
Exploring Pro-Inflammatory Immunological Mediators: Unraveling the Mechanisms of Neuroinflammation in Lysosomal Storage Diseases


Abstract
:1. Introduction
2. Gaucher-Disease-Associated Neuroinflammation: Deciphering the Complex Interactions between Neurological and Immune Systems
3. Fabry-Disease-Associated Neuroinflammation: Deciphering the Complex Interactions between Neurological and Immune Systems
4. GM1-Gangliosidosis-Associated Neuroinflammation: Deciphering the Complex Interactions between Neurological and Immune Systems
5. GM2-Gangliosidosis-Associated Neuroinflammation: Deciphering the Complex Interactions between Neurological and Immune Systems
6. Niemann–Pick Type C-Disease-Associated Neuroinflammation: Deciphering the Complex Interactions between Neurological and Immune Systems
7. Farber-Disease-Associated Neuroinflammation: Deciphering the Complex Interactions between Neurological and Immune Systems
8. Krabbe-Disease-Associated Neuroinflammation: Deciphering the Complex Interactions between Neurological and Immune Systems
9. Wolman Disease—Associated Neuroinflammation: Deciphering the Complex Interactions between Neurological and Immune Systems
10. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bosch, M.E.; Kielian, T. Neuroinflammatory paradigms in lysosomal storage diseases. Front. Neurosci. 2015, 9, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Crooke, S.T. A call to arms against ultra-rare diseases. Nat. Biotechnol. 2021, 39, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Grabowski, G.A. Immunological cells and functions in Gaucher disease. Crit. Rev. Oncog. 2013, 18, 197–220. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23510064 (accessed on 28 February 2023). [CrossRef] [Green Version]
- Pandey, M.K.; Jabre, N.A.; Xu, Y.H.; Zhang, W.; Setchell, K.D.; Grabowski, G.A. Gaucher disease: Chemotactic factors and immunological cell invasion in a mouse model. Mol. Genet. Metab. 2014, 111, 163–171. [Google Scholar] [CrossRef]
- Pandey, M.K.; Rani, R.; Zhang, W.; Setchell, K.; Grabowski, G.A. Immunological cell type characterization and Th1-Th17 cytokine production in a mouse model of Gaucher disease. Mol. Genet. Metab. 2012, 106, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Waldek, S.; Feriozzi, S. Fabry nephropathy: A review—How can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014, 15, 72. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.S.; Meng, X.L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Rozenfeld, P.; Agriello, E.; De Francesco, N.; Martinez, P.; Fossati, C. Leukocyte perturbation associated with Fabry disease. J. Inherit. Metab. Dis. 2009, 32, S67–S77. [Google Scholar] [CrossRef]
- De Francesco, P.N.; Mucci, J.M.; Ceci, R.; Fossati, C.A.; Rozenfeld, P.A. Fabry disease peripheral blood immune cells release inflammatory cytokines: Role of globotriaosylceramide. Mol. Genet. Metab. 2013, 109, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Cigna, D.; D’Anna, C.; Zizzo, C.; Francofonte, D.; Sorrentino, I.; Colomba, P.; Albeggiani, G.; Armini, A.; Bianchi, L.; Bini, L.; et al. Alteration of proteomic profiles in PBMC isolated from patients with Fabry disease: Preliminary findings. Mol. Biosyst. 2013, 9, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.F.; Krokhin, O.V.; Beavis, R.C.; Ries, M.; Robinson, C.; Goldin, E.; Brady, R.O.; Wilkins, J.A.; Schiffmann, R. Proteomics of specific treatment-related alterations in Fabry disease: A strategy to identify biological abnormalities. Proc. Natl. Acad. Sci. USA 2007, 104, 2873–2878. [Google Scholar] [CrossRef] [Green Version]
- Hollander, Z.; Dai, D.L.; Putko, B.N.; Yogasundaram, H.; Wilson-McManus, J.E.; Thompson, R.B.; Khan, A.; West, M.L.; McManus, B.M.; Oudit, G.Y. Gender-specific plasma proteomic biomarkers in patients with Anderson-Fabry disease. Eur. J. Heart Fail. 2015, 17, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [Green Version]
