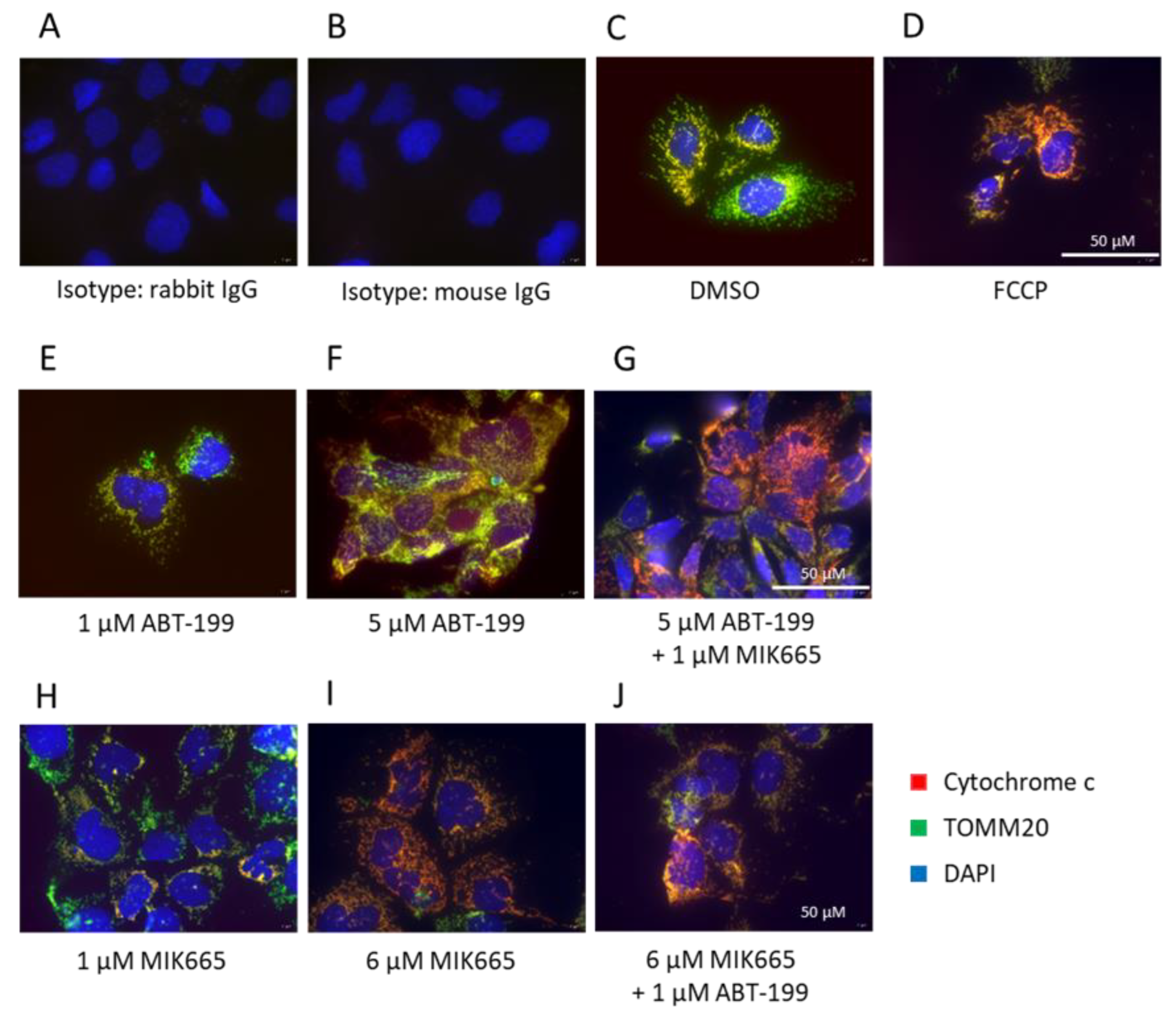
3.2. Combination of BH3-Mimetics Induces Release of Cytochrome c into the Cytosol
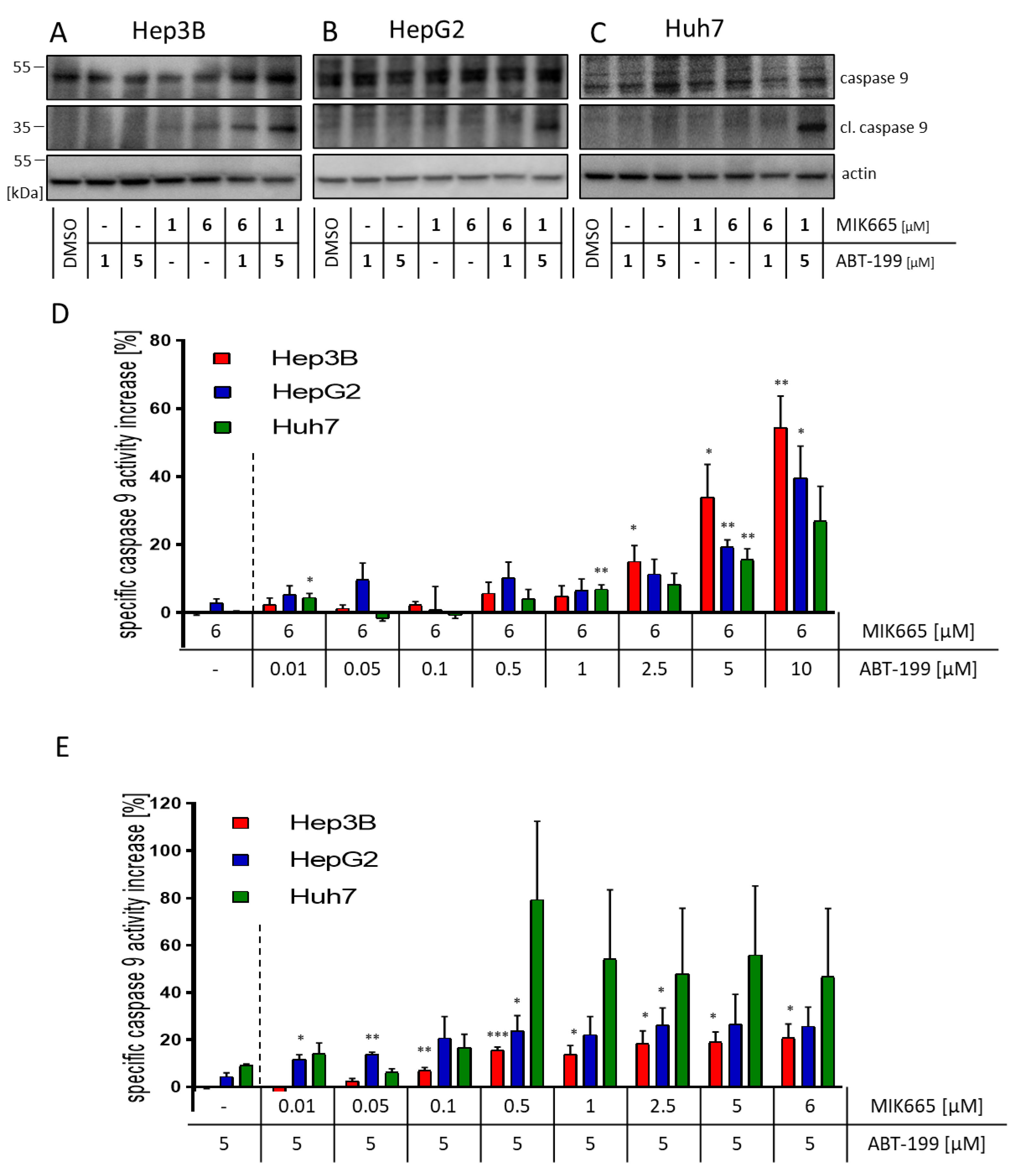
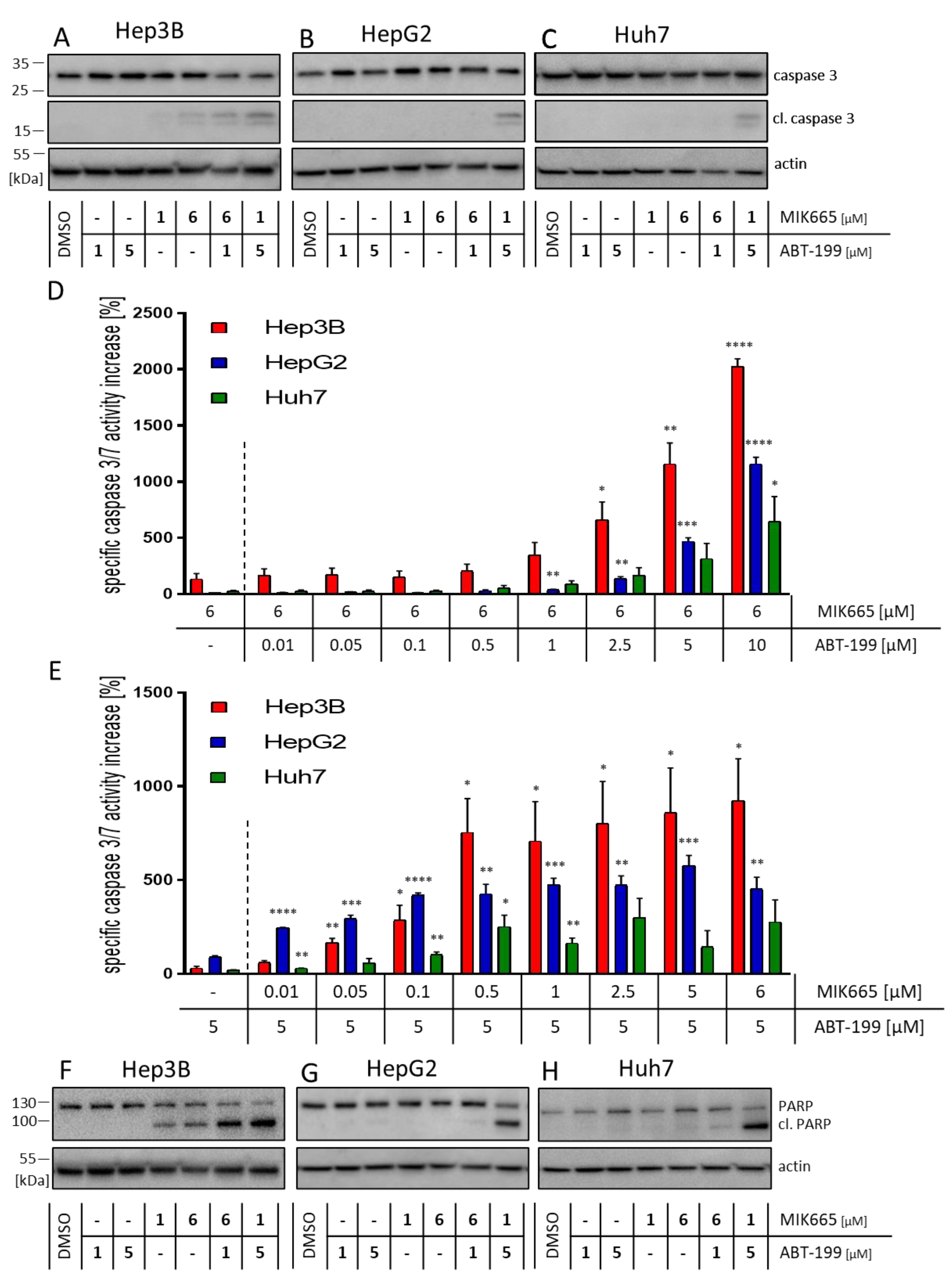
3.3. Combination Treatment of ABT-199 and MIK665 Induces a Caspase Cascade
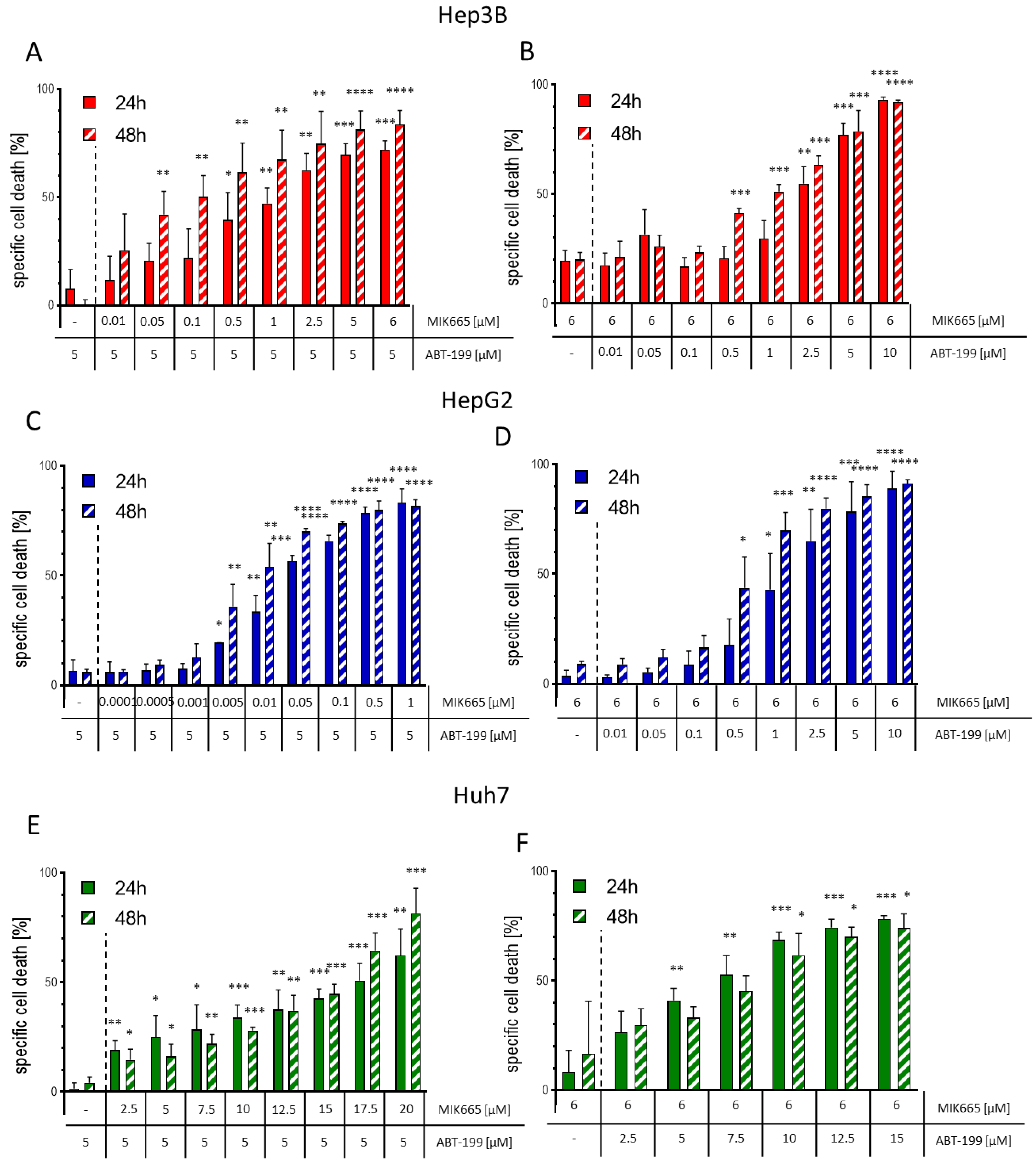
3.4. The Combination of ABT-199 and MIK665 Effectively Induces Cell Death in HCC Cell Lines
