

DAPTA, a C-C Chemokine Receptor 5 (CCR5), Leads to the Downregulation of Notch/NF-κB Signaling and Proinflammatory Mediators in CD40+ Cells in Experimental Autoimmune Encephalomyelitis Model in SJL/J Mice
 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
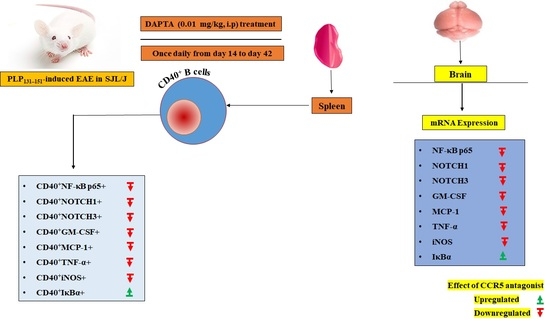
2.3. EAE Induction and DAPTA Treatment
2.4. Flow Cytometry Analysis
2.5. Real-Time PCR Analysis
2.6. Statistical Analysis
3. Results
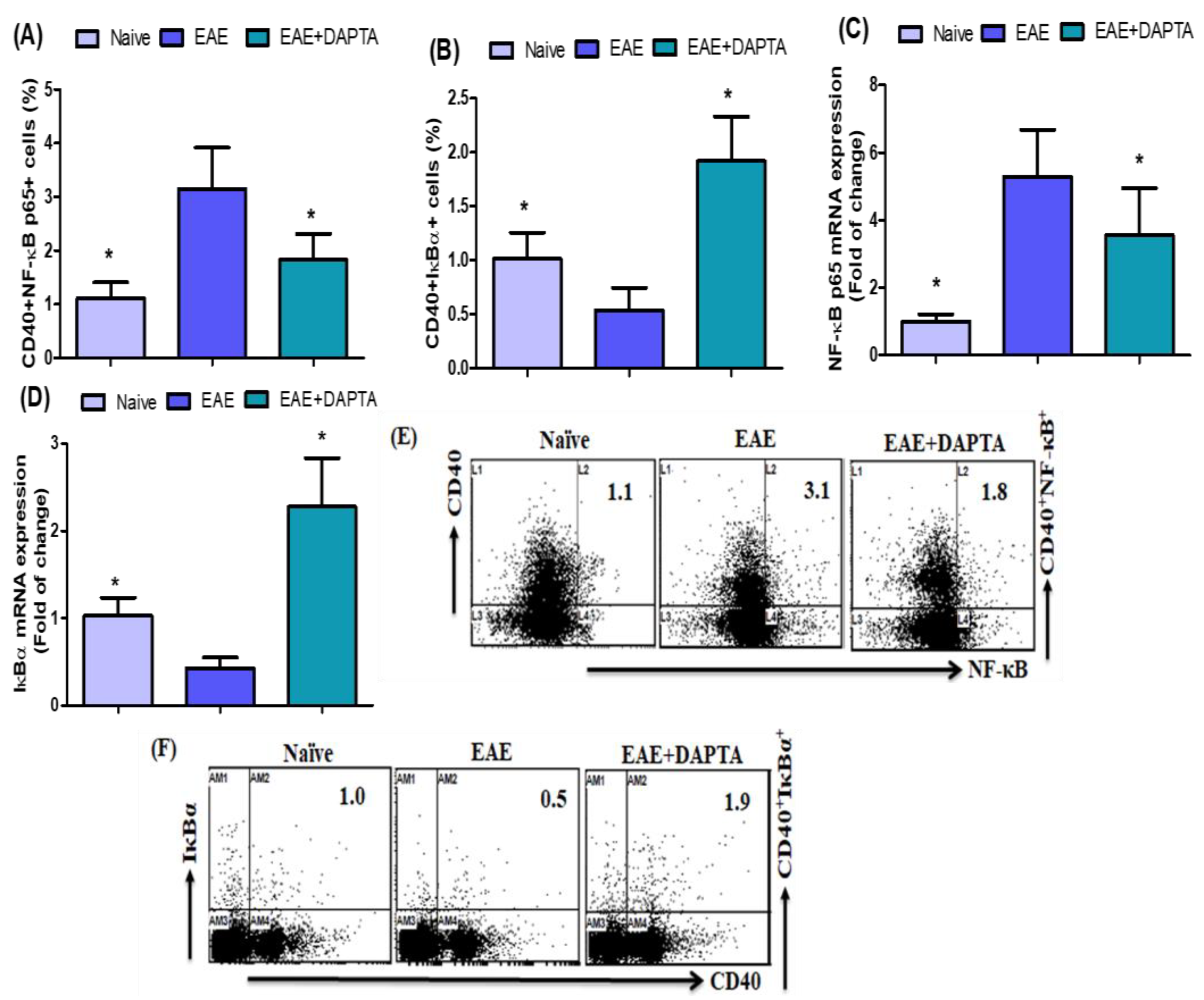
3.1. DAPTA Treatment Regulates NF-κB p65/IκBα Expression in EAE Mice
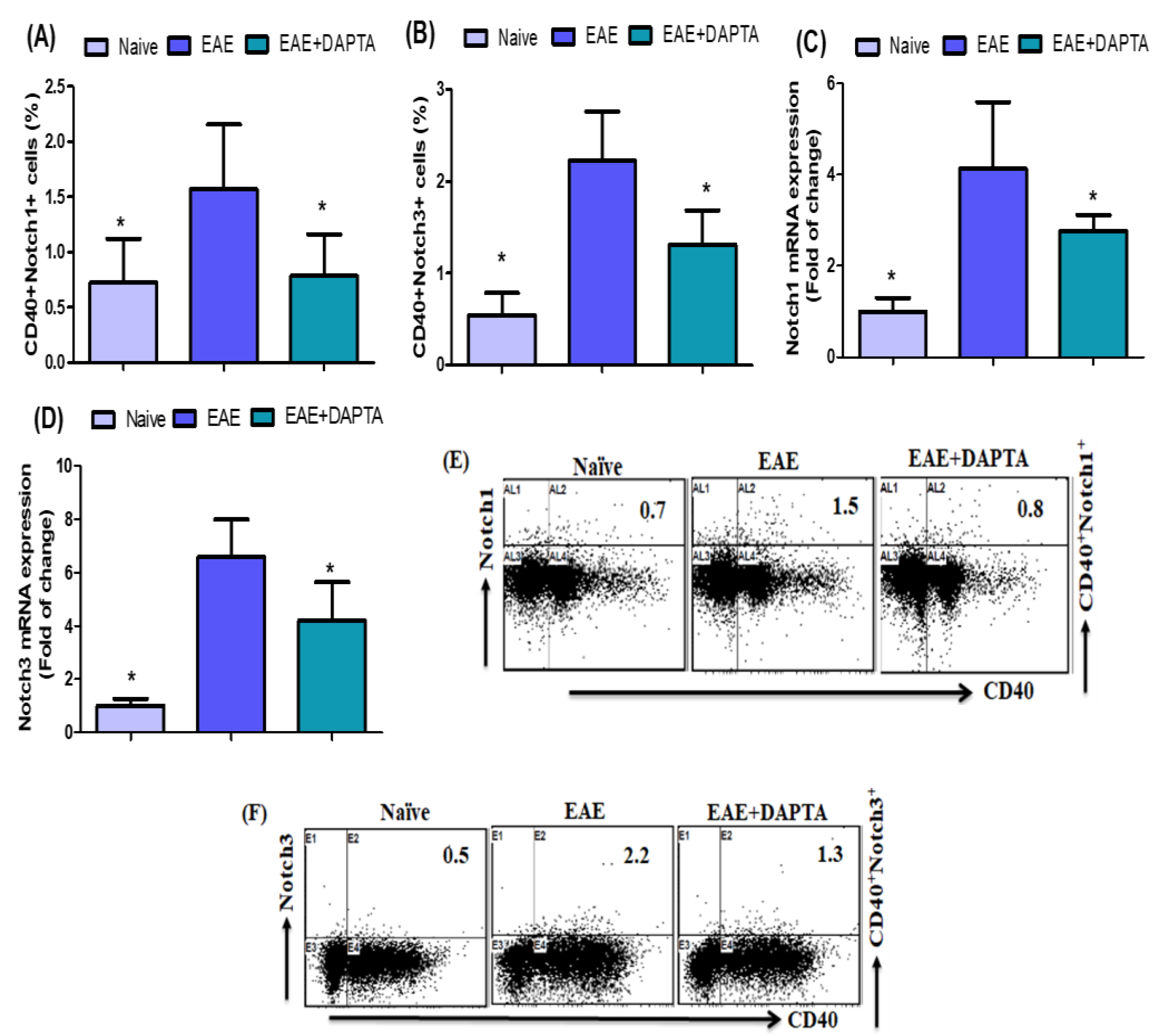
3.2. Effect of DAPTA on Notch1 and Notch3 Signaling
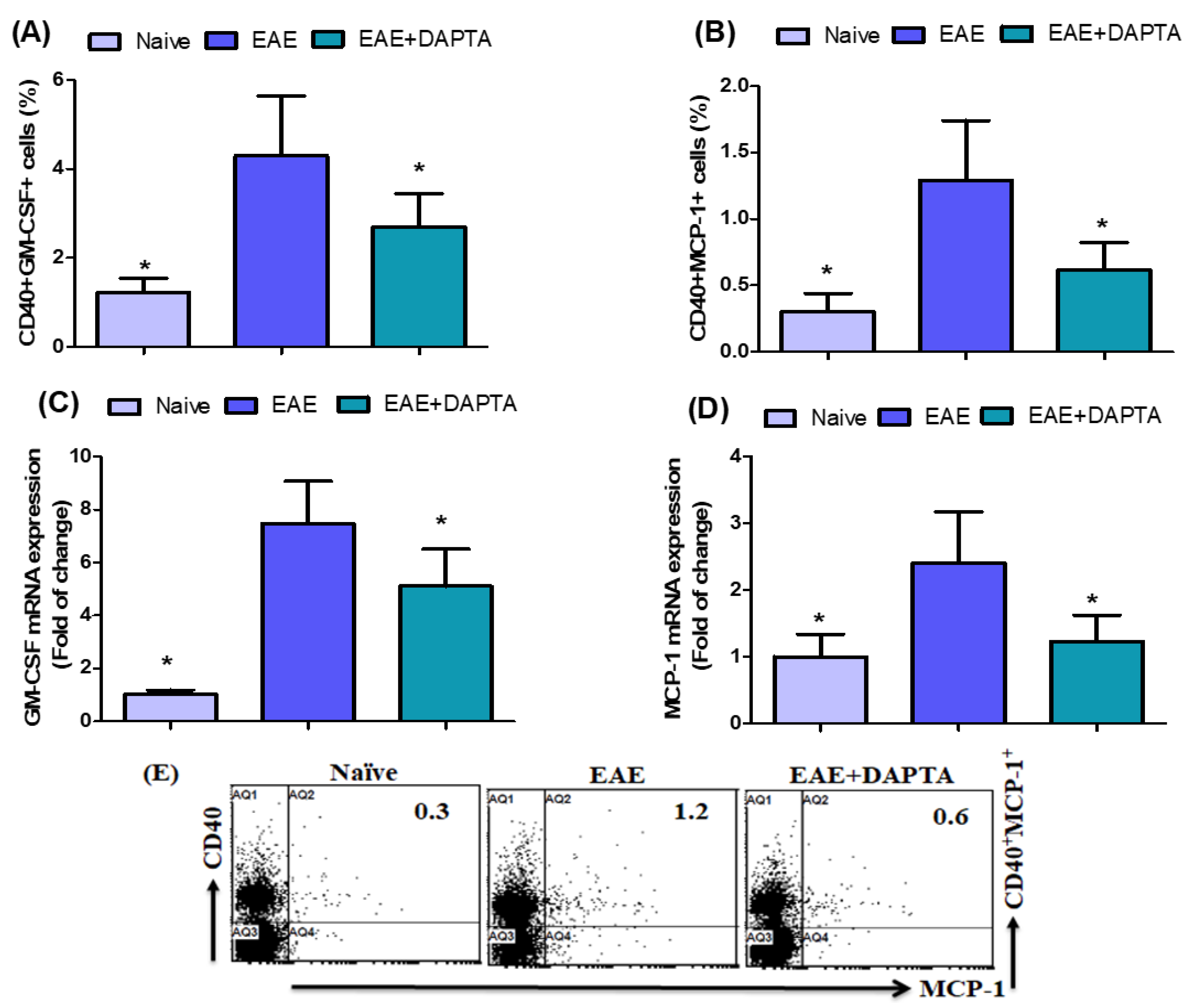
3.3. DAPTA Downregulates GM-CSF and MCP-1 Expression in EAE Mice
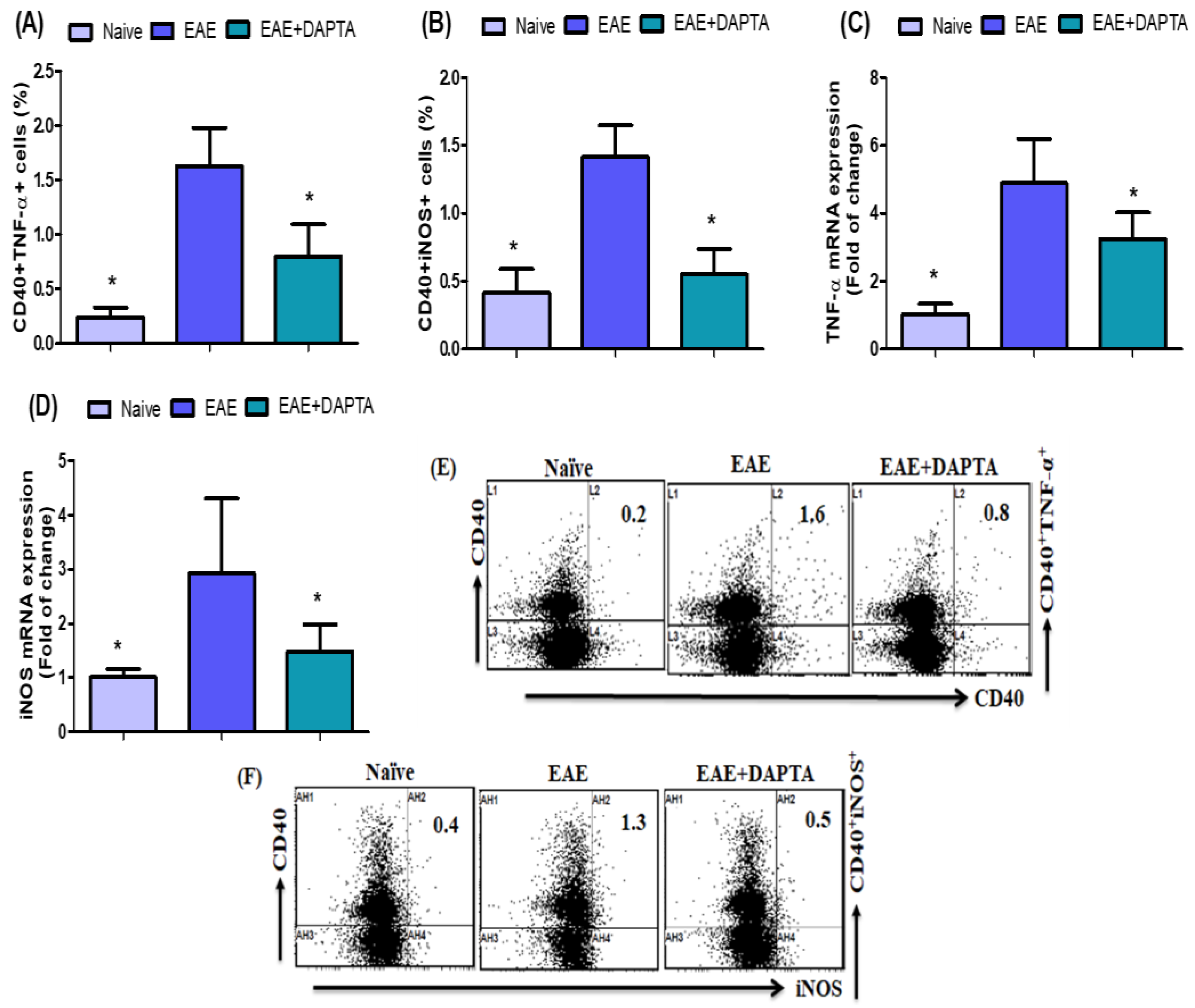
3.4. DAPTA Downregulates Proinflammatory Mediators in EAE Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lassmann, H.; Bruck, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Kahana, E. Multiple sclerosis: Geoepidemiology, genetics, and the environment. Autoimmun. Rev. 2010, 9, A387–A394. [Google Scholar] [CrossRef] [PubMed]
- Gu, C. KIR4.1: K+ Channel Illusion or Reality in the Autoimmune Pathogenesis of Multiple Sclerosis. Front. Mol. Neurosci. 2016, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Zhornitsky, S.; Johnson, T.A.; Metz, L.M.; Weiss, S.; Yong, V.W. Prolactin in combination with interferon-β reduces disease severity in an animal model of multiple sclerosis. J. Neuroinflammation 2015, 12, 55. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Handunnetthi, L.; Giovannoni, G.; Ramagopalan, S.V. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS ONE 2010, 5, e12496. [Google Scholar] [CrossRef]
- Cepok, S.; von Geldern, G.; Grummel, V.; Hochgesand, S.; Celik, H.; Hartung, H.; Hemmer, B. Accumulation of class switched IgD-IgM-memory B cells in the cerebrospinal fluid during neuroinflammation. J. Neuroimmunol. 2006, 180, 33–39. [Google Scholar] [CrossRef]
- von Budingen, H.C.; Harrer, M.D.; Kuenzle, S.; Meier, M.; Goebels, N. Clonally expanded plasma cells in the cerebrospinal fluid of MS patients produce myelin-specific antibodies. Eur. J. Immunol. 2008, 38, 2014–2023. [Google Scholar] [CrossRef]
- Hartung, D.M.; Bourdette, D.N.; Ahmed, S.M.; Whitham, R.H. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: Too big to fail? Neurology 2015, 84, 2185–2192. [Google Scholar] [CrossRef]
- Baird, J.F.; Sandroff, B.M.; Motl, R.W. Therapies for mobility disability in persons with multiple sclerosis. Expert Rev. Neurother. 2018, 18, 493–502. [Google Scholar] [CrossRef]