- Brunetti-Pierri, N.; Scaglia, F. GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 2008, 94, 391–396. [Google Scholar] [CrossRef]
- Przybilla, M.J.; Ou, L.; Tabaran, A.F.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; O’Sullivan, M.G.; Whitley, C.B. Comprehensive behavioral and biochemical outcomes of novel murine models of GM1-gangliosidosis and Morquio syndrome type B. Mol. Genet. Metab. 2019, 126, 139–150. [Google Scholar] [CrossRef]
- Son, M.Y.; Kwak, J.E.; Seol, B.; Lee, D.Y.; Jeon, H.; Cho, Y.S. A novel human model of the neurodegenerative disease GM1 gangliosidosis using induced pluripotent stem cells demonstrates inflammasome activation. J. Pathol. 2015, 237, 98–110. [Google Scholar] [CrossRef]
- Myerowitz, R. Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene. Hum. Mutat. 1997, 9, 195–208. [Google Scholar] [CrossRef]
- Myerowitz, R.; Lawson, D.; Mizukami, H.; Mi, Y.; Tifft, C.J.; Proia, R.L. Molecular pathophysiology in Tay-Sachs and Sandhoff diseases as revealed by gene expression profiling. Hum. Mol. Genet. 2002, 11, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.P.; Proia, R.L. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc. Natl. Acad. Sci. USA 2004, 101, 8425–8430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, R.; Tifft, C.J.; Proia, R.L. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA 2000, 97, 10954–10959. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Sano, T.; Irisa, M.; Kodama, T.; Saito, T.; Furusawa, E.; Kaizu, K.; Yanagi, Y.; Tsukimura, T.; Togawa, T.; et al. FcRgamma-dependent immune activation initiates astrogliosis during the asymptomatic phase of Sandhoff disease model mice. Sci. Rep. 2017, 7, 40518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef] [Green Version]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet. J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 121. [Google Scholar] [CrossRef] [Green Version]
- Li, C.M.; Park, J.H.; He, X.; Levy, B.; Chen, F.; Arai, K.; Adler, D.A.; Disteche, C.M.; Koch, J.; Sandhoff, K.; et al. The human acid ceramidase gene (ASAH): Structure, chromosomal location, mutation analysis, and expression. Genomics 1999, 62, 223–231. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Y.; Benitez, B.A.; Nagree, M.S.; Dearborn, J.T.; Jiang, X.; Guzman, M.A.; Woloszynek, J.C.; Giaramita, A.; Yip, B.K.; et al. Genetic ablation of acid ceramidase in Krabbe disease confirms the psychosine hypothesis and identifies a new therapeutic target. Proc. Natl. Acad. Sci. USA 2019, 116, 20097–20103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins-Salsbury, J.A.; Shea, L.; Jiang, X.; Hunter, D.A.; Guzman, A.M.; Reddy, A.S.; Qin, E.Y.; Li, Y.; Gray, S.J.; Ory, D.S.; et al. Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J. Neurosci. 2015, 35, 6495–6505. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, D.L.; Hulkova, H.; Bialer, M.G.; Desnick, R.J. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. J. Hepatol. 2013, 58, 1230–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bychkov, I.O.; Kamenets, E.A.; Filatova, A.Y.; Skoblov, M.Y.; Mikhaylova, S.V.; Strokova, T.V.; Gundobina, O.S.; Zakharova, E.Y. The novel synonymous variant in LIPA gene affects splicing and causes lysosomal acid lipase deficiency. Mol. Genet. Metab. 2019, 127, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Pluvinage, J.V.; Sun, J.; Claes, C.; Flynn, R.A.; Haney, M.S.; Iram, T.; Meng, X.; Lindemann, R.; Riley, N.M.; Danhash, E.; et al. The CD22-IGF2R interaction is a therapeutic target for microglial lysosome dysfunction in Niemann-Pick type C. Sci. Transl. Med. 2021, 13, eabg2919. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Dinkel, L.; Müller, S.A.; Sebastian Monasor, L.; Schifferer, M.; Cantuti-Castelvetri, L.; König, J.; Vidatic, L.; Bremova-Ertl, T.; Lieberman, A.P.; et al. Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia. Nat. Commun. 2021, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Magnusen, A.F.; Rani, R.; McKay, M.A.; Hatton, S.L.; Nyamajenjere, T.C.; Magnusen, D.N.A.; Köhl, J.; Grabowski, G.A.; Pandey, M.K. C-X-C Motif Chemokine Ligand 9 and Its CXCR3 Receptor Are the Salt and Pepper for T Cells Trafficking in a Mouse Model of Gaucher Disease. Int. J. Mol. Sci. 2021, 22, 12712. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; McKay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef]