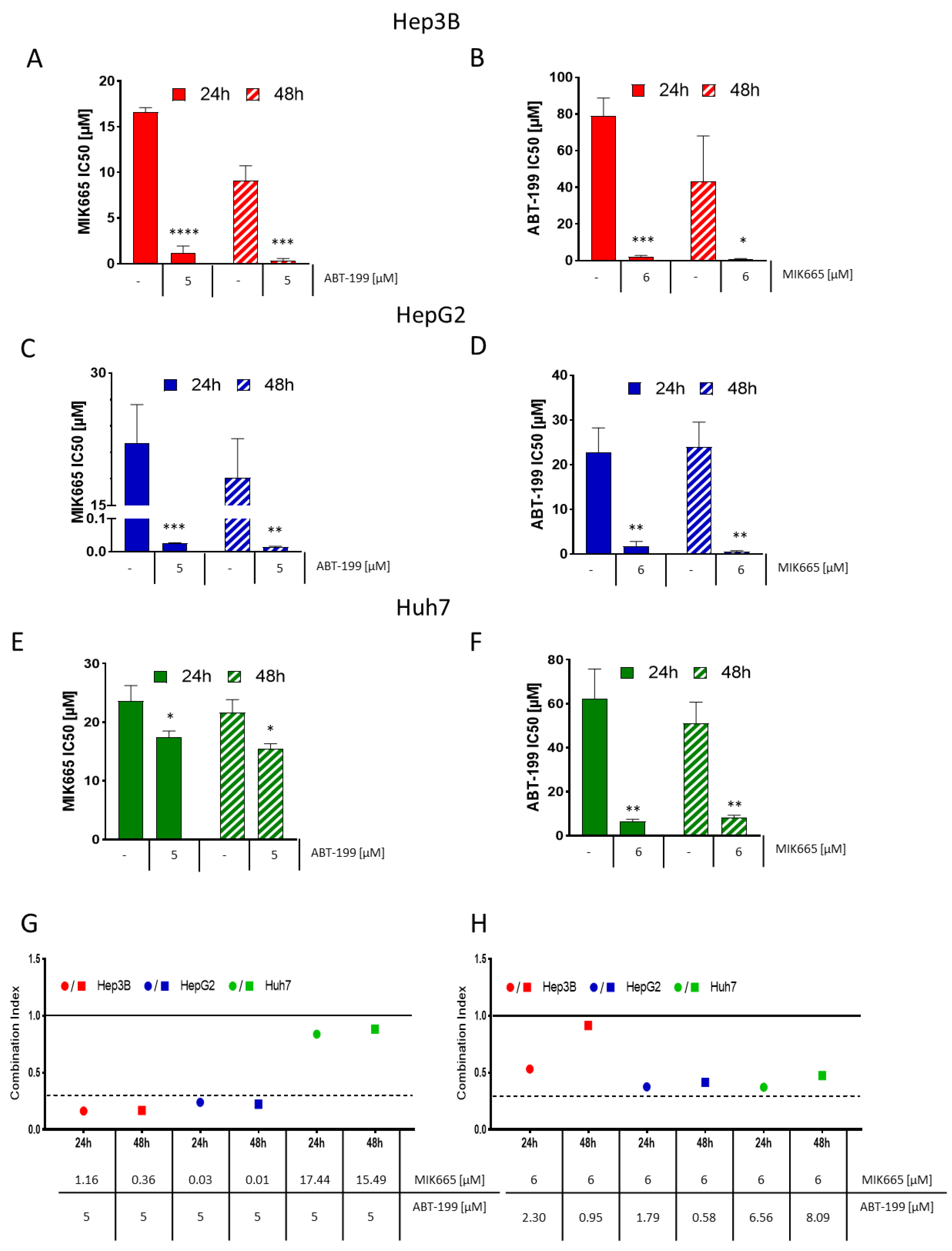
3.5. ABT-199 and MIK665 Act Synergistically in HCC Cell Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Townsend, P.A.; Kozhevnikova, M.V.; Cexus, O.N.F.; Zamyatnin, A.A.; Soond, S.M. BH3-mimetics: Recent developments in cancer therapy. J. Exp. Clin. Cancer Res. 2021, 40, 355. [Google Scholar] [CrossRef] [PubMed]
- Happo, L.; Strasser, A.; Cory, S. BH3-only proteins in apoptosis at a glance. J. Cell Sci. 2012, 125, 1081–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar] [CrossRef] [Green Version]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Müllauer, F.; Böck, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef] [Green Version]
- Pflaum, J.; Schlosser, S.; Müller, M. p53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef] [Green Version]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.K.; Smith, M.L.; Hessler, P.; Rapp, L.R.; Idler, K.B.; Park, C.H.; Leverson, J.D.; Lam, L.T. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 2017, 17, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderquist, R.S.; Crawford, L.; Liu, E.; Lu, M.; Agarwal, A.; Anderson, G.R.; Lin, K.H.; Winter, P.S.; Cakir, M.; Wood, K.C. Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nat. Commun. 2018, 9, 3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA Medication Guide Venclexta. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/208573s026lbl.pdf#page=54 (accessed on 25 August 2022).
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 2022, 22, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, F.; Merino, D.; Lee, L.; Breslin, K.; Pal, B.; Ritchie, M.E.; Smyth, G.K.; Christie, M.; Phillipson, L.J.; Burns, C.J.; et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell 2013, 24, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liu, H.; Xue, R.; Tang, W.; Zhang, S. BH3 Mimetic ABT-199 Enhances the Sensitivity of Gemcitabine in Pancreatic Cancer in vitro and in vivo. Dig. Dis. Sci. 2018, 63, 3367–3375. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Michels, J.; Obrist, F.; Vitale, I.; Lissa, D.; Garcia, P.; Behnam-Motlagh, P.; Kohno, K.; Wu, G.S.; Brenner, C.; Castedo, M.; et al. MCL-1 dependency of cisplatin-resistant cancer cells. Biochem. Pharmacol. 2014, 92, 55–61. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; van Dao, T.; de Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Chan, S.L.; Galle, P.R.; Rimassa, L.; Sangro, B. Systemic treatment of hepatocellular carcinoma: An EASL position paper. J. Hepatol. 2021, 75, 960–974. [Google Scholar] [CrossRef]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv238–iv255. [Google Scholar] [CrossRef] [PubMed]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.-L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Tümen, D.; Heumann, P.; Gülow, K.; Demirci, C.-N.; Cosma, L.-S.; Müller, M.; Kandulski, A. Pathogenesis and Current Treatment Strategies of Hepatocellular Carcinoma. Biomedicines 2022, 10, 3202. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting MCL-1 in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 67. [Google Scholar] [CrossRef]
- Fleischer, B.; Schulze-Bergkamen, H.; Schuchmann, M.; Weber, A.; Biesterfeld, S.; Müller, M.; Krammer, P.H.; Galle, P.R. Mcl-1 is an anti-apoptotic factor for human hepatocellular carcinoma. Int. J. Oncol. 2006, 28, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Kehr, S.; Vogler, M. It’s time to die: BH3 mimetics in solid tumors. Biochim. Et Biophys. Acta Mol. Cell Res. 2021, 1868, 118987. [Google Scholar] [CrossRef]
- Algarín, E.M.; Díaz-Tejedor, A.; Mogollón, P.; Hernández-García, S.; Corchete, L.A.; San-Segundo, L.; Martín-Sánchez, M.; González-Méndez, L.; Schoumacher, M.; Banquet, S.; et al. Preclinical evaluation of the simultaneous inhibition of MCL-1 and BCL-2 with the combination of S63845 and venetoclax in multiple myeloma. Haematologica 2020, 105, e116–e120. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, T.; van Vliet, A.; Garg, A.D.; Dewaele, M.; Rubio, N.; Gupta, S.; de Witte, P.; Samali, A.; Agostinis, P. Pro-apoptotic signaling induced by photo-oxidative ER stress is amplified by Noxa, not Bim. Biochem. Biophys. Res. Commun. 2013, 438, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Yokosuka, O.; Imazeki, F.; Arai, M.; Saisho, H. Enhanced Sensitivity of Human Hepatoma Cells to 5-Fluorouracil by Small Interfering RNA Targeting Bcl-2. DNA Cell Biol. 2005, 24, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-F.; Lin, J.-P.; Shiau, C.-W.; Tai, W.-T.; Liu, C.-Y.; Yu, H.-C.; Chen, P.-J.; Cheng, A.-L. Inhibition of Bcl-2 improves effect of LCL161, a SMAC mimetic, in hepatocellular carcinoma cells. Biochem. Pharmacol. 2012, 84, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Hikita, H.; Takehara, T.; Shimizu, S.; Kodama, T.; Shigekawa, M.; Iwase, K.; Hosui, A.; Miyagi, T.; Tatsumi, T.; Ishida, H.; et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology 2010, 52, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Cucarull, B.; Tutusaus, A.; Subías, M.; Stefanovic, M.; Hernáez-Alsina, T.; Boix, L.; Reig, M.; García de Frutos, P.; Marí, M.; Colell, A.; et al. Regorafenib Alteration of the BCL-xL/MCL-1 Ratio Provides a Therapeutic Opportunity for BH3-Mimetics in Hepatocellular Carcinoma Models. Cancers 2020, 12, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, F.; Lieber, J.; Dewerth, A.; Hoh, A.; Fuchs, J.; Armeanu-Ebinger, S. BH3 mimetics reduce adhesion and migration of hepatoblastoma and hepatocellular carcinoma cells. Exp. Cell Res. 2013, 319, 1443–1450. [Google Scholar] [CrossRef]