- Tur, C.; Carbonell-Mirabent, P.; Cobo-Calvo, Á.; Otero-Romero, S.; Arrambide, G.; Midaglia, L.; Castilló, J.; Vidal-Jordana, Á.; Rodríguez-Acevedo, B.; Zabalza, A.; et al. Association of Early Progression Independent of Relapse Activity With Long-term Disability After a First Demyelinating Event in Multiple Sclerosis. JAMA Neurol. 2023, 80, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Mc Guire, C.; Prinz, M.; Beyaert, R.; van Loo, G. Nuclear factor kappa B (NF-κB) in multiple sclerosis pathology. Trends Mol. Med. 2013, 19, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, B.; Stegagno, C.; Cannella, B.; Rizzuto, N.; Moretto, G.; Raine, C.S. Activation of NF-kappaB and c-jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am. J. Pathol. 1999, 155, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Stone, S.; Lin, W. Role of nuclear factor κB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural. Regen. Res. 2018, 13, 1507–1515. [Google Scholar]
- Chen, D.; Ireland, S.J.; Remington, G.; Alvarez, E.; Racke, M.K.; Greenberg, B.; Frohman, E.M.; Monson, N.L. CD40-Mediated NF-κB Activation in B Cells Is Increased in Multiple Sclerosis and Modulated by Therapeutics. J. Immunol. 2016, 197, 4257–4265. [Google Scholar] [CrossRef]
- Jurynczyk, M.; Jurewicz, A.; Bielecki, B.; Raine, C.S.; Selmaj, K. Inhibition of Notch signaling enhances tissue repair in an animal model of multiple sclerosis. J. Neuroimmunol. 2005, 170, 3–10. [Google Scholar] [CrossRef]
- Jurynczyk, M.; Jurewicz, A.; Raine, C.S.; Selmaj, K. Notch3 inhibition in myelin-reactive T cells down-regulates protein kinase Cθ and attenuates experimental autoimmune encephalomyelitis. J. Immunol. 2008, 180, 2634–2640. [Google Scholar] [CrossRef]
- Grandbarbe, L.; Michelucci, A.; Heurtaux, T.; Hemmer, K.; Morga, E.; Heuschling, P. Notch signaling modulates the activation of microglial cells. Glia 2007, 55, 1519–1530. [Google Scholar] [CrossRef]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–755. [Google Scholar] [CrossRef]
- Ifergan, I.; Davidson, T.S.; Kebir, H.; Xu, D.; Palacios-Macapagal, D.; Cann, J.; Rodgers, J.M.; Hunter, Z.N.; Pittet, C.L.; Beddow, S.; et al. Targeting the GM-CSF receptor for the treatment of CNS autoimmunity. J. Autoimmun. 2017, 84, 1–11. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Shriver, L.P.; Maresz, K.; Pedras-Vasconcelos, J.; Verthelyi, D.; Dittel, B.N. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 178, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Duncker, P.C.; Stoolman, J.S.; Huber, A.K.; Segal, B.M. GM-CSF Promotes Chronic Disability in Experimental Autoimmune Encephalomyelitis by Altering the Composition of Central Nervous System-Infiltrating Cells, but Is Dispensable for Disease Induction. J. Immunol. 