
DCLK1 Drives EGFR-TKI-Acquired Resistance in Lung Adenocarcinoma by Remodeling the Epithelial–Mesenchymal Transition Status

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Transwell Assays
2.3. CRISPR-Cas9-Mediated Knockout of DCLK1
2.4. DCLK1 Rescue in Knockout Cell Lines
2.5. IC50 Determination
2.6. Western Blotting and Antibodies
2.7. RNA Extraction and Real-Time PCR
2.8. Xenograft Tumor Model in Nude Mice
2.9. Multiple Immunofluorescence Staining
2.10. Data and Statistical Analysis
3. Results
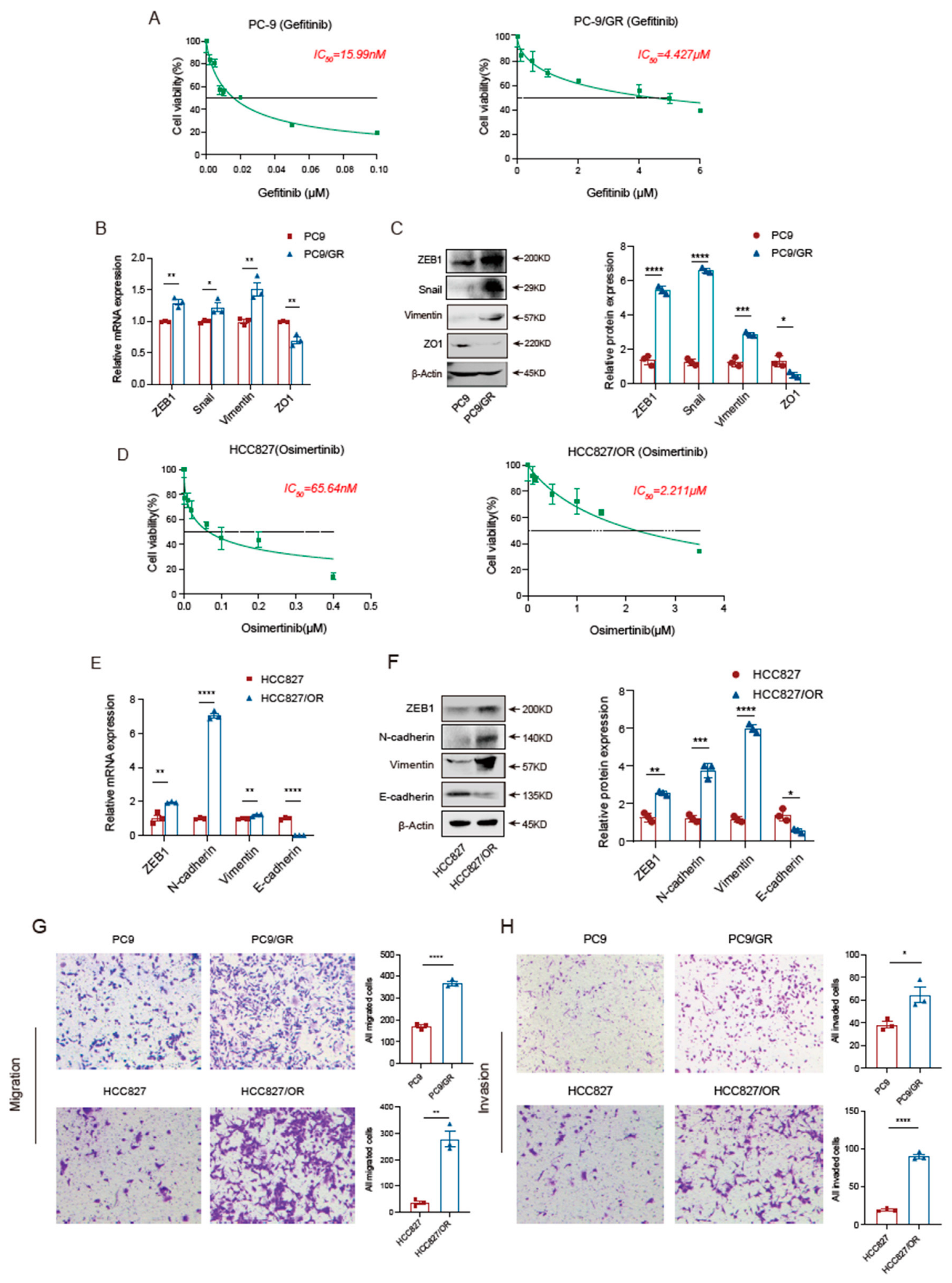
3.1. EGFR-TKI-Resistant Cells Have More Robust Migration and Invasion Abilities Than Sensitive Cells
3.2. Knockout of DCLK1 Suppresses the EMT Program of TKI-Resistant Cells
3.3. DCLK1 Rescue Restores the Malignant Phenotype of TKI-Resistant Cells
3.4. DCLK1 Mediates EMT Program Activation in TKI-Resistant Cells In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verstappe, J.; Berx, G. A role for partial epithelial-to-mesenchymal transition in enabling stemness in homeostasis and cancer. Semin. Cancer Biol. 2023, 90, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Hsu, W.H.; Yang, J.C.; Mok, T.S.; Loong, H.H. Overview of current systemic management of EGFR-mutant NSCLC. Ann. Oncol. 2018, 29, i3–i9. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Passaro, A.; Janne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Miyauchi, E.; Morita, S.; Nakamura, A.; Hosomi, Y.; Watanabe, K.; Ikeda, S.; Seike, M.; Fujita, Y.; Minato, K.; Ko, R.; et al. Updated Analysis of NEJ009: Gefitinib-Alone Versus Gefitinib Plus Chemotherapy for Non-Small-Cell Lung Cancer with Mutated EGFR. J. Clin. Oncol. 2022, 40, 3587–3592. [Google Scholar] [CrossRef]
- Noronha, V.; Patil, V.M.; Joshi, A.; Menon, N.; Chougule, A.; Mahajan, A.; Janu, A.; Purandare, N.; Kumar, R.; More, S.; et al. Gefitinib Versus Gefitinib Plus Pemetrexed and Carboplatin Chemotherapy in EGFR-Mutated Lung Cancer. J. Clin. Oncol. 2020, 38, 124–136. [Google Scholar] [CrossRef]
- Tang, Z.H.; Lu, J.J. Osimertinib resistance in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Lett. 2018, 420, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.G.; Shih, J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef]
