
Apparent Opportunities and Hidden Pitfalls: The Conflicting Results of Restoring NRF2-Regulated Redox Metabolism in Friedreich’s Ataxia Pre-Clinical Models and Clinical Trials


Abstract
:1. Introduction
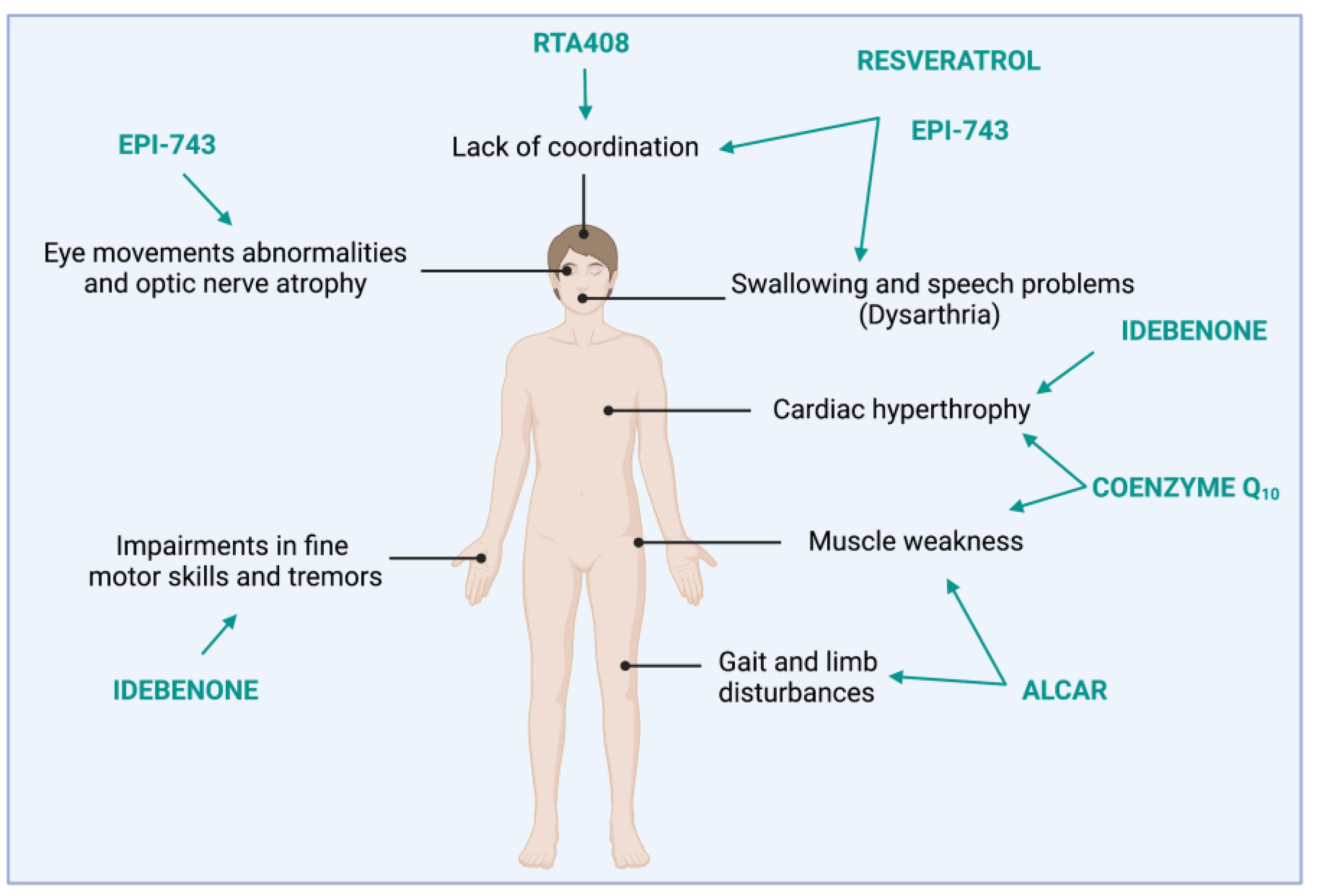
2. FRDA: Clinical and Molecular Features
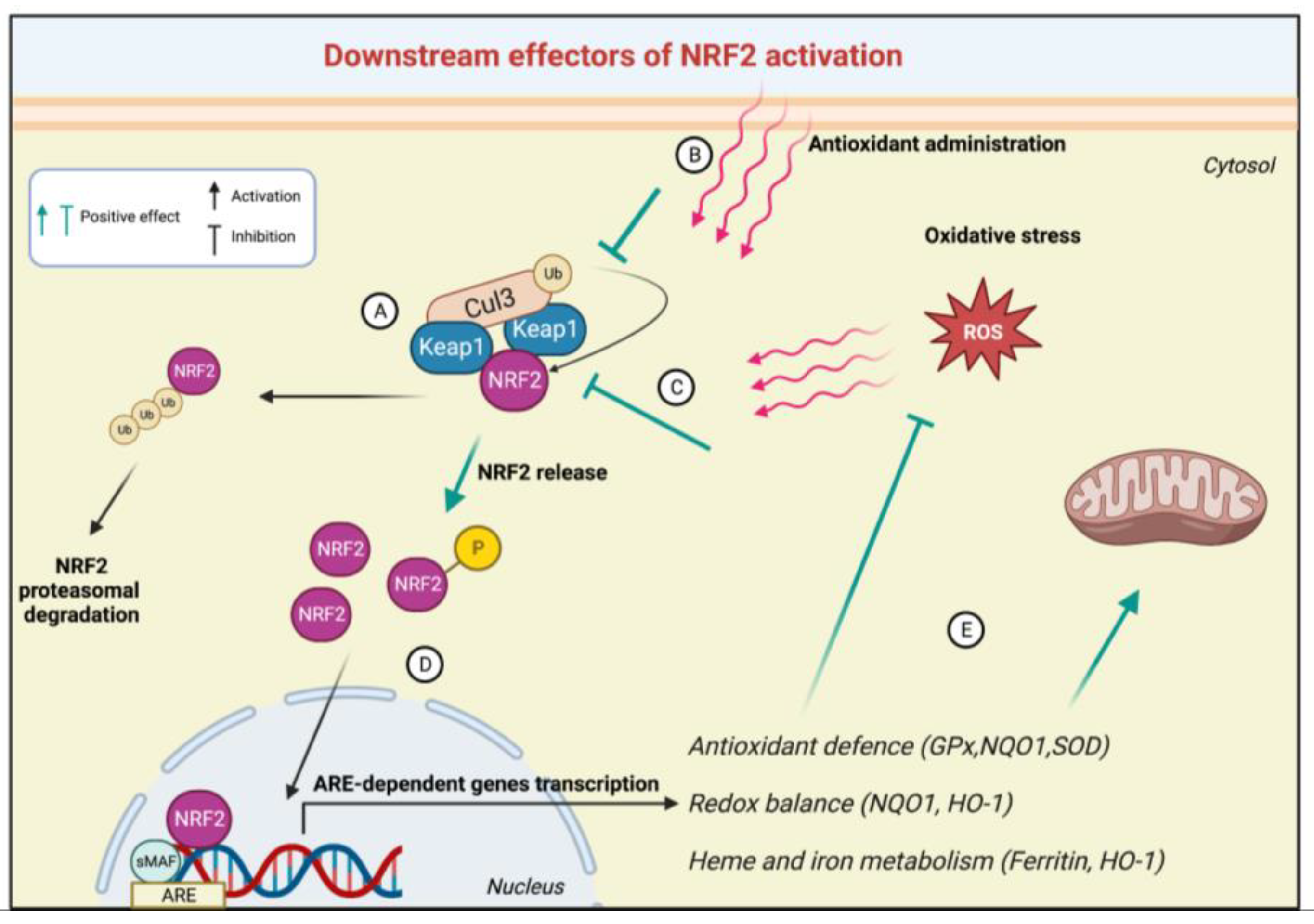
3. The NRF2 Signalling Pathway in FRDA
4. NRF2 Activators: Antioxidant Therapeutic Approach to Mitigate Oxidative Stress in FRDA
4.1. Natural Compounds
4.2. Synthetic Compounds
5. Constraints of Current Therapeutic Approaches and Future Prospects
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koeppen, A.H.; Morral, J.A.; Davis, A.N.; Qian, J.; Petrocine, S.V.; Knutson, M.D.; Gibson, W.M.; Cusack, M.J.; Li, D. The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 2009, 118, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich Ataxia: Neuropathology Revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Marmolino, D. Friedreich’s ataxia: Past, present and future. Brain Res. Rev. 2011, 67, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Br. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Bürk, K. Friedreich Ataxia: Current status and future prospects. Cerebellum Ataxias 2017, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Indelicato, E.; Nachbauer, W.; Eigentler, A.; Amprosi, M.; Gothe, R.M.; Giunti, P.; Mariotti, C.; Arpa, J.; Durr, A.; Klopstock, T.; et al. Onset features and time to diagnosis in Friedreich’s Ataxia. Orphanet J. Rare Dis. 2020, 15, 198. [Google Scholar] [CrossRef] [PubMed]
- Bidichandani, S.I.; Delatycki, M.B. Friedreich Ataxia. In GeneReviews® [Internet]; December 1998; University of Washington: Seattle, WA, USA, 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1281/ (accessed on 25 February 2023).
- Berciano, J.; Infante, J.; Garcia, A.; Polo, J.M.; Volpini, V.; Combarros, O. Very late-onset Friedreich’s ataxia with minimal GAA1 expansion mimicking multiple system atrophy of cerebellar type. Mov. Disord. 2005, 20, 1643–1645. [Google Scholar] [CrossRef]
- Parkinson, M.H.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Giunti, P. Clinical features of Friedreich’s ataxia: Classical and atypical phenotypes. J. Neurochem. 2013, 126 (Suppl. S1), 103–117. [Google Scholar] [CrossRef]
- Lecocq, C.; Charles, P.; Azulay, J.-P.; Meissner, W.; Rai, M.; N’Guyen, K.; Péréon, Y.; Fabre, N.; Robin, E.; Courtois, S.; et al. Delayed-onset Friedreich’s ataxia revisited. Mov. Disord. 2016, 31, 62–69. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef]
- Bencze, K.Z.; Kondapalli, K.C.; Cook, J.D.; McMahon, S.; Millan-Pacheco, C.; Pastor, N.; Stemmler, T. The Structure and Function of Frataxin. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 269–291. [Google Scholar] [CrossRef]
- Chiang, S.; Huang, M.; Park, K.; Richardson, D. Antioxidant defense mechanisms and its dysfunctional regulation in the mitochondrial disease, Friedreich’s ataxia. Free. Radic. Biol. Med. 2020, 159, 177–188. [Google Scholar] [CrossRef]
- Yoon, T.; Cowan, J.A. Frataxin-mediated Iron Delivery to Ferrochelatase in the Final Step of Heme Biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef]
- Stemmler, T.L.; Lesuisse, E.; Pain, D.; Dancis, A. Frataxin and Mitochondrial FeS Cluster Biogenesis. J. Biol. Chem. 2010, 285, 26737–26743. [Google Scholar] [CrossRef]
- Jasoliya, M.J.; McMackin, M.Z.; Henderson, C.K.; Perlman, S.L.; Cortopassi, G.A. Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans. Hum. Mol. Genet. 2017, 26, 2627–2633. [Google Scholar] [CrossRef]
- Clark, E.; Johnson, J.; Na Dong, Y.; Mercado-Ayon, E.; Warren, N.; Zhai, M.; McMillan, E.; Salovin, A.; Lin, H.; Lynch, D.R. Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease. Neuronal Signal. 2018, 2, NS20180060. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999, 8, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchère, F.; Lönn, M.; Holmgren, A.; Rustin, P. Impaired Nuclear Nrf2 Translocation Undermines the Oxidative Stress Response in Friedreich Ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin Deficiency Leads to Defects in Expression of Antioxidants and Nrf2 Expression in Dorsal Root Ganglia of the Friedreich’s Ataxia YG8R Mouse Model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin Deficiency Leads to Reduced Expression and Impaired Translocation of NF-E2-Related Factor (Nrf2) in Cultured Motor Neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef] [PubMed]
- Maciejczyk, M.; Mikoluc, B.; Pietrucha, B.; Pliszka, E.H.; Pac, M.; Motkowski, R.; Car, H. Oxidative stress, mitochondrial abnormalities and antioxidant defense in Ataxia-telangiectasia, Bloom syndrome and Nijmegen breakage syndrome. Redox Biol. 2017, 11, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int. J. Mol. Sci. 2017, 18, 2173. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Russo, M.; D’amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front. Cell. Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef]
- Rodríguez, L.R.; Lapeña, T.; Calap-Quintana, P.; Moltó, M.D.; Gonzalez-Cabo, P.; Langa, J.A.N. Antioxidant Therapies and Oxidative Stress in Friedreich’s Ataxia: The Right Path or Just a Diversion? Antioxidants 2020, 9, 664. [Google Scholar] [CrossRef]
- Foury, F.; Cazzalini, O. Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett. 1997, 411, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Dehmer, T.; Schols, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Burk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2017, 592, 718–727. [Google Scholar] [CrossRef]
- La Rosa, P.; Petrillo, S.; Bertini, E.S.; Piemonte, F. Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement. Biomolecules 2020, 10, 702. [Google Scholar] [CrossRef]
- Metz, G.; Coppard, N.; Cooper, J.M.; Delatycki, M.B.; Dürr, A.; Di Prospero, N.A.; Giunti, P.; Lynch, D.R.; Schulz, J.B.; Rummey, C.; et al. Rating disease progression of Friedreich’s ataxia by the International Cooperative Ataxia Rating Scale: Analysis of a 603-patient database. Brain 2013, 136, 259–268. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial Diseases of the Brain. Free. Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef]
- Wollmann, T.; Barroso, J.; Monton, F.I.; Nieto, A. Neuropsychological Test Performance of Patients With Friedreich’s Ataxia. J. Clin. Exp. Neuropsychol. 2002, 24, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Corben, L.A.; Georgiou-Karistianis, N.; Fahey, M.C.; Storey, E.; Churchyard, A.; Horne, M.; Bradshaw, J.L.; Delatycki, M.B. Towards an understanding of cognitive function in Friedreich ataxia. Brain Res. Bull. 2006, 70, 197–202. [Google Scholar] [CrossRef]
- Corben, L.; Akhlaghi, H.; Georgiou-Karistianis, N.; Bradshaw, J.; Egan, G.; Storey, E.; Churchyard, A.; Delatycki, M. Impaired inhibition of prepotent motor tendencies in Friedreich ataxia demonstrated by the Simon interference task. Brain Cogn. 2011, 76, 140–145. [Google Scholar] [CrossRef]
- Pousset, F.; Legrand, L.L.; Monin, M.L.M.; Ewenczyk, C.C.; Charles, P.P.; Komajda, M.M.; Brice, A.; Pandolfo, M.; Isnard, R.; du Montcel, S.T.; et al. A 22-Year Follow-up Study of Long-term Cardiac Outcome and Predictors of Survival in Friedreich Ataxia. JAMA Neurol. 2015, 72, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Störk, S.; Liu, D.; Hu, K.; Herrmann, S.; Ertl, G.; Niemann, M. Cardiomyopathy of Friedreich Ataxia. J. Neurochem. 2013, 126, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Perdomini, M.; Belbellaa, B.; Monassier, L.; Reutenauer, L.; Messaddeq, N.; Cartier, N.; Crystal, R.G.; Aubourg, P.; Puccio, H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med. 2014, 20, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Milbrandt, T.A.; Kunes, J.R.; Karol, L.A. Friedreich’s Ataxia and Scoliosis. J. Pediatr. Orthop. 2008, 28, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Mulder, H.; Igoillo-Esteve, M. Diabetes in Friedreich Ataxia. J. Neurochem. 2013, 126, 94–102. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Corben, L.A. Clinical Features of Friedreich Ataxia. J. Child Neurol. 2012, 27, 1133–1137. [Google Scholar] [CrossRef]
- Bürk, K.; Mälzig, U.; Wolf, S.; Heck, S.; Dimitriadis, K.; Schmitz-Hübsch, T.; Hering, S.; Lindig, T.M.; Haug, V.; Timmann, D.; et al. Comparison of three clinical rating scales in Friedreich ataxia (FRDA). Mov. Disord. 2009, 24, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Bürk, K.; Schulz, S.R.; Schulz, J.B. Monitoring progression in Friedreich ataxia (FRDA): The use of clinical scales. J. Neurochem. 2013, 126, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Trouillas, P.; Takayanagi, T.; Hallett, M.; Currier, R.; Subramony, S.; Wessel, K.; Bryer, A.; Diener, H.; Massaquoi, S.; Gomez, C.; et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neu-ropharmacology Committee of the World Federation of Neurology. J. Neurol. Sci. 1997, 145, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Storey, E.; Tuck, K.; Hester, R.; Hughes, A.; Churchyard, A. Inter-rater reliability of the International Cooperative Ataxia Rating Scale (ICARS). Mov. Disord. 2004, 19, 190–192. [Google Scholar] [CrossRef]
- Cano, S.J.; Hobart, J.C.; Hart, P.E.; Korlipara, L.P.; Schapira, A.H.; Cooper, J.M. International cooperative ataxia rating scale (ICARS): Appropriate for studies of Friedreich’s ataxia? Mov. Disord. 2005, 20, 1585–1591. [Google Scholar] [CrossRef]
- Schmitz-Hübsch, T.; Du Montcel, S.T.; Baliko, L.; Berciano, J.; Boesch, S.; Depondt, C.; Giunti, P.; Globas, C.; Infante, J.; Kang, J.-S.; et al. Scale for the assessment and rating of ataxia: Development of a new clinical scale. Neurology 2006, 66, 1717–1720. [Google Scholar] [CrossRef]
- Subramony, S.H.; May, W.; Lynch, D.; Gomez, C.; Fischbeck, K.; Hallett, M.; Taylor, P.; Wilson, R.; Ashizawa, T. Measuring Friedreich ataxia: Interrater reliability of a neurologic rating scale. Neurology 2005, 64, 1261–1262. [Google Scholar] [CrossRef]
- Galea, C.A.; Huq, A.; Lockhart, P.J.; Tai, G.; Corben, L.A.; Yiu, E.M.; Gurrin, L.C.; Lynch, D.R.; Gelbard, S.; Durr, A.; et al. Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 2016, 79, 485–495. [Google Scholar] [CrossRef]
- Li, Y.; Lu, Y.; Polak, U.; Lin, K.; Shen, J.; Farmer, J.; Seyer, L.; Bhalla, A.D.; Rozwadowska, N.; Lynch, D.R.; et al. Expanded GAA repeats impede transcription elongation through the FXN gene and induce transcriptional silencing that is restricted to the FXN locus. Hum. Mol. Genet. 2015, 24, 6932–6943. [Google Scholar] [CrossRef]
- Butler, J.S.; Napierala, J. Friedreich’s ataxia—A case of aberrant transcription termination? Transcription 2015, 6, 33–36. [Google Scholar] [CrossRef]
- Gottesfeld, J.M. Molecular Mechanisms and Therapeutics for the GAA·TTC Expansion Disease Friedreich Ataxia. Neurotherapeutics 2019, 16, 1032–1049. [Google Scholar] [CrossRef]
- Chutake, Y.K.; Costello, W.N.; Lam, C.; Bidichandani, S.I. Altered Nucleosome Positioning at the Transcription Start Site and Deficient Transcriptional Initiation in Friedreich Ataxia. J. Biol. Chem. 2014, 289, 15194–15202. [Google Scholar] [CrossRef]
- Grabczyk, E.; Mancuso, M.; Sammarco, M. A persistent RNA·DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res. 2007, 35, 5351–5359. [Google Scholar] [CrossRef]
- Groh, M.; Lufino, M.M.P.; Wade-Martins, R.; Gromak, N. R-loops Associated with Triplet Repeat Expansions Promote Gene Silencing in Friedreich Ataxia and Fragile X Syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Chastain, P.D.; Parniewski, P.; Ohshima, K.; Pandolfo, M.; Griffith, J.D.; Wells, R.D. Sticky DNA: Self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol. Cell 1999, 3, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Vetcher, A.A.; Napierala, M.; Iyer, R.R.; Chastain, P.D.; Griffith, J.D.; Wells, R.D. Sticky DNA, a Long GAA·GAA·TTC Triplex That Is Formed Intramolecularly, in the Sequence of Intron 1 of the Frataxin Gene. J. Biol. Chem. 2002, 277, 39217–39227. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikova, M.M.; Mirkin, S.M. Replication Stalling at Friedreich’s Ataxia (GAA)n Repeats In Vivo. Mol. Cell. Biol. 2004, 24, 2286–2295. [Google Scholar] [CrossRef]
- Wells, R.D. DNA triplexes and Friedreich ataxia. FASEB J. 2008, 22, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is Reduced in Friedreich Ataxia Patients and is Associated with Mitochondrial Membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol. 2009, 256, 3–8. [Google Scholar] [CrossRef]
- Silva, A.M.; Brown, J.M.; Buckle, V.J.; Wade-Martins, R.; Lufino, M.M. Expanded GAA repeats impair FXN gene expression and reposition the FXN locus to the nuclear lamina in single cells. Hum. Mol. Genet. 2015, 24, 3457–3471. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahdawi, S.; Pinto, R.M.; Ismail, O.; Varshney, D.; Lymperi, S.; Sandi, C.; Trabzuni, D.; Pook, M.A. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 2007, 17, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Oliveira, A.F.; Cosentino, C.; Fantuzzi, F.; Demarez, C.; Toivonen, S.; Hu, A.; Chintawar, S.; Lopes, M.; Pachera, N.; et al. Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. J. Clin. Investig. 2020, 5, e134221. [Google Scholar] [CrossRef] [PubMed]
- Filla, A.; De Michele, G.; Cavalcanti, F.; Pianese, L.; Monticelli, A.; Campanella, G.; Cocozza, S. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 1996, 59, 554–560. [Google Scholar] [PubMed]
- Dürr, A.; Cossee, M.; Agid, Y.; Campuzano, V.; Mignard, C.; Penet, C.; Mandel, J.-L.; Brice, A.; Koenig, M. Clinical and Genetic Abnormalities in Patients with Friedreich’s Ataxia. N. Engl. J. Med. 1996, 335, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Gellera, C.; Pareyson, D.; Castellotti, B.; Mazzucchelli, F.; Zappacosta, B.; Pandolfo, M.; Di Donato, S. Very late onset Friedreich’s ataxia without cardiomyopathy is associated with limited GAA expansion in the X25 gene. Neurology 1997, 49, 1153–1155. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Morral, J.A.; McComb, R.D.; Feustel, P. The neuropathology of late-onset Friedreich’s ataxia. Cerebellum 2010, 10, 96–103. [Google Scholar] [CrossRef]
- Santos, R.; Lefevre, S.D.; Sliwa, D.; Seguin, A.; Camadro, J.-M.; Lesuisse, E. Friedreich Ataxia: Molecular Mechanisms, Redox Considerations, and Therapeutic Opportunities. Antioxid. Redox Signal. 2010, 13, 651–690. [Google Scholar] [CrossRef]
- Harding, I.H.; Lynch, D.R.; Koeppen, A.H.; Pandolfo, M. Central Nervous System Therapeutic Targets in Friedreich Ataxia. Hum. Gene Ther. 2020, 31, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Milani, M.; D’ambrosi, N. Neuroinflammation in Friedreich’s Ataxia. Int. J. Mol. Sci. 2022, 23, 6297. [Google Scholar] [CrossRef]
- Pérez-Luz, S.; Gimenez-Cassina, A.; Fernández-Frías, I.; Wade-Martins, R.; Díaz-Nido, J. Delivery of the 135 kb human FXN genomic DNA locus gives rise to different FXN isoforms. Genomics 2015, 106, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Abruzzo, P.M.; Marini, M.; Bolotta, A.; Malisardi, G.; Manfredini, S.; Ghezzo, A.; Pini, A.; Tasco, G.; Casadio, R. FXN mRNA isoforms in FRDA patients and normal subjects: Effect of tocotrienol supplementation. BioMed Res. Int. 2013, 2013, 276808. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, Q.; Weng, L.; Hauser, L.A.; Strawser, C.J.; Mesaros, C.; Lynch, D.R.; Blair, I.A. Characterization of a new N-terminally acetylated extra-mitochondrial isoform of FXN in human erythrocytes. Sci. Rep. 2018, 8, 17043. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of Mitochondrial Iron Accumulation by Yfh1p, a Putative Homolog of Frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef]
- Adamec, J.; Rusnak, F.; Owen, W.G.; Naylor, S.; Benson, L.M.; Gacy, A.M.; Isaya, G. Iron-dependent self-assembly of recombinant yeast FXN: Implications for Friedreich ataxia. Am. J. Hum. Genet. 2000, 67, 549. [Google Scholar] [CrossRef]
- Adinolfi, S.; Trifuoggi, M.; Politou, A.S.; Martin, S.; Pastore, A. A structural approach to understanding the iron-binding properties of phylogenetically different frataxins. Hum. Mol. Genet. 2002, 11, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F.; Adinolfi, S.; Pastore, A.; Laue, T.M.; Chasteen, N.D. Iron binding and oxidation kinetics in FXN CyaY of Escherichia coli. J. Mol. Biol. 2004, 341, 605. [Google Scholar] [CrossRef]
- Cook, J.D.; Bencze, K.Z.; Jankovic, A.D.; Crater, A.K.; Busch, C.N.; Bradley, P.B.; Stemmler, A.J.; Spaller, M.R.; Stemmler, T.L. Monomeric yeast FXN is an iron binding protein. Biochemistry 2006, 45, 7767. [Google Scholar] [CrossRef]
- Yau, W.Y.; O’Connor, E.; Sullivan, R.; Akijian, L.; Wood, N.W. DNA repair in trinucleotide repeat ataxias. FEBS J. 2018, 285, 3669–3682. [Google Scholar] [CrossRef]
- Uceda, A.B.; Donoso, J.; Frau, J.; Vilanova, B.; Adrover, M. Frataxins Emerge as New Players of the Intracellular Antioxidant Machinery. Antioxidants 2021, 10, 315. [Google Scholar] [CrossRef]
- Doni, D.; Rigoni, G.; Palumbo, E.; Baschiera, E.; Peruzzo, R.; De Rosa, E.; Caicci, F.; Passerini, L.; Bettio, D.; Russo, A.; et al. The displacement of FXN from the mitochondrial cristae correlates with abnormal respiratory supercomplexes formation and bioenergetic defects in cells of Friedreich ataxia patients. FASEB J. 2021, 35, e21362. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.-L.; O’Neill, H.A.; Kennedy, M.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Frataxin Acts as an Iron Chaperone Protein to Modulate Mitochondrial Aconitase Activity. Science 2004, 305, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Potdar, P.D.; Raghu, A. Review on Molecular Diagnostic Techniques in Friedreich’s Ataxia. Annu. Res. Rev. Biol. 2013, 3, 659–677. Available online: https://journalarrb.com/index.php/ARRB/article/view/24839 (accessed on 25 February 2023).
- Mladěnka, P.; Šimůnek, T.; Hübl, M.; Hrdina, R. The role of reactive oxygen and nitrogen species in cellular iron metabolism. Free. Radic. Res. 2006, 40, 263–272. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, G. Mitochondrial and metabolic dysfunction in Friedreich ataxia: Update on pathophysiological relevance and clinical interventions. Neuronal Signal. 2021, 5, NS20200093. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.; Wilson, R.B. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. Experiment 2019, 369, 47–54. [Google Scholar] [CrossRef]
- Turchi, R.; Faraonio, R.; Lettieri-Barbato, D.; Aquilano, K. An Overview of the Ferroptosis Hallmarks in Friedreich’s Ataxia. Biomolecules 2020, 10, 1489. [Google Scholar] [CrossRef]
- La Rosa, P.; Petrillo, S.; Fiorenza, M.T.; Bertini, E.S.; Piemonte, F. Ferroptosis in Friedreich’s Ataxia: A Metal-Induced Neurodegenerative Disease. Biomolecules 2020, 10, 1551. [Google Scholar] [CrossRef]
- Karunatilleke, N.C.; Fast, C.S.; Ngo, V.; Brickenden, A.; Duennwald, M.L.; Konermann, L.; Choy, W.-Y. Nrf2, the Major Regulator of the Cellular Oxidative Stress Response, is Partially Disordered. Int. J. Mol. Sci. 2021, 22, 7434. [Google Scholar] [CrossRef]
- Andrews, N.C.; Erdjument-Bromage, H.; Davidson, M.B.; Tempst, P.; Orkin, S.H. Erythroid transcription factor NF-E2 is a haematopoietic-specific basic–leucine zipper protein. Nature 1993, 362, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, A.V.; Rosenkranz, A.A.; Georgiev, G.P.; Sobolev, A.S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci. 2022, 291, 120111. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Huang, H.-C.; Yang, C.S.; Pickett, C.B. Increased Protein Stability as a Mechanism That Enhances Nrf2-mediated Transcriptional Activation of the Antioxidant Response Element. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef]
- Kaundal, R.K.; Datusalia, A.K.; Sharma, S.S. Posttranscriptional regulation of Nrf2 through miRNAs and their role in Alzheimer’s disease. Pharmacol. Res. 2022, 175, 106018. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A. The Keap1-Nrf2pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/β-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): History, structure and function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef]
- Liu, S.; Pi, J.; Zhang, Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol. 2022, 54, 102389. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef]
- Katsuoka, F.; Motohashi, H.; Engel, J.D.; Yamamoto, M. Nrf2 transcriptionally activates the mafG gene through an antioxidant response element. J. Biol. Chem. 2005, 280, 4483–4490. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Gambhir, L.; Tyagi, G.; Bhardwaj, R.; Kapoor, N.; Sharma, G. Perturbation of Cellular Redox Status: Role of Nrf2, a Master Regulator of Cellular Redox. In Reactive Oxygen Species; IntechOpen: Jaipur, Rajasthan, India, 2022. [Google Scholar] [CrossRef]
- Tejo, F.V.; Quintanilla, R. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10, 1069. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription Factors NRF2 and NF-κB Are Coordinated Effectors of the Rho Family, GTP-binding Protein RAC1 during Inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Zhu, H. Regulation of Nrf2 Signaling. React. Oxyg. Species (Apex) 2019, 8, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.-H.; Jain, A.K.; Papusha, V.; Jaiswal, A.K. An Auto-regulatory Loop between Stress Sensors INrf2 and Nrf2 Controls Their Cellular Abundance. J. Biol. Chem. 2007, 282, 36412–36420. [Google Scholar] [CrossRef] [PubMed]
- Anzovino, A.; Chiang, S.; Brown, B.E.; Hawkins, C.L.; Richardson, D.R.; Huang, M.L. FXN-Deficiency in the Heart Results in an Impaired Nrf2 Response: A Dual Mechanism Mediated via Up-Regulation of Keap1 and GSK3β Axis. Free Radic. Biol. Med. 2016, 100, S143–S144. [Google Scholar] [CrossRef]
- La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. Int. J. Mol. Sci. 2020, 21, 916. [Google Scholar] [CrossRef]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; DI Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.M.; Kosman, D.J. Molecular Defects in Friedreich’s Ataxia: Convergence of Oxidative Stress and Cytoskeletal Abnormalities. Front. Mol. Biosci. 2020, 7, 569293. [Google Scholar] [CrossRef]
- Sahdeo, S.; Scott, B.D.; McMackin, M.Z.; Jasoliya, M.; Brown, B.; Wulff, H.; Perlman, S.L.; Pook, M.A.; Cortopassi, G.A. Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum. Mol. Genet. 2014, 23, 6848–6862. [Google Scholar] [CrossRef]
- Weaver, L.C.; Richards, A.B.; Abreu, B.E. Central nervous system effects of a local anesthetic dyclonine. Toxicol. Appl. Pharmacol. 1960, 2, 616–627. [Google Scholar] [CrossRef]
- Abeti, R.; Jasoliya, M.; Al-Mahdawi, S.; Pook, M.; Gonzalez-Robles, C.; Hui, C.K.; Cortopassi, G.; Giunti, P. A Drug Combination Rescues FXN-Dependent Neural and Cardiac Pathophysiology in FA Models. Front. Mol. Biosci. 2022, 9, 830650, Erratum in Front. Mol. Biosci. 2022, 9, 968121. [Google Scholar] [CrossRef] [PubMed]
- Jauslin, M.L.; Meier, T.; Smith, R.A.J.; Murphy, P.M. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003, 17, 1972–1974. [Google Scholar] [CrossRef]
- Abeti, R.; Uzun, E.; Renganathan, I.; Honda, T.; Pook, M.A.; Giunti, P. Targeting lipid peroxidation and mitochondrial imbalance in Friedreich’s ataxia. Pharmacol. Res. 2015, 99, 344–350. [Google Scholar] [CrossRef]
- Petrillo, S.; D’amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the Treatment of Friedreich’s Ataxia: A Comparison among Drugs. Int. J. Mol. Sci. 2019, 20, 5211. [Google Scholar] [CrossRef]
- Jasoliya, M.; Sacca, F.; Sahdeo, S.; Chedin, F.; Pane, C.; Brescia Morra, V.; Filla, A.; Pook, M.; Cortopassi, G. Dimethyl fumarate dosing in humans increases FXN expression: A potential therapy for Friedreich’s Ataxia. PLoS ONE 2019, 14, e0217776. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.K.; Dedkova, E.N.; Montgomery, C.; Cortopassi, G. Dimethyl fumarate dose-dependently increases mitochondrial gene expression and function in muscle and brain of Friedreich’s ataxia model mice. Hum. Mol. Genet. 2020, 29, 3954–3965. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, Z.; Xing, Z.; Liu, Y.; Zhao, H.; Tang, Z.; Luo, Y.; Hao, S.; Li, K. Cur@SF NPs alleviate Friedreich’s ataxia in a mouse model through synergistic iron chelation and antioxidation. J. Nanobiotechnol. 2022, 20, 118. [Google Scholar] [CrossRef]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef]
- Yiu, E.M.; Tai, G.; Peverill, R.E.; Lee, K.J.; Croft, K.D.; Mori, T.A.; Scheiber-Mojdehkar, B.; Sturm, B.; Praschberger, M.; Vogel, A.P.; et al. An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on FXN levels. J. Neurol. 2015, 262, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Korlipara, L.V.P.; Hart, P.E.; Bradley, J.L.; Schapira, A.H.V. Coenzyme Q10and vitamin E deficiency in Friedreich’s ataxia: Predictor of efficacy of vitamin E and coenzyme Q10therapy. Eur. J. Neurol. 2008, 15, 1371–1379. [Google Scholar] [CrossRef]
- Lodi, R.; Hart, P.E.; Rajagopalan, B.; Taylor, D.J.; Crilley, J.G.; Bradley, J.L.; Blamire, A.M.; Manners, D.; Styles, P.; Schapira, A.H.; et al. Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich’s ataxia. Ann. Neurol. 2001, 49, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Sorbi, S.; Forleo, P.; Fani, C.; Piacentini, S. Double-Blind, Crossover, Placebo-Controlled Clinical Trial with L-Acetylcarnitine in Patients with Degenerative Cerebellar Ataxia. Clin. Neuropharmacol. 2000, 23, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Rustin, P.; Munnich, A.; Rotig, A. Quinone analogs prevent enzymes targeted in Friedreich ataxia from iron-induced injury in vitro. Biofactors 1999, 9, 247–251. [Google Scholar] [CrossRef]
- Buyse, G.; Mertens, L.; Di Salvo, G.; Matthijs, I.; Weidemann, F.; Eyskens, B.; Goossens, W.; Goemans, N.; Sutherland, G.R.; Van Hove, J.L.K. Idebenone treatment in Friedreich’s ataxia: Neurological, cardiac, and biochemical monitoring. Neurology 2003, 60, 1569–1570. [Google Scholar] [CrossRef]
- Pineda, M.; Arpa, J.; Montero, R.; Aracil, A.; Domínguez, F.; Galván, M.; Mas, A.; Martorell, L.; Sierra, C.; Brandi, N.; et al. Idebenone treatment in paediatric and adult patients with Friedreich ataxia: Long-term follow-up. Eur. J. Paediatr. Neurol. 2008, 12, 470–475. [Google Scholar] [CrossRef]
- Lynch, D.R.; Perlman, S.L.; Meier, T. A Phase 3, Double-blind, Placebo-Controlled Trial of Idebenone in Friedreich Ataxia. Arch. Neurol. 2010, 67, 941–947. [Google Scholar] [CrossRef]
- Lagedrost, S.J.; Sutton, M.S.J.; Cohen, M.S.; Satou, G.M.; Kaufman, B.D.; Perlman, S.L.; Rummey, C.; Meier, T.; Lynch, D.R. Idebenone in Friedreich ataxia cardiomyopathy—Results from a 6-month phase III study (IONIA). Am. Heart J. 2011, 161, 639–645.e1. [Google Scholar] [CrossRef]
- Artuch, R.; Aracil, A.; Mas, A.; Colomé, C.; Rissech, M.; Monrós, E.; Pineda, M. Friedreich’s Ataxia: Idebenone Treatment in Early Stage Patients. Neuropediatrics 2002, 33, 190–193. [Google Scholar] [CrossRef]
- Cook, A.; Boesch, S.; Heck, S.; Brunt, E.; Klockgether, T.; Schöls, L.; Schulz, A.; Giunti, P. Patient-reported outcomes in Friedreich’s ataxia after withdrawal from idebenone. Acta Neurol. Scand. 2019, 139, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Fuentes, A.J.; Cesar, S.; Montero, R.; Latre, C.; Genovès, J.; Martorell, L.; Cuadras, D.; Colom, H.; Pineda, M.; O’callaghan, M.D.M.; et al. Plasma idebenone monitoring in Friedreich’s ataxia patients during a long-term follow-up. Biomed. Pharmacother. 2021, 143, 112143. [Google Scholar] [CrossRef]
- Mariotti, C.; Fancellu, R.; Caldarazzo, S.; Nanetti, L.; Di Bella, D.; Plumari, M.; Lauria, G.; Cappellini, M.D.; Duca, L.; Solari, A.; et al. Erythropoietin in Friedreich ataxia: No effect on FXN in a randomized controlled trial. Mov. Disord. 2012, 27, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Bolotta, A.; Pini, A.; Abruzzo, P.M.; Ghezzo, A.; Modesti, A.; Gamberi, T.; Ferreri, C.; Bugamelli, F.; Fortuna, F.; Vertuani, S.; et al. Effects of tocotrienol supplementation in Friedreich’s ataxia: A model of oxidative stress pathology. Exp. Biol. Med. 2019, 245, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef]
- Zesiewicz, T.A.; Hancock, J.; Ghanekar, S.D.; Kuo, S.H.; Dohse, C.A.; Vega, J. Emerging therapies in Friedreich’s Ataxia. Expert Rev. Neurother. 2020, 20, 1215–1228. [Google Scholar] [CrossRef]
- Zesiewicz, T.; Salemi, J.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2018, 6, 15–26. [Google Scholar] [CrossRef]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; et al. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann. Neurol. 2020, 89, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Bath, S.; Elbarbry, F. An Organ System Approach to Explore the Antioxidative, Anti-Inflammatory, and Cytoprotective Actions of Resveratrol. Oxid. Med. Cell. Longev. 2015, 2015, 803971. [Google Scholar] [CrossRef]
- Jardim, F.R.; De Rossi, F.T.; Nascimento, M.X.; Barros, R.G.d.S.; Borges, P.A.; Prescilio, I.C.; De Oliveira, M.R. Resveratrol and Brain Mitochondria: A Review. Mol. Neurobiol. 2018, 55, 2085–2101. [Google Scholar] [CrossRef]
- Varoni, E.M.; Lo Faro, A.F.; Sharifi-Rad, J.; Iriti, M. Anticancer Molecular Mechanisms of Resveratrol. Front. Nutr. 2016, 3, 8. [Google Scholar] [CrossRef]
- Magyar, K.; Halmosi, R.; Palfi, A.; Feher, G.; Czopf, L.; Fulop, A.; Battyany, I.; Sumegi, B.; Toth, K.; Szabados, E. Cardioprotection by resveratrol: A human clinical trial in patients with stable coronary artery disease. Clin. Hemorheol. Microcirc. 2012, 50, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Truong, V.-L.; Jun, M.; Jeong, W.-S. Role of resveratrol in regulation of cellular defense systems against oxidative stress. BioFactors 2018, 44, 36–49. [Google Scholar] [CrossRef]
- Benayahoum, A.; Amira-Guebailia, H.; Houache, O. Homolytic and heterolytic O–H bond cleavage in trans-resveratrol and some phenantrene analogs: A theoretical study. Comput. Theor. Chem. 2014, 1037, 1–9. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Baer-Dubowska, W. Modulation of Nrf2 and NF-κB Signaling Pathways by Naturally Occurring Compounds in Relation to Cancer Prevention and Therapy. Are Combinations Better Than Single Compounds? Int. J. Mol. Sci. 2021, 22, 8223. [Google Scholar] [CrossRef]
- Wang, G.; Xie, X.; Yuan, L.; Qiu, J.; Duan, W.; Xu, B.; Chen, X. Resveratrol ameliorates rheumatoid arthritis via activation of SIRT1-Nrf2 signaling pathway. Biofactors 2020, 46, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Iside, C.; Scafuro, M.; Nebbioso, A.; Altucci, L. SIRT1 Activation by Natural Phytochemicals: An Overview. Front. Pharmacol. 2020, 11, 1225. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Gao, X.; Wei, W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-β1 expressions in rat glomerular mesangial cells. Exp. Cell Res. 2017, 361, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.M.; Martins, L.A.M.; Souza, D.O.; Quincozes-Santos, A. Glioprotective Effect of Resveratrol: An Emerging Therapeutic Role for Oligodendroglial Cells. Mol. Neurobiol. 2018, 55, 2967–2978. [Google Scholar] [CrossRef]
- Yang, J.; Huang, J.; Shen, C.; Cheng, W.; Yu, P.; Wang, L.; Tang, F.; Guo, S.; Yang, Q.; Zhang, J. Resveratrol Treatment in Different Time-Attenuated Neuronal Apoptosis After Oxygen and Glucose Deprivation/Reoxygenation via Enhancing the Activation of Nrf-2 Signaling Pathway In Vitro. Cell Transplant. 2018, 27, 1789–1797. [Google Scholar] [CrossRef]
- Yadav, A.; Sunkaria, A.; Singhal, N.; Sandhir, R. Resveratrol loaded solid lipid nanoparticles attenuate mitochondrial oxidative stress in vascular dementia by activating Nrf2/HO-1 pathway. Neurochem. Int. 2018, 112, 239–254. [Google Scholar] [CrossRef]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial Respiratory Complex I: Structure, Function and Implication in Human Diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-Q.; Wu, P.-F.; Long, L.-H.; Chen, Y.; Hu, Z.-L.; Ni, L.; Wang, F.; Chen, J.-G. Resveratrol promotes cellular glucose utilization in primary cultured cortical neurons via calcium-dependent signaling pathway. J. Nutr. Biochem. 2013, 24, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-K.; Pokharel, Y.R.; Lim, S.C.; Han, H.-K.; Ryu, C.S.; Kim, S.K.; Kwak, M.K.; Kang, K.W. Inhibition of liver fibrosis by solubilized coenzyme Q10: Role of Nrf2 activation in inhibiting transforming growth factor-β1 expression. Toxicol. Appl. Pharmacol. 2009, 240, 377–384. [Google Scholar] [CrossRef]
- Li, L.; Du, J.; Lian, Y.; Zhang, Y.; Li, X.; Liu, Y.; Zou, L.; Wu, T. Protective Effects of Coenzyme Q10 Against Hydrogen Peroxide-Induced Oxidative Stress in PC12 Cell: The Role of Nrf2 and Antioxidant Enzymes. Cell. Mol. Neurobiol. 2015, 36, 103–111. [Google Scholar] [CrossRef]
- Pala, R.; Orhan, C.; Tuzcu, M.; Sahin, N.; Ali, S.; Cinar, V.; Atalay, M.; Sahin, K. Coenzyme Q10 Supplementation Modulates NFκB and Nrf2 Pathways in Exercise Training. J. Sport. Sci. Med. 2016, 15, 196–203. [Google Scholar]
- Quinzii, C.M.; López, L.C.; Naini, A.; DiMauro, S.; Hirano, M. Human CoQ10deficiencies. Biofactors 2008, 32, 113–118. [Google Scholar] [CrossRef]
- Hart, P.E.; Lodi, R.; Rajagopalan, B.; Bradley, J.L.; Crilley, J.G.; Turner, C.D.; Blamire, A.; Manners, D.; Styles, P.; Schapira, A.; et al. Antioxidant treatment of patients with Friedreich ataxia: Four-year follow-up. Arch. Neurol. 2005, 62, 621–626. [Google Scholar] [CrossRef]
- Ferreira, G.C.; McKenna, M.C. l-Carnitine and Acetyl-l-carnitine Roles and Neuroprotection in Developing Brain. Neurochem. Res. 2017, 42, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Bioenergetic approaches for neuroprotection in Parkinson’s disease. Ann. Neurol. 2003, 53, S39–S48. [Google Scholar] [CrossRef]
- Goety, C.G.; Tanner, C.M.; Cohen, J.A.; Thelen, J.A.; Carroll, V.S.; Klawans, H.L.; Fariello, R.G. L-acetyl-carnitine in Huntington’s disease: Double-blind placebo controlled crossover study of drug effects on movement dis-order and dementia. Mov. Disord. 1990, 5, 263–265. [Google Scholar] [CrossRef]
- Calabrese, V.; Ravagna, A.; Colombrita, C.; Scapagnini, G.; Guagliano, E.; Calvani, M.; Butterfield, D.A.; Stella, A.M.G. Acetylcarnitine induces heme oxygenase in rat astrocytes and protects against oxidative stress: Involvement of the transcription factor Nrf2. J. Neurosci. Res. 2005, 79, 509–521. [Google Scholar] [CrossRef]
- Yang, S.-P.; Yang, X.-Z.; Cao, G.-P. Acetyl-l-carnitine prevents homocysteine-induced suppression of Nrf2/Keap1 mediated antioxidation in human lens epithelial cells. Mol. Med. Rep. 2015, 12, 1145–1150. [Google Scholar] [CrossRef]
- Schöls, L.; Zange, J.; Abele, M.; Schillings, M.; Skipka, G.; Kuntz-Hehner, S.; Van Beekvelt, M.; Colier, W.N.J.M.; Müller, K.; Klockgether, T.; et al. L-carnitine and creatine in Friedreich’s ataxia. A randomized, placebo-controlled crossover trial. J. Neural Transm. 2005, 112, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J. Leukoc. Biol. 2015, 99, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Schepici, G.; Bramanti, P.; Mazzon, E. Efficacy of Sulforaphane in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8637. [Google Scholar] [CrossRef] [PubMed]
- Houghton, C.A. Sulforaphane: Its “Coming of Age” as a Clinically Relevant Nutraceutical in the Prevention and Treatment of Chronic Disease. Oxid. Med. Cell. Longev. 2019, 2019, 2716870. [Google Scholar] [CrossRef]
- Santín-Márquez, R.; Alarcón-Aguilar, A.; López-Diazguerrero, N.E.; Chondrogianni, N.; Königsberg, M. Sulforaphane—Role in aging and neurodegeneration. Geroscience 2019, 41, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Mangla, B.; Javed, S.; Sultan, M.H.; Kumar, P.; Kohli, K.; Najmi, A.; Alhazmi, H.A.; Al Bratty, M.; Ahsan, W. Sulforaphane: A review of its therapeutic potentials, advances in its nanodelivery, recent patents, and clinical trials. Phytother. Res. 2021, 35, 5440–5458. [Google Scholar] [CrossRef]
- Janowiak, B.E.; Hayward, M.A.; Peterson, F.C.; Volkman, B.F.; Griffith, O.W. γ-Glutamylcysteine Synthetase−Glutathione Synthetase: Domain Structure and Identification of Residues Important in Substrate and Glutathione Binding. Biochemistry 2006, 45, 10461–10473. [Google Scholar] [CrossRef]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef]
- Shang, G.; Tang, X.; Gao, P.; Guo, F.; Liu, H.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. Sulforaphane attenuation of experimental diabetic nephropathy involves GSK-3 beta/Fyn/Nrf2 signaling pathway. J. Nutr. Biochem. 2015, 26, 596–606. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Mamun, A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2020, 707, 135624. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Nucifora, L.G.; Koga, M.; Shaffer, L.S.; Higgs, C.; Tanaka, T.; Wang, A.M.; Coughlin, J.M.; Barker, P.B.; Fahey, J.W.; et al. Sulforaphane Augments Glutathione and Influences Brain Metabolites in Human Subjects: A Clinical Pilot Study. Mol. Neuropsychiatry 2018, 3, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Connors, S.L.; Macklin, E.A.; Smith, K.D.; Fahey, J.W.; Talalay, P.; Zimmerman, A.W. Sulforaphane treatment of autism spectrum disorder (ASD). Proc. Natl. Acad. Sci. USA 2014, 111, 15550–15555. [Google Scholar] [CrossRef]
- Zimmerman, A.W.; Singh, K.; Connors, S.L.; Liu, H.; Panjwani, A.A.; Lee, L.-C.; Diggins, E.; Foley, A.; Melnyk, S.; Singh, I.N.; et al. Randomized controlled trial of sulforaphane and metabolite discovery in children with Autism Spectrum Disorder. Mol. Autism. 2021, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sun, G.; Zhang, J.; Ting, S.-M.; Gonzales, N.; Aronowski, J. Dimethyl Fumarate Protects Brain From Damage Produced by Intracerebral Hemorrhage by Mechanism Involving Nrf2. Stroke 2015, 46, 1923–1928. [Google Scholar] [CrossRef]
- Della Giustina, A.; Bonfante, S.; Zarbato, G.F.; Danielski, L.G.; Mathias, K.; De Oliveira, A.N.; Garbossa, L.; Cardoso, T.; Fileti, M.E.; De Carli, R.J.; et al. Dimethyl Fumarate Modulates Oxidative Stress and Inflammation in Organs After Sepsis in Rats. Inflammation 2018, 41, 315–327. [Google Scholar] [CrossRef]
- Andersen, J.L.; Gesser, B.; Funder, E.D.; Nielsen, C.J.F.; Gotfred-Rasmussen, H.; Rasmussen, M.K.; Toth, R.; Gothelf, K.V.; Arthur, J.S.C.; Iversen, L.; et al. Dimethyl fumarate is an allosteric covalent inhibitor of the p90 ribosomal S6 kinases. Nat. Commun. 2018, 9, 4344. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chuikov, S.; Taitano, S.; Wu, Q.; Rastogi, A.; Tuck, S.J.; Corey, J.M.; Lundy, S.K.; Mao-Draayer, Y. Dimethyl Fumarate Protects Neural Stem/Progenitor Cells and Neurons from Oxidative Damage through Nrf2-ERK1/2 MAPK Pathway. Int. J. Mol. Sci. 2015, 16, 13885–13907. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; García-Yagüe, Á.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef]
- Dubey, D.; Kieseier, B.C.; Hartung, H.P.; Hemmer, B.; Warnke, C.; Menge, T.; Miller-Little, W.A.; Stuve, O. Dimethyl fumarate in relapsing–remitting multiple sclerosis: Rationale, mechanisms of action, pharmacokinetics, efficacy and safety. Expert Rev. Neurother. 2015, 15, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; El Rayess, Y.; Rizk, A.A.; Sadaka, C.; Zgheib, R.; Zam, W.; Sestito, S.; Rapposelli, S.; Neffe-Skocińska, K.; Zielińska, D.; et al. Turmeric and Its Major Compound Curcumin on Health: Bioactive Effects and Safety Profiles for Food, Pharmaceutical, Biotechnological and Medicinal Applications. Front. Pharmacol. 2020, 11, 01021. [Google Scholar] [CrossRef]
- Kulkarni, S.; Dhir, A. An overview of curcumin in neurological disorders. Indian J. Pharm. Sci. 2010, 72, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Adami, R.; Bottai, D. Curcumin and neurological diseases. Nutr. Neurosci. 2022, 25, 441–461. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Mohamamdinejad, R.; Farkhondeh, T.; Samarghandian, S. Curcumin Activates the Nrf2 Pathway and Induces Cellular Protection Against Oxidative Injury. Curr. Mol. Med. 2020, 20, 116–133. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Zhong, P.; Zhao, Y.; Kanchana, K.; Zhang, Y.; Khan, Z.A.; Chakrabarti, S.; Wu, L.; Wang, J.; Liang, G. Curcumin protects hearts from FFA-induced injury by activating Nrf2 and inactivating NF-κB both in vitro and in vivo. J. Mol. Cell. Cardiol. 2015, 79, 1–12. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, X.; Lai, S.; Liu, J.; Qi, J. Curcumin activates Nrf2/HO-1 signaling to relieve diabetic cardiomyopathy injury by reducing ROS in vitro and in vivo. FASEB J. 2022, 36, e22505. [Google Scholar] [CrossRef]
- Wang, H.; Muhammad, I.; Li, W.; Sun, X.; Cheng, P.; Zhang, X. Sensitivity of Arbor Acres broilers and chemoprevention of aflatoxin B1-induced liver injury by curcumin, a natural potent inducer of phase-II enzymes and Nrf2. Environ. Toxicol. Pharmacol. 2018, 59, 94–104. [Google Scholar] [CrossRef]
- Lu, C.; Xu, W.; Zhang, F.; Shao, J.; Zheng, S. Nrf2 Knockdown Disrupts the Protective Effect of Curcumin on Alcohol-Induced Hepatocyte Necroptosis. Mol. Pharm. 2016, 13, 4043–4053. [Google Scholar] [CrossRef]
- Lopresti, A.L. The Problem of Curcumin and Its Bioavailability: Could Its Gastrointestinal Influence Contribute to Its Overall Health-Enhancing Effects? Adv. Nutr. Int. Rev. J. 2018, 9, 41–50. [Google Scholar] [CrossRef]
- Madeo, L.F.; Sarogni, P.; Cirillo, G.; Vittorio, O.; Voliani, V.; Curcio, M.; Shai-Hee, T.; Büchner, B.; Mertig, M.; Hampel, S. Curcumin and Graphene Oxide Incorporated into Alginate Hydrogels as Versatile Devices for the Local Treatment of Squamous Cell Carcinoma. Materials 2022, 15, 1648. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hao, Z.; Yu, S.; De Moraes, C.G.; Suh, L.H.; Zhao, X.; Lin, Q. An Ultraflexible and Stretchable Aptameric Graphene Nanosensor for Biomarker Detection and Monitoring. Adv. Funct. Mater. 2019, 29, 1905202. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Fujita, T.; Matsumoto, M.; Okamoto, K.; Imada, I. Effects of idebenone (CV-2619) and its metabolites on respiratory activity and lipid peroxidation in brain mitochondria from rats and dogs. J. Pharm.-Dyn. 1985, 8, 1006–1017. [Google Scholar] [CrossRef]
- James, A.M.; Cochemé, H.M.; Smith, R.A.J.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive ox-ygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Lu, L.; Yang, H. Therapeutic Effects of Idebenone on Leber Hereditary Optic Neuropathy. Curr. Eye Res. 2020, 45, 1315–1323. [Google Scholar] [CrossRef]
- Mercuţ, M.F.; Tănasie, C.A.; Dan, A.O.; Nicolcescu, A.M.; Ică, O.M.; Mocanu, C.L.; Ştefănescu-Dima, A. Retinal morphological and functional response to Idebenone therapy in Leber hereditary optic neuropathy. Rom. J. Morphol. Embryol. 2022, 63, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Kotov, S.; Sidorova, O.; Borodataya, E. A pilot study of idebenone in the treatment of patients with hereditary myopathies. Zhurnal Nevrol. Psikhiatrii Im. S. S. Korsakova 2022, 122, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Clementi, M.E.; Pizzoferrato, M.; Bianchetti, G.; Brancato, A.; Sampaolese, B.; Maulucci, G.; Tringali, G. Cytoprotective Effect of Idebenone through Modulation of the Intrinsic Mitochondrial Pathway of Apoptosis in Human Retinal Pigment Epithelial Cells Exposed to Oxidative Stress Induced by Hydrogen Peroxide. Biomedicines 2022, 10, 503. [Google Scholar] [CrossRef]
- Sturm, B.; Stupphann, D.; Kaun, C.; Boesch, S.; Schranzhofer, M.; Wojta, J.; Goldenberg, H.; Scheiber-Mojdehkar, B. Recombinant human erythropoietin: Effects on FXN expression in vitro. Eur. J. Clin. Investig. 2005, 35, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Shrader, W.D.; Amagata, A.; Barnes, A.; Enns, G.M.; Hinman, A.; Jankowski, O.; Kheifets, V.; Komatsuzaki, R.; Lee, E.; Mollard, P.; et al. α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg. Med. Chem. Lett. 2011, 21, 3693–3698. [Google Scholar] [CrossRef]
- Chan, S.S.; Copeland, W.C. DNA polymerase gamma and mitochondrial disease: Understanding the consequence of POLG mutations. Biochim. Biophys. Acta 2009, 1787, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Shekh-Ahmad, T.; Eckel, R.; Naidu, S.D.; Higgins, M.; Yamamoto, M.; Dinkova-Kostova, A.T.; Kovac, S.; Abramov, A.Y.; Walker, M.C. KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain 2018, 141, 1390–1403. [Google Scholar] [CrossRef]
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; Pook, M.A.; Giunti, P.; Abramov, A.Y. ‘Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia’. Cell Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef]
- Monfort, B.; Want, K.; Gervason, S.; D’autréaux, B. Recent Advances in the Elucidation of Frataxin Biochemical Function Open Novel Perspectives for the Treatment of Friedreich’s Ataxia. Front. Neurosci. 2022, 16, 838335. [Google Scholar] [CrossRef]
- Sandi, C.; Pinto, R.M.; Al-Mahdawi, S.; Ezzatizadeh, V.; Barnes, G.; Jones, S.; Rusche, J.R.; Gottesfeld, J.M.; Pook, M.A. Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol. Dis. 2011, 42, 496–505. [Google Scholar] [CrossRef]
- Sivakumar, A.; Cherqui, S. Advantages and Limitations of Gene Therapy and Gene Editing for Friedreich’s Ataxia. Front. Genome Ed. 2022, 4, 903139. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Perel, P.; Roberts, I.; Sena, E.; Wheble, P.; Briscoe, C.; Sandercock, P.; Macleod, M.; Mignini, L.E.; Jayaram, P.; Khan, K.S. Comparison of treatment effects between animal experiments and clinical trials: Systematic review. BMJ 2006, 334, 197. [Google Scholar] [CrossRef] [PubMed]
- Perdomini, M.; Hick, A.; Puccio, H.; Pook, M.A. Animal and cellular models of Friedreich ataxia. J. Neurochem. 2013, 126, 65–79. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.D.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.J.; Santos, M.M.; Ohshima, K.; Smith, J.; Li, L.; Bunting, M.; Cossée, M.; Koenig, M.; Sequeiros, J.; Kaplan, J.; et al. Frataxin knockin mouse. FEBS Lett. 2002, 512, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Gérard, C.; Archambault, A.F.; Bouchard, C.; Tremblay, J.P. A promising mouse model for Friedreich Ataxia progressing like human patients. Behav. Brain Res. 2023, 436, 114107. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.S.; Farmer, J.M.; Perlman, S.; Wilmot, G.; Gomez, C.M.; Bushara, K.O.; Mathews, K.D.; Subramony, S.H.; Ashizawa, T.; Balcer, L.J.; et al. Measuring the rate of progression in Friedreich ataxia: Implications for clinical trial design. Mov. Disord. 2010, 25, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Magrané, J.; Clark, E.M.; Halawani, S.M.; Warren, N.; Rattelle, A.; Lynch, D.R. Early VGLUT1-specific parallel fiber synaptic deficits and dysregulated cerebellar circuit in the KIKO mouse model of Friedreich ataxia. Dis. Model. Mech. 2017, 10, 1529–1538. [Google Scholar] [CrossRef]
- Lin, H.; Magrané, J.; Rattelle, A.; Stepanova, A.; Galkin, A.; Clark, E.M.; Na Dong, Y.; Halawani, S.M.; Lynch, D.R. Early cerebellar deficits in mitochondrial biogenesis and respiratory chain complexes in the KIKO mouse model of Friedreich ataxia. Dis. Model. Mech. 2017, 10, 1343–1352. [Google Scholar] [CrossRef]
- Khonsari, H.; Schneider, M.; Al-Mahdawi, S.; Chianea, Y.G.; Themis, M.; Parris, C.; Pook, M.A. Lentivirus-meditated FXN gene delivery reverses genome instability in Friedreich ataxia patient and mouse model fibroblasts. Gene Ther. 2016, 23, 846–856. [Google Scholar] [CrossRef]
- Shroff, G. A novel approach of human embryonic stem cells therapy in treatment of Friedrich’s Ataxia. Int. J. Case Rep. Images 2015, 6, 261–266. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | Mode of Action | Model | Dosage | Effect | Ref. |
|---|---|---|---|---|---|
| RESVERATROL | Induction of NRF2 phosphorylation (PI3K/Akt-dependent mechanism) | In vitro Fibroblasts derived from FRDA patients; Cardiomyocytes and cerebellar granule neurons | 100 μM RV/30 μM DMF | Increase in FXN mRNA transcription and mitochondrial biogenesis | [123] |
| Promotion of SIRT1 activation, which leads to reduced KEAP1 expression and increased NRF2 nuclear translocation | In vivo YG8LR mice | 200 mg/kg/d RV + DMF | Decrease in ROS levels; Improvement in rotarod performance | ||
| COENZYME Q10 | Inhibition of NRF2 proteasomal degradation by promoting its stabilization and nuclear translocation | In vitro Fibroblasts derived from FRDA patients | 0.1 pM–50 μM | Prevention of cell death in GSH-depleted cells | [124] |
| SULFORAPHANE | Prevention of NRF2 proteasomal degradation by interaction with KEAP1, thereby promoting NRF2; Nuclear translocation and reinforcing NRF2 binding to ARE sequences | In vitro KIKO and YG8R mice fibroblasts | 50 nM | Prevention of lipid peroxidation and cell death | [125] |
| Frataxin-silenced NSC34 motor neurons | 5 μM | Increase in FXN protein; Increase in Nrf2 transcript and protein expressions; Increase in NQO1, SOD, and GSH content; Axonal re-growth and increased neurites’ numbers | [24] | ||
| Fibroblasts derived from FRDA patients | 10 μM | Increase in NRF2 transcript and protein expressions; Increase in NRF2-target genes (NQO1 and HO-1) expressions | [126] | ||
| Neural Stem Cells from KIKO mice | 5 μM | Reduction of ROS levels; Re-establishment of a proper differentiation program | [25] | ||
| DMF | Promotion of covalent modification of NRF2 DNA binding domain, leading to NRF2 activation | In vitro Lymphocytes derived from FRDA patients | 10–30 μM | Increase in FXN mRNA and protein expression and mitochondrial biogenesis; Reduce of R-loop at GAA sites in FRDA patients | [127] |
| In vivo YG8R mouse model | 5 and 10 mg/kg | Increase in FXN mRNA and protein expression | |||
| FXNKD mouse model of FRDA | 110–160 mg/day | Rescue of brain mitochondria-related enzymes (Complex II, Complex IV, and aconitase) | [128] | ||
| CURCUMIN | Inhibition of NRF2 proteasomal degradation by promoting its stabilization and nuclear translocation | In vivo YG8R FRDA mice | 150 mg/kg 5 days | Increase in Fe-S biogenesis; Elimination of iron deposits from heart | [129] |
| IDEBENONE | Inhibition of NRF2 proteasomal degradation by promoting its stabilization and nuclear translocation | In vitro Fibroblasts derived from FRDA patients | 1 μM | Increase in Nrf2 transcript and protein expression; Increase in NQO1 (Nrf2-target gene) transcript expression | [126] |
| EPI-743 | Inhibition of NRF2 proteasomal degradation by promoting its stabilization and nuclear translocation | In vitro Fibroblasts derived from FRDA patients | 1 μM | Increase in FXN mRNA andNrf2 transcript and protein expression; | [126] |
| 1 μM | Increase in Nrf2 nuclear translocation;Rescue of mitochondrial tubular network | [118] | |||
| RTA408 | Inhibition of KEAP1 by direct binding to its Cys151 | In vitro KIKO and YG8R mice cerebellar granule neurons (CGNs) | 50 nM | Restoration of OXPHOS complex; Prevention of lipid peroxidation; Reduction of mROS and increase in GSH content | [130] |
| Fibroblasts derived from FRDA patients | 50 nM | ||||
| Fibroblasts derived from FRDA patients | 100 nM | Increase in Nrf2 transcript and protein expression; Increase in NQO1, GCL, and HO-1 (Nrf2-target genes) transcript expressions and in GSH content | [126] |
| Compound | Mode of Action | Patients | Dosage and Time | Effect | Ref. |
|---|---|---|---|---|---|
| RESVERATROL | Induction of NRF2 phosphorylation (PI3K/Akt-dependent mechanism); Promotion of SIRT1 activation, which leads to reduced KEAP1 expression and increased NRF2 nuclear translocation | 24 | 1 or 5 g/daily | Significant neurologic, audiologic, and speech improvements in the high-dose group | [131] |
| 12 months | No improvement in cardiac outcomes or FXN expression | ||||
| COENZYME Q10 | Inhibition of NRF2 proteasomal degradation by promoting its stabilization and nuclear translocation | 43 | CoQ10: 600 mg/d (2× 100 mg for three times/day) + Vitamin E 2100 IU/day supplementation | Restoration of CoQ10 serum levels | [132] |
| 10 | 2 years CoQ10: 400 mg/d + Vitamin E 2100 IU/day supplementation 6 months | Improvement in cardiac and skeletal muscle bioenergetics; Improvements in ICARS score (post hoc analysis) in 49% of patients | [133] | ||
| ALCAR | Prevention of NRF2 proteasomal degradation by interaction with KEAP1, NRF2 nuclear translocation, and reinforcement of NRF2 binding to ARE sequences | 11 | 1000 mg/d twice a day 6 months | Improvements in coordination after 3 and 6 months and significant effect on muscle tone after 6 months | [134] |
| IDEBENONE | Promotion of covalent modification of NRF2 DNA binding domain, leading to NRF2 activation | 3 | 5 mg/kg/d 4–9 months | Decrease in myocardial hypertrophy; Improvements in fine movements | [135] |
| 8 | 5–20 mg/kg/d 3–5 years | Significant reduction of cardiac hypertrophy in six of eight patients. | [136] | ||
| 24 (10 paediatric, 14 adults) | 5–20 mg/kg/d 3–5 years | Prevention of progression of cardiomyopathy in both paediatric and adult patients; Stabilizing effect on neurological dysfunction only in paediatric patients | [137] | ||
| 70 | 10–54 mg/kg/d 6 months | No improvements in neurological outcomes and no assessment of cardiac outcomes | [138] | ||
| 70 | 450/900 mg/d or 1350/2250 mg/d 6 months | No decrease in hypertrophy or improved cardiac function | [139] | ||
| 9 | 5 mg/kg/d 1 year | Cerebellar improvement (after 3 months); Significant reduction of ICARS scores | [140] | ||
| 29 | 1350–2250 mg/d 2 months | No improvements in ICARS score or in cardiac outcomes | [141] | ||
| 27 | 5–20 mg/kg/d 4–11 years | No improvements in neurologic or cardiac outcomes | [142] | ||
| IDEBENONE +Erythropoietin | Erythropoietin: increases FXN mRNA levels | 16 | IDE 5 mg/kg/d EPO: 20,000–40,000 IU | No significant hematologic, clinical, or biochemical impact | [143] |
| IDEBENONE +Tocotrienol | Tocotrienol: enhances FXN-3 mRNA expression | 14 | IDE + Tocotrienol mixture 5 mg/kg/d 2 months (expected 1 year) | Decrease in oxidative stress indexes (GSH/GSSG ratio; carbonyl group) | [144] |
| EPI-743 | Inhibition of NRF2 proteasomal degradation by promoting its stabilization and nuclear translocation | 14 (2 FRDA patients) | 100 mg, two times per day, increased to three times per day on day 29, 12 weeks | Improved strength, exercise tolerance, speech fluency, sleep, increased social interaction; Partial rescue of complete cortical blindness | [145] |
| 3 FRDA patients (rare variant) | 400 mg 18 months | Significant improvement in neurological functions (FARS score) already at 6 months | [146] | ||
| 63 | 200–400 mg 2 years | No improvements in visual acuity, 25-foot walk test, peg-hole test, or echocardiography; Post hoc analysis showed significant improvement in FARS score | [147] | ||
| RTA408 | Inhibition of KEAP1 by direct binding to its Cys151 | 69 | 160 mg/d/12 weeks | Significant improvements in FARS score; Increase in Nrf2-target gene expression. | [148] |
| 155 | 150 mg/d/48 weeks | Second part of the trial: results confirmed even at a lower dosage | [149] | ||
| Extension phase | Estimated to be completed in December 2024 | NCT02255435 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiberi, J.; Segatto, M.; Fiorenza, M.T.; La Rosa, P. Apparent Opportunities and Hidden Pitfalls: The Conflicting Results of Restoring NRF2-Regulated Redox Metabolism in Friedreich’s Ataxia Pre-Clinical Models and Clinical Trials. Biomedicines 2023, 11, 1293. https://doi.org/10.3390/biomedicines11051293
Tiberi J, Segatto M, Fiorenza MT, La Rosa P. Apparent Opportunities and Hidden Pitfalls: The Conflicting Results of Restoring NRF2-Regulated Redox Metabolism in Friedreich’s Ataxia Pre-Clinical Models and Clinical Trials. Biomedicines. 2023; 11(5):1293. https://doi.org/10.3390/biomedicines11051293
Chicago/Turabian StyleTiberi, Jessica, Marco Segatto, Maria Teresa Fiorenza, and Piergiorgio La Rosa. 2023. "Apparent Opportunities and Hidden Pitfalls: The Conflicting Results of Restoring NRF2-Regulated Redox Metabolism in Friedreich’s Ataxia Pre-Clinical Models and Clinical Trials" Biomedicines 11, no. 5: 1293. https://doi.org/10.3390/biomedicines11051293