2018, 200, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.E.; Newcombe, J.; Cuzner, M.L.; Woodroofe, M.N. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J. Neuroimmunol. 1998, 84, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Van Der Voorn, P.; Tekstra, J.; Beelen, R.H.; Tensen, C.P.; Van Der Valk, P.; De Groot, C.J. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am. J. Pathol. 1999, 154, 45–51. [Google Scholar] [CrossRef]
- Huang, D.R.; Wang, J.; Kivisakk, P.; Rollins, B.J.; Ransohoff, R.M. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 2001, 193, 713–726. [Google Scholar] [CrossRef]
- McManus, C.; Berman, J.W.; Brett, F.M.; Staunton, H.; Farrell, M.; Brosnan, C.F. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: An immunohistochemical and in situ hybridization study. J. Neuroimmunol. 1998, 86, 20–29. [Google Scholar] [CrossRef]
- Alam, M.S.; Gaida, M.M.; Bergmann, F.; Lasitschka, F.; Giese, T.; Giese, N.A.; Hackert, T.; Hinz, U.; Hussain, S.P.; Kozlov, S.V.; et al. Selective inhibition of the p38 alternative activation pathway in infiltrating T cells inhibits pancreatic cancer progression. Nat. Med. 2015, 21, 1337–1343. [Google Scholar] [CrossRef]
- Bałkowiec-Iskra, E.; Vermehren-Schmaedick, A.; Balkowiec, A. Tumor necrosis factor-α increases brain-derived neurotrophic factor expression in trigeminal ganglion neurons in an activity-dependent manner. Neuroscience 2011, 180, 322–333. [Google Scholar] [CrossRef]
- Sharief, M.K.; Hentges, R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 1991, 325, 467–472. [Google Scholar] [CrossRef]
- Fresegna, D.; Bullitta, S.; Musella, A.; Rizzo, F.R.; De Vito, F.; Guadalupi, L.; Caioli, S.; Balletta, S.; Sanna, K.; Dolcetti, E.; et al. Re-examining the role of TNF in MS pathogenesis and therapy. Cells 2020, 9, 2290. [Google Scholar] [CrossRef]
- Jung, K.; Kim, J.; Ahn, G.; Matsuda, H.; Akane, T.; Ahn, M.; Shin, T. Alendronate alleviates the symptoms of experimental autoimmune encephalomyelitis. Int. Immunopharmacol. 2020, 84, 106534. [Google Scholar] [CrossRef] [PubMed]
- Shin, T.; Kim, S.; Moon, C.; Wie, M.; Kim, H. Aminoguanidine-induced amelioration of autoimmune encephalomyelitis is mediated by reduced expression of inducible nitric oxide synthase in the spinal cord. Immunol. Investig. 2000, 29, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.; Ahn, M.; Jee, Y.; Heo, S.; Kim, S.; Kim, H.; Sim, K.B.; Koh, C.S.; Shin, Y.G.; Shin, T. Sodium salicylate-induced amelioration of experimental autoimmune encephalomyelitis in Lewis rats is associated with the suppression of inducible nitric oxide synthase and cyclooxygenases. Neurosci. Lett. 2004, 356, 123–126. [Google Scholar] [CrossRef]