- Jakobsen, K.R.; Demuth, C.; Sorensen, B.S.; Nielsen, A.L. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 172–182. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Iwata, T.; Onitsuka, T.; Shimokawa, H.; Hanagiri, T.; Oyama, T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010, 30, 2513–2517. [Google Scholar]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Koizumi, H.; Fujioka, H.; Togashi, K.; Thompson, J.; Yates, J.R., 3rd; Gleeson, J.G.; Emoto, K. DCLK1 phosphorylates the microtubule-associated protein MAP7D1 to promote axon elongation in cortical neurons. Dev. Neurobiol. 2017, 77, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, H.; Tanaka, T.; Gleeson, J.G. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron 2006, 49, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Patel, O.; Dai, W.; Mentzel, M.; Griffin, M.D.; Serindoux, J.; Gay, Y.; Fischer, S.; Sterle, S.; Kropp, A.; Burns, C.J.; et al. Biochemical and Structural Insights into Doublecortin-like Kinase Domain 1. Structure 2016, 24, 1550–1561. [Google Scholar] [CrossRef]
- Cheng, L.; Yang, Z.; Guo, W.; Wu, C.; Liang, S.; Tong, A.; Cao, Z.; Thorne, R.F.; Yang, S.Y.; Yu, Y.; et al. DCLK1 autoinhibition and activation in tumorigenesis. Innovation 2022, 3, 100191. [Google Scholar] [CrossRef]
- Agulto, R.L.; Rogers, M.M.; Tan, T.C.; Ramkumar, A.; Downing, A.M.; Bodin, H.; Castro, J.; Nowakowski, D.W.; Ori-McKenney, K.M. Autoregulatory control of microtubule binding in doublecortin-like kinase 1. Elife 2021, 10. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef]
- Bailey, J.M.; Alsina, J.; Rasheed, Z.A.; McAllister, F.M.; Fu, Y.Y.; Plentz, R.; Zhang, H.; Pasricha, P.J.; Bardeesy, N.; Matsui, W.; et al. DCLK1 Marks a Morphologically Distinct Subpopulation of Cells with Stem Cell Properties in Preinvasive Pancreatic Cancer. Gastroenterology 2014, 146, 245–256. [Google Scholar] [CrossRef]
- Nevi, L.; Di Matteo, S.; Carpino, G.; Zizzari, I.G.; Samira, S.; Ambrosino, V.; Costantini, D.; Overi, D.; Giancotti, A.; Monti, M.; et al. DCLK1, a Putative Stem Cell Marker in Human Cholangiocarcinoma. Hepatology 2021, 73, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Weygant, N.; Qu, D.; Berry, W.L.; May, R.; Chandrakesan, P.; Owen, D.B.; Sureban, S.M.; Ali, N.; Janknecht, R.; Houchen, C.W. Small molecule kinase inhibitor LRRK2-IN-1 demonstrates potent activity against colorectal and pancreatic cancer through inhibition of doublecortin-like kinase 1. Mol. Cancer 2014, 13, 103. [Google Scholar] [CrossRef]
- Chandrakesan, P.; Panneerselvam, J.; Qu, D.; Weygant, N.; May, R.; Bronze, M.S.; Houchen, C.W. Regulatory Roles of Dclk1 in Epithelial Mesenchymal Transition and Cancer Stem Cells. J. Carcinog. Mutagen. 2016, 7, 257. [Google Scholar] [CrossRef]
- Tao, H.; Tanaka, T.; Okabe, K. Doublecortin and CaM kinase-like-1 expression in pathological stage I non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2017, 143, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.Q.; Zhao, W.J.; Zhu, L.L.; Xu, S.J.; Zhang, X.L.; Liang, Y.; Ding, X.F.; Kiselyov, A.; Chen, G. XMD-17-51 Inhibits DCLK1 Kinase and Prevents Lung Cancer Progression. Front. Pharmacol. 2021, 12, 603453. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, J.; Mohandoss, P.; Patel, R.; Gillan, H.; Li, M.; Kumar, K.; Nguyen, D.; Weygant, N.; Qu, D.; Pitts, K.; et al. DCLK1 Regulates Tumor Stemness and Cisplatin Resistance in Non-small Cell Lung Cancer via ABCD-Member-4. Mol. Oncolytics 2020, 18, 24–36. [Google Scholar] [CrossRef]
- Yan, R.; Fan, X.; Xiao, Z.; Liu, H.; Huang, X.; Liu, J.; Zhang, S.; Yao, J.; An, G.; Ge, Y. Inhibition of DCLK1 sensitizes resistant lung adenocarcinomas to EGFR-TKI through suppression of Wnt/beta-Catenin activity and cancer stemness. Cancer Lett. 2022, 531, 83–97. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Pan, J.; Xiong, D.; Zhang, Q.; Palen, K.; Shoemaker, R.H.; Johnson, B.; Sei, S.; Wang, Y.; You, M. Precision immunointerception of EGFR-driven tumorigenesis for lung cancer prevention. Front. Immunol. 2023, 14, 1036563. [Google Scholar] [CrossRef]
- Shah, M.P.; Neal, J.W. Targeting Acquired and Intrinsic Resistance Mechanisms in Epidermal Growth Factor Receptor Mutant Non-Small-Cell Lung Cancer. Drugs 2022, 82, 649–662. [Google Scholar] [CrossRef]
- Recondo, G.; Facchinetti, F.; Olaussen, K.A.; Besse, B.; Friboulet, L. Making the first move in EGFR-driven or ALK-driven NSCLC: First-generation or next-generation TKI? Nat. Rev. Clin. Oncol. 2018, 15, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Lin, Y.C.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 29–39. [Google Scholar] [CrossRef]
- Patel, S.A.; Nilsson, M.B.; Yang, Y.; Le, X.; Tran, H.T.; Elamin, Y.Y.; Yu, X.; Zhang, F.; Poteete, A.; Ren, X.; et al. IL6 Mediates Suppression of T- and NK-cell Function in EMT-associated TKI-resistant EGFR-mutant NSCLC. Clin. Cancer. Res. 2023, 29, 1292–1304. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Yang, Y.; Heeke, S.; Patel, S.A.; Poteete, A.; Udagawa, H.; Elamin, Y.Y.; Moran, C.A.; Kashima, Y.; Arumugam, T.; et al. CD70 is a therapeutic target upregulated in EMT-associated EGFR tyrosine kinase inhibitor resistance. Cancer Cell 2023, 41, 340–355.e346. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Ramalingam, S.; Subramaniam, D.; Natarajan, G.; Anant, S.; Houchen, C.W. Selective blockade of DCAMKL-1 results in tumor growth arrest by a Let-7a MicroRNA-dependent mechanism. Gastroenterology 2009, 137, 649–659.e2. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Lightfoot, S.A.; Hoskins, A.B.; Lerner, M.; Brackett, D.J.; Postier, R.G.; Ramanujam, R.; Mohammed, A.; Rao, C.V.; et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011, 71, 2328–2338. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Mondalek, F.G.; Qu, D.; Ponnurangam, S.; Pantazis, P.; Anant, S.; Ramanujam, R.P.; Houchen, C.W. Nanoparticle-based delivery of siDCAMKL-1 increases microRNA-144 and inhibits colorectal cancer tumor growth via a Notch-1 dependent mechanism. J. Nanobiotechnol. 2011, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Fan, X.; Huang, X.; Weygant, N.; Xiao, Z.; Yan, R.; Liu, H.; Liu, J.; An, G.; Yao, J. DCLK1-Short Splice Variant Promotes Esophageal Squamous Cell Carcinoma Progression via the MAPK/ERK/MMP2 Pathway. Mol. Cancer Res. 2021, 19, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wen, T.; Zhou, Y.; Fan, X.; Du, T.; Gao, T.; Li, L.; Liu, J.; Yang, L.; Yao, J.; et al. DCLK1 Plays a Metastatic-Promoting Role in Human Breast Cancer Cells. Biomed. Res. Int. 2019, 2019, 1061979. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, R.; Huang, X.; Liu, H.; Xiao, Z.; Liu, J.; An, G.; Ge, Y. DCLK1 Drives EGFR-TKI-Acquired Resistance in Lung Adenocarcinoma by Remodeling the Epithelial–Mesenchymal Transition Status. Biomedicines 2023, 11, 1490. https://doi.org/10.3390/biomedicines11051490
Yan R, Huang X, Liu H, Xiao Z, Liu J, An G, Ge Y. DCLK1 Drives EGFR-TKI-Acquired Resistance in Lung Adenocarcinoma by Remodeling the Epithelial–Mesenchymal Transition Status. Biomedicines. 2023; 11(5):1490. https://doi.org/10.3390/biomedicines11051490
Chicago/Turabian StyleYan, Rui, Xuying Huang, Heshu Liu, Zeru Xiao, Jian Liu, Guangyu An, and Yang Ge. 2023. "DCLK1 Drives EGFR-TKI-Acquired Resistance in Lung Adenocarcinoma by Remodeling the Epithelial–Mesenchymal Transition Status" Biomedicines 11, no. 5: 1490. https://doi.org/10.3390/biomedicines11051490