- Arnoult, D.; Petit, F.; Lelièvre, J.D.; Lecossier, D.; Hance, A.; Monceaux, V.; Hurtrel, B.; Ho Tsong Fang, R.; Ameisen, J.C.; Estaquier, J. Caspase-dependent and -independent T-cell death pathways in pathogenic simian immunodeficiency virus infection: Relationship to disease progression. Cell Death Differ. 2003, 10, 1240–1252. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, R.; Celis, E. T helper lymphocytes rescue CTL from activation-induced cell death. J. Immunol. 2006, 177, 2862–2872. [Google Scholar] [CrossRef] [Green Version]
- Neumann, L.; Pforr, C.; Beaudouin, J.; Pappa, A.; Fricker, N.; Krammer, P.H.; Lavrik, I.N.; Eils, R. Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol. Syst. Biol. 2010, 6, 352. [Google Scholar] [CrossRef]
- Kaminski, M.; Kiessling, M.; Süss, D.; Krammer, P.H.; Gülow, K. Novel role for mitochondria: Protein kinase Ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol. Cell. Biol. 2007, 27, 3625–3639. [Google Scholar] [CrossRef] [Green Version]
- Haderer, M.; Neubert, P.; Rinner, E.; Scholtis, A.; Broncy, L.; Gschwendtner, H.; Kandulski, A.; Pavel, V.; Mehrl, A.; Brochhausen, C.; et al. Novel pathomechanism for spontaneous bacterial peritonitis: Disruption of cell junctions by cellular and bacterial proteases. Gut 2022, 71, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Amato, C.M.; Skees, J.; Todd, K.J.; Lambert, K.A.; Robinson, W.A.; van Gulick, R.; Weight, R.M.; Dart, C.R.; Tobin, R.P.; et al. Simultaneously Inhibiting BCL2 and MCL1 Is a Therapeutic Option for Patients with Advanced Melanoma. Cancers 2020, 12, 2182. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Inoue-Yamauchi, A.; Jeng, P.S.; Kim, K.; Chen, H.-C.; Han, S.; Ganesan, Y.T.; Ishizawa, K.; Jebiwott, S.; Dong, Y.; Pietanza, M.C.; et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat. Commun. 2017, 8, 16078. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef] [Green Version]
- Chiou, J.-T.; Huang, N.-C.; Huang, C.-H.; Wang, L.-J.; Lee, Y.-C.; Shi, Y.-J.; Chang, L.-S. NOXA-mediated degradation of MCL1 and BCL2L1 causes apoptosis of daunorubicin-treated human acute myeloid leukemia cells. J. Cell. Physiol. 2021, 236, 7356–7375. [Google Scholar] [CrossRef]
- Bose, P.; Gandhi, V.; Konopleva, M. Pathways and mechanisms of venetoclax resistance. Leuk. Lymphoma 2017, 58, 2026–2039. [Google Scholar] [CrossRef]
- Fischer, U.; Jänicke, R.U.; Schulze-Osthoff, K. Many cuts to ruin: A comprehensive update of caspase substrates. Cell Death Differ. 2003, 10, 76–100. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Kudo, M.; Merle, P.; Meyer, T.; Qin, S.; Ikeda, M.; Xu, R.; Edeline, J.; Ryoo, B.-Y.; Ren, Z.; et al. LBA34 Primary results from the phase III LEAP-002 study: Lenvatinib plus pembrolizumab versus lenvatinib as first-line (1L) therapy for advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2022, 33, S1401. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Phase I Study of MIK665, a Mcl-1 Inhibitor, in Patients with Refractory or Relapsed Lymphoma or Multiple Myeloma—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02992483 (accessed on 23 May 2023).
- Phase I Study of S64315 Administred Intravenously in Patients with Acute Myeloid Leukaemia or Myelodysplastic Syndrome—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02979366?term=NCT02979366&draw=2&rank=1 (accessed on 23 May 2023).
- AMG 176 First in Human Trial in Participants with Relapsed or Refractory Multiple Myeloma and Participants with Relapsed or Refractory Acute Myeloid Leukemia—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02675452?term=NCT02675452&draw=2&rank=1 (accessed on 23 May 2023).
- A Study of PRT1419 in Patients with Relapsed/Refractory Hematologic Malignancies—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04543305?term=NCT04543305&draw=2&rank=1 (accessed on 23 May 2023).
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Lasica, M.; Anderson, M.A. Review of Venetoclax in CLL, AML and Multiple Myeloma. J. Pers. Med. 2021, 11, 463. [Google Scholar] [CrossRef]
- Institut de Recherches Internationales Servier|ADIR, a Servier Group company|Servier. Phase I Dose Escalation Study of Intravenously Administered S64315 in Combination with Orally Administered Venetoclax in Patients with Acute Myeloid Leukaemia: NCT03672695, CL1-64315-002|2018-001809-88. Available online: https://ClinicalTrials.gov/show/NCT03672695 (accessed on 26 May 2023).
- Li, Z.; He, S.; Look, A.T. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia 2019, 33, 262–266. [Google Scholar] [CrossRef]
- Fairlie, W.D.; Lee, E.F. Targeting the BCL-2-regulated apoptotic pathway for the treatment of solid cancers. Biochem. Soc. Trans. 2021, 49, 2397–2410. [Google Scholar] [CrossRef]
- Seiller, C.; Maiga, S.; Touzeau, C.; Bellanger, C.; Kervoëlen, C.; Descamps, G.; Maillet, L.; Moreau, P.; Pellat-Deceunynck, C.; Gomez-Bougie, P.; et al. Dual targeting of BCL2 and MCL1 rescues myeloma cells resistant to BCL2 and MCL1 inhibitors associated with the formation of BAX/BAK hetero-complexes. Cell Death Dis. 2020, 11, 316. [Google Scholar] [CrossRef]
- Sieghart, W.; Losert, D.; Strommer, S.; Cejka, D.; Schmid, K.; Rasoul-Rockenschaub, S.; Bodingbauer, M.; Crevenna, R.; Monia, B.P.; Peck-Radosavljevic, M.; et al. Mcl-1 overexpression in hepatocellular carcinoma: A potential target for antisense therapy. J. Hepatol. 2006, 44, 151–157. [Google Scholar] [CrossRef]
- Akgul, C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell. Mol. Life Sci. 2009, 66, 1326–1336. [Google Scholar] [CrossRef]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef] [PubMed]
- Touzeau, C.; Ryan, J.; Guerriero, J.; Moreau, P.; Chonghaile, T.N.; Le Gouill, S.; Richardson, P.; Anderson, K.; Amiot, M.; Letai, A. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 2016, 30, 761–764. [Google Scholar] [CrossRef]
- Anderson, M.A.; Deng, J.; Seymour, J.F.; Tam, C.; Kim, S.Y.; Fein, J.; Yu, L.; Brown, J.R.; Westerman, D.; Si, E.G.; et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 2016, 127, 3215–3224. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Wang, A.; Bai, Y.; Lin, J.; Yang, X.; Wang, D.; Yang, X.; Jiang, Y.; Zhao, H. Development and validation of a TP53-associated immune prognostic model for hepatocellular carcinoma. EBioMedicine 2019, 42, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michalski, M.; Bauer, M.; Walz, F.; Tümen, D.; Heumann, P.; Stöckert, P.; Gunckel, M.; Kunst, C.; Kandulski, A.; Schmid, S.; et al. Simultaneous Inhibition of Mcl-1 and Bcl-2 Induces Synergistic Cell Death in Hepatocellular Carcinoma. Biomedicines 2023, 11, 1666. https://doi.org/10.3390/biomedicines11061666
Michalski M, Bauer M, Walz F, Tümen D, Heumann P, Stöckert P, Gunckel M, Kunst C, Kandulski A, Schmid S, et al. Simultaneous Inhibition of Mcl-1 and Bcl-2 Induces Synergistic Cell Death in Hepatocellular Carcinoma. Biomedicines. 2023; 11(6):1666. https://doi.org/10.3390/biomedicines11061666
Chicago/Turabian StyleMichalski, Marlen, Magdalena Bauer, Franziska Walz, Deniz Tümen, Philipp Heumann, Petra Stöckert, Manuela Gunckel, Claudia Kunst, Arne Kandulski, Stephan Schmid, and et al. 2023. "Simultaneous Inhibition of Mcl-1 and Bcl-2 Induces Synergistic Cell Death in Hepatocellular Carcinoma" Biomedicines 11, no. 6: 1666. https://doi.org/10.3390/biomedicines11061666