- Banisadr, G.; Gosselin, R.D.; Mechighel, P.; Kitabgi, P.; Rostene, W.; Parsadaniantz, S.M. Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: Evidence for its colocalization with neurotransmitters and neuropeptides. J. Comp. Neurol. 2005, 489, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Godiska, R.; Chantry, D.; Dietsch, G.N.; Gray, P.W. Chemokine expression in murine experimental allergic encephalomyelitis. J. Neuroimmunol. 1995, 58, 167–176. [Google Scholar] [CrossRef]
- Muller, D.M.; Pender, M.P.; Greer, J.M. Chemokines and chemokine receptors: Potential therapeutic targets in multiple sclerosis. Curr. Drug Targets Inflamm. Allergy 2004, 3, 279–290. [Google Scholar] [CrossRef]
- Gu, S.M.; Park, M.H.; Yun, H.M.; Han, S.B.; Oh, K.W.; Son, D.J.; Yun, J.S.; Hong, J.T. CCR5 knockout suppresses experimental autoimmune encephalomyelitis in C57BL/6 mice. Oncotarget 2016, 7, 15382–15393. [Google Scholar] [CrossRef]
- dos Santos, A.C.; Barsante, M.M.; Arantes, R.M.E.; Bernard, C.C.; Teixeira, M.M.; Carvalho-Tavares, J. CCL2 and CCL5 mediate leukocyte adhesion in experimental autoimmune encephalomyelitis–an intravital microscopy study. J. Neuroimmunol. 2005, 162, 122–129. [Google Scholar] [CrossRef]
- Balashov, K.E.; Rottman, J.B.; Weiner, H.L.; Hancock, W.W. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6873–6878. [Google Scholar] [CrossRef]
- Fischer, F.R.; Santambrogio, L.; Luo, Y.; Berman, M.A.; Hancock, W.W.; Dorf, M.E. Modulation of experimental autoimmune encephalomyelitis: Effect of altered peptide ligand on chemokine and chemokine receptor expression. J. Neuroimmunol. 2000, 110, 195–208. [Google Scholar] [CrossRef]
- Moriguchi, K.; Miyamoto, K.; Tanaka, N.; Ueno, R.; Nakayama, T.; Yoshie, O.; Kusunoki, S. C-C chemokine receptor type 4 antagonist Compound ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2016, 291, 54–58. [Google Scholar] [CrossRef]
- Redwine, L.S.; Pert, C.B.; Rone, J.D.; Nixon, R.; Vance, M.; Sandler, B.; Lumpkin, M.D.; Dieter, D.J.; Ruff, M.R. Peptide T blocks GP120/CCR5 chemokine receptor-mediated chemotaxis. Clin. Immunol. 1999, 93, 124–131. [Google Scholar] [CrossRef]
- Rosi, S.; Pert, C.B.; Ruff, M.R.; McGann-Gramling, K.; Wenk, G.L. Chemokine receptor 5 antagonist D-Ala-peptide T-amide reduces microglia and astrocyte activation within the hippocampus in a neuroinflammatory rat model of Alzheimer’s disease. Neuroscience 2005, 134, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Choi, D.Y.; Jung, Y.Y.; Yun, Y.W.; Lee, B.J.; Han, S.B.; Hong, J.T. Decreased pain responses of C-C chemokine receptor 5 knockout mice to chemical or inflammatory stimuli. Neuropharmacology 2013, 67, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Alotaibi, M.R.; Alasmari, A.F.; Alshammari, M.A.; Al-Mazroua, H.A.; Attia, S.M. DAPTA, a C-C chemokine receptor 5 (CCR5) antagonist attenuates immune aberrations by downregulating Th9/Th17 immune responses in BTBR T+ Itpr3tf/J mice. Eur. J. Pharmacol. 2019, 846, 100–108. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Nadeem, A.; Ansari, M.A.; Bakheet, S.A.; Shahid, M.; Al-Mazroua, H.A.; Sobeai, H.M.A.; Alasmari, A.F.; Alanazi, M.M.; Alhamed, A.S.; et al. CC chemokine receptor 5 antagonist alleviates inflammation by regulating IFN-γ/IL-10 and STAT4/Smad3 signaling in a mouse model of autoimmune encephalomyelitis. Cell Immunol. 2022, 379, 104580. [Google Scholar] [CrossRef] [PubMed]
- Furlan, R.; Cuomo, C.; Martino, G. Animal models of multiple sclerosis. Methods Mol. Biol. 2009, 549, 157–173. [Google Scholar]
- Jolivel, V.; Luessi, F.; Masri, J.; Kraus, S.H.; Hubo, M.; Poisa-Beiro, L.; Klebow, S.; Paterka, M.; Yogev, N.; Tumani, H.; et al. Modulation of dendritic cell properties by laquinimod as a mechanism for modulating multiple sclerosis. Brain 2013, 136 Pt 4, 1048–1066. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple sclerosis: Mechanisms and immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Hohlfeld, R.; Dornmair, K.; Meinl, E.; Wekerle, H. The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. 2016, 15, 317–331. [Google Scholar] [CrossRef]
- Barclay, W.; Shinohara, M.L. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol. 2017, 27, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Eggers, E.L.; Michel, B.A.; Wu, H.; Wang, S.Z.; Bevan, C.J.; Abounasr, A.; Pierson, N.S.; Bischof, A.; Kazer, M.; Leitner, E.; et al. Clonal relationships of CSF B cells in treatment-naive multiple sclerosis patients. JCI Insight 2017, 2, e92724. [Google Scholar] [CrossRef]
- Greenfield, A.L.; Dandekar, R.; Ramesh, A.; Eggers, E.L.; Wu, H.; Laurent, S.; Harkin, W.; Pierson, N.S.; Weber, M.S.; Henry, R.G.; et al. Longitudinally persistent cerebrospinal fluid B cells can resist treatment in multiple sclerosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Bar-Or, A.; Fawaz, L.; Fan, B.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.; et al. Abnormal B-cell cytokine responses: A trigger of T-cell–mediated disease in MS? Ann. Neurol. 2010, 67, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Aarts, S.A.B.M.; Seijkens, T.T.P.; van Dorst, K.J.F.; Dijkstra, C.D.; Kooij, G.; Lutgens, E. The CD40-CD40L dyad in experimental autoimmune encephalomyelitis and multiple sclerosis. Front. Immunol. 2017, 8, 1791. [Google Scholar] [CrossRef]
- Aarts, S.A.; Seijkens, T.T.; Kusters, P.J.; van Tiel, C.M.; Reiche, M.E.; den Toom, M.; Beckers, L.; van Roomen, C.P.; de Winther, M.P.; Kooij, G.; et al. Macrophage CD40 signaling drives experimental autoimmune encephalomyelitis. J. Pathol. 2019, 247, 471–480. [Google Scholar] [CrossRef]
- Iezzi, G.; Sonderegger, I.; Ampenberger, F.; Schmitz, N.; Marsland, B.J.; Kopf, M. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 876–881. [Google Scholar] [CrossRef]
- Becher, B.; Durell, B.G.; Miga, A.V.; Hickey, W.F.; Noelle, R.J. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J. Exp. Med. 2001, 193, 967–974. [Google Scholar] [CrossRef]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-kB signaling as a therapeutic target in autoimmunity. J. Biomol. Screen. 2016, 21, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Lin, Y.; Li, J.; Fenstermaker, A.G.; Way, S.W.; Clayton, B.; Jamison, S.; Harding, H.P.; Ron, D.; Popko, B. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J. Neurosci. 2013, 33, 5980–5991. [Google Scholar] [CrossRef]
- Leibowitz, S.M.; Yan, J. NF-κB Pathways in the Pathogenesis of Multiple Sclerosis and the Therapeutic Implications. Front. Mol. Neurosci. 2016, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Greer, J.M.; NF-kappa, B. A potential therapeutic target for the treatment of multiple sclerosis. CNS Neurol. Disord. Drug Targets 2008, 7, 536–557. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Fang, T.C.; Artis, D.; Shestova, O.; Pross, S.E.; Maillard, I.; Pear, W.S. Notch signaling is an important regulator of type 2 immunity. J. Exp. Med. 2005, 202, 1037–1042. [Google Scholar] [CrossRef]
- Sandy, A.R.; Stoolman, J.; Malott, K.; Pongtornpipat, P.; Segal, B.M.; Maillard, I. Notch signaling regulates T cell accumulation and function in the central nervous system during experimental autoimmune encephalomyelitis. J. Immunol. 2013, 191, 1606–1613. [Google Scholar] [CrossRef]
- Fernández, M.; Monsalve, E.M.; López-López, S.; Ruiz-García, A.; Mellado, S.; Caminos, E.; García-Ramírez, J.J.; Laborda, J.; Tranque, P.; Díaz-Guerra, M.J.M. Absence of Notch1 in murine myeloid cells attenuates the development of experimental autoimmune encephalomyelitis by affecting Th1 and Th17 priming. Eur. J. Immunol. 2017, 47, 2090–2100. [Google Scholar] [CrossRef]
- Dishowitz, M.I.; Mutyaba, P.L.; Takacs, J.D.; Barr, A.M.; Engiles, J.B.; Ahn, J.; Hankenson, K.D. Systemic inhibition of canonical Notch signaling results in sustained callus inflammation and alters multiple phases of fracture healing. PLoS ONE 2013, 8, e68726. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Gaignage, M.; Donckers, D.; Nasr, Z.; Cheou, P.; van Snick, J.; D’Auria, L.; van Pesch, V. Prophylactic treatment against GM-CSF, but not IL-17, abolishes relapses in a chronic murine model of multiple sclerosis. Eur. J. Immunol. 2018, 48, 1883–1891. [Google Scholar] [CrossRef]
- Rasouli, J.; Ciric, B.; Imitola, J.; Gonnella, P.; Hwang, D.; Mahajan, K.; Mari, E.R.; Safavi, F.; Leist, T.P.; Zhang, G.X.; et al. Expression of GM-CSF in T Cells Is Increased in Multiple Sclerosis and Suppressed by IFN-beta Therapy. J. Immunol. 2015, 194, 5085–5093. [Google Scholar] [CrossRef]
- McQualter, J.L.; Darwiche, R.; Ewing, C.; Onuki, M.; Kay, T.W.; Hamilton, J.A.; Reid, H.H.; Bernard, C.C. Granulocyte macrophage colony-stimulating factor: A new putative therapeutic target in multiple sclerosis. J. Exp. Med. 2001, 194, 873–882. [Google Scholar] [CrossRef]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, K.J.; Strieter, R.M.; Kunkel, S.L.; Lukacs, N.W.; Karpus, W.J. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1alpha and monocyte chemotactic protein-1. J. Neuroimmunol. 1998, 92, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Berman, J.W.; Guida, M.P.; Warren, J.; Amat, J.; Brosnan, C.F. Localization of monocyte chemoattractant peptide-1 expression in the central nervous system in experimental autoimmune encephalomyelitis and trauma in the rat. J. Immunol. 1996, 156, 3017–3023. [Google Scholar] [CrossRef]
- Nygårdas, P.T.; Määttä, J.A.; Hinkkanen, A.E. Chemokine expression by central nervous system resident cells and infiltrating neutrophils during experimental autoimmune encephalomyelitis in the BALB/c mouse. Eur. J. Immunol. 2000, 30, 1911–1918. [Google Scholar] [CrossRef]
- Glabinski, A.R.; Ransohoff, R.M. Chemokines and chemokine receptors in CNS pathology. J. Neurovirol. 1999, 5, 3–12. [Google Scholar] [CrossRef]
- Youssef, S.; Wildbaum, G.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by MIP-1alpha and MCP-1 naked DNA vaccines. J. Autoimmun. 1999, 13, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Zong, Y.; Mohammad, A.; Ajit, D.; Cui, J.; Han, D.; Hamilton, J.L.; Simonyi, A.; Sun, A.Y.; Gu, Z.; et al. Pro-inflammatory cytokines and lipopolysaccharide induce changes in cell morphology, and upregulation of ERK1/2, iNOS and sPLA(2)-IIA expression in astrocytes and microglia. J. Neuroinf. 2011, 8, 121. [Google Scholar] [CrossRef]
- Bagasra, O.; Michaels, F.H.; Zheng, Y.M.; Bobroski, L.E.; Spitsin, S.V.; Fu, Z.F.; Tawadros, R.; Koprowski, H. Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 12041–12045. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Du, M.; Gao, Y.; Liu, H.; Wang, H.; Wu, X.; Wang, Z. Astragaloside IV Attenuates Experimental Autoimmune Encephalomyelitis of Mice by Counteracting Oxidative Stress at Multiple Levels. PLoS ONE 2013, 8, e76495. [Google Scholar] [CrossRef] [PubMed]
- Danilov, A.I.; Andersson, M.; Bavand, N.; Wiklund, N.P.; Olsson, T.; Brundin, L. Nitric oxide metabolite determinations reveal continuous inflammation in multiple sclerosis. J. Neuroimmunol. 2003, 136, 112–118. [Google Scholar] [CrossRef]
- Willenborg, D.O.; Staykova, M.; Fordham, S.; O’Brien, N.; Linares, D. The contribution of nitric oxide and interferon gamma to the regulation of the neuro-inflammation in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2007, 191, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Selak, M.; O’Connor, J.; Croul, S.; Lorenzana, C.; Butunoi, C.; Kalman, B. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J. Neurol. Sci. 2000, 177, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Renno, T.; Krakowski, M.; Piccirillo, C.; Lin, J.Y.; Owens, T. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis: Regulation by Th1 cytokines. J. Immunol. 1995, 154, 944–953. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alghibiwi, H.; Ansari, M.A.; Nadeem, A.; Algonaiah, M.A.; Attia, S.M.; Bakheet, S.A.; Albekairi, T.H.; Almudimeegh, S.; Alhamed, A.S.; Shahid, M.; et al. DAPTA, a C-C Chemokine Receptor 5 (CCR5), Leads to the Downregulation of Notch/NF-κB Signaling and Proinflammatory Mediators in CD40+ Cells in Experimental Autoimmune Encephalomyelitis Model in SJL/J Mice. Biomedicines 2023, 11, 1511. https://doi.org/10.3390/biomedicines11061511
Alghibiwi H, Ansari MA, Nadeem A, Algonaiah MA, Attia SM, Bakheet SA, Albekairi TH, Almudimeegh S, Alhamed AS, Shahid M, et al. DAPTA, a C-C Chemokine Receptor 5 (CCR5), Leads to the Downregulation of Notch/NF-κB Signaling and Proinflammatory Mediators in CD40+ Cells in Experimental Autoimmune Encephalomyelitis Model in SJL/J Mice. Biomedicines. 2023; 11(6):1511. https://doi.org/10.3390/biomedicines11061511
Chicago/Turabian StyleAlghibiwi, Hanan, Mushtaq A. Ansari, Ahmed Nadeem, Majed Ali Algonaiah, Sabry M. Attia, Saleh A. Bakheet, Thamer H. Albekairi, Sultan Almudimeegh, Abdullah S. Alhamed, Mudassar Shahid, and et al. 2023. "DAPTA, a C-C Chemokine Receptor 5 (CCR5), Leads to the Downregulation of Notch/NF-κB Signaling and Proinflammatory Mediators in CD40+ Cells in Experimental Autoimmune Encephalomyelitis Model in SJL/J Mice" Biomedicines 11, no. 6: 1511. https://doi.org/10.3390/biomedicines11061511