- Magnusen, A.F.; Hatton, S.L.; Rani, R.; Pandey, M.K. Genetic Defects and Pro-inflammatory Cytokines in Parkinson’s Disease. Front. Neurol. 2021, 12, 636139. [Google Scholar] [CrossRef]
- Hatton, S.L.; Pandey, M.K. Fat and Protein Combat Triggers Immunological Weapons of Innate and Adaptive Immune Systems to Launch Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 1089. [Google Scholar] [CrossRef]
- Bradbury, A.M.; Bongarzone, E.R.; Sands, M.S. Krabbe disease: New hope for an old disease. Neurosci. Lett. 2021, 752, 135841. [Google Scholar] [CrossRef]
- Vitner, E.B. The role of brain innate immune response in lysosomal storage disorders: Fundamental process or evolutionary side effect? FEBS Lett. 2020, 594, 3619–3631. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—Challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef]
- Pandey, M.K.; Grabowski, G.A.; Kohl, J. An unexpected player in Gaucher disease: The multiple roles of complement in disease development. Semin. Immunol. 2018, 37, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, V.S.; Magnusen, A.F.; Rani, R.; Marsili, L.; Slavotinek, A.M.; Prows, D.R.; Hopkin, R.J.; McKay, M.A.; Pandey, M.K. Targeting the Complement-Sphingolipid System in COVID-19 and Gaucher Diseases: Evidence for a New Treatment Strategy. Int. J. Mol. Sci. 2022, 23, 14340. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ran, M.; Li, H.; Lin, Y.; Ma, K.; Yang, Y.; Fu, X.; Yang, S. New insight into neurological degeneration: Inflammatory cytokines and blood–brain barrier. Front. Mol. Neurosci. 2022, 15, 1013933. [Google Scholar] [CrossRef] [PubMed]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Moriarty, R.A.; Stroka, K.M. Recent progress and new challenges in modeling of human pluripotent stem cell-derived blood-brain barrier. Theranostics 2021, 11, 10148–10170. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Daneman, R. Formation and maintenance of the BBB. Mech. Dev. 2015, 138, 8–16. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Ubogu, E.E. Biology of the human blood-nerve barrier in health and disease. Exp. Neurol. 2020, 328, 113272. [Google Scholar] [CrossRef]
- Stubbs, E.B., Jr. Targeting the blood-nerve barrier for the management of immune-mediated peripheral neuropathies. Exp. Neurol. 2020, 331, 113385. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Weerasuriya, A.; Mizisin, A.P. The blood-nerve barrier: Structure and functional significance. Methods Mol. Biol. 2011, 686, 149–173. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Y.; Stirnemann, J.; Belmatoug, N. Gaucher disease: A review. Rev. Med. Interne. 2019, 40, 313–322. [Google Scholar] [CrossRef]
- van Meer, G.; Wolthoorn, J.; Degroote, S. The fate and function of glycosphingolipid glucosylceramide. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 869–873. [Google Scholar] [CrossRef] [Green Version]
- Reza, S.; Ugorski, M.; Suchański, J. Glucosylceramide and galactosylceramide, small glycosphingolipids with significant impact on health and disease. Glycobiology 2021, 31, 1416–1434. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Zheng, S.J.; Mao, X.R.; Li, J.F. The role of glucosylceramide and glucosylceramide synthase in liver disease: From bench to bedside—Review. Acta Biochim. Pol. 2020, 68, 33–39. [Google Scholar] [CrossRef]
- Bleicher, R.J.; Cabot, M.C. Glucosylceramide synthase and apoptosis. Biochim. Biophys. Acta 2002, 1585, 172–178. [Google Scholar] [CrossRef]
- Pandey, M.K. Pre-existing humoral immune comebacks control the development of the severe form of coronavirus disease 2019 in Gaucher patients. Clin. Transl. Discov. 2022, 2, e96. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Jia, L.; Quinn, B.; Zamzow, M.; Stringer, K.; Aronow, B.; Sun, Y.; Zhang, W.; Setchell, K.D.; Grabowski, G.A. Global gene expression profile progression in Gaucher disease mouse models. BMC Genom. 2011, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Cogan, D.G.; Chu, F.C.; Reingold, D.; Barranger, J. Ocular Motor Signs in Some Metabolic Diseases. Arch. Ophthalmol. 1981, 99, 1802–1808. [Google Scholar] [CrossRef]
- Sidransky, E.; Tsujl, S.; Stubblefield, B.K.; Gurrie, J.; FitzGibbon, E.J.; Glnns, E.I. Gaudier patients with oculomotor abnormalities do not have a unique genotype. Clin. Genet. 1992, 41, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Horowitz, M.; Abel, R.B.; Currie, J.N.; Yu, K.-T.; Kaneski, C.; Higgins, J.J.; O’Neill, R.R.; Fedio, P.; Pikus, A.; et al. Isolated horizontal supranuclear gaze palsy as a marker of severe systemic involvement in Gaucher’s disease. Neurology 1993, 43, 1993. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.F.; Glew, R.H.; Peters, S.P. Familial Psychosis and Diverse Neurologic Abnormalities in Adult-Onset Gaucher’s Disease. Arch. Neurol. 1979, 36, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Fueki, N.; Sasaki, M.; Sakuragawa, N. Uncoupling of blood flow and oxygen metabolism in the cerebellum in type 3 Gaucher disease. Brain Dev. 1991, 13, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.J.; Finckh, U.; Höppner, J.; Lakner, V.; Liebisch, I.; Grau, G.; Rolfs, A. Two new missense mutations in a non-Jewish Caucasian family with type 3 Gaucher disease. Neurology 1996, 46, 1102–1107. [Google Scholar] [CrossRef]
- Grover, W.D.; Tucker, S.H.; Wenger, D.A. Clinical variation in 2 related children with neuronopathic Gaucher disease. Ann. Neurol. 1978, 3, 281–283. [Google Scholar] [CrossRef]
- Conradi, N.; Kyllerman, M.; Månsson, J.E.; Percy, A.K.; Svennerholm, L. Late-infantile Gaucher disease in a child with myoclonus and bulbar signs: Neuropathological and neurochemical findings. Acta Neuropathol. 1991, 82, 152–157. [Google Scholar] [CrossRef]
- Dobbelaere, D.; Sukno, S.; Defoort-Dhellemmes, S.; Lamblin, M.D.; Largillière, C. Neurological outcome of a patient with Gaucher disease type III treated by enzymatic replacement therapy. J. Inherit. Metab. Dis. 1998, 21, 74–76. [Google Scholar] [CrossRef]
- Verghese, J.; Goldberg, R.F.; Desnick, R.J.; Grace, M.E.; Goldman, J.E.; Lee, S.C.; Dickson, D.W.; Rapin, I. Myoclonus from Selective Dentate Nucleus Degeneration in Type 3 Gaucher Disease. Arch. Neurol. 2000, 57, 389–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erikson, A. Gaucher disease-Norrbottnian type (III). Acta Paediatr. 1986, 75, 1–42. [Google Scholar] [CrossRef]
- Park, J.K.; Orvisky, E.; Tayebi, N.; Kaneski, C.; Lamarca, M.E.; Stubblefield, B.K.; Martin, B.M.; Schiffmann, R.; Sidransky, E. Myoclonic Epilepsy in Gaucher Disease: Genotype-Phenotype Insights from a Rare Patient Subgroup. Pediatr. Res. 2003, 53, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, N.; Xu, Y.H.; Oh, S.; Sun, Y.; Jia, L.; Keddache, M.; Grabowski, G.A. Gaucher disease: Transcriptome analyses using microarray or mRNA sequencing in a Gba1 mutant mouse model treated with velaglucerase alfa or imiglucerase. PLoS ONE 2013, 8, e74912. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Grabowski, G.A. Advances in Gaucher Disease: Basic and Clinical Perspectives; Future Medicine Ltd.: London, UK, 2013; pp. 78–93. [Google Scholar]
- Xu, Y.H.; Quinn, B.; Witte, D.; Grabowski, G.A. Viable mouse models of acid beta-glucosidase deficiency: The defect in Gaucher disease. Am. J. Pathol. 2003, 163, 2093–2101. [Google Scholar] [CrossRef]
- Dasgupta, N.; Xu, Y.H.; Li, R.; Peng, Y.; Pandey, M.K.; Tinch, S.L.; Liou, B.; Inskeep, V.; Zhang, W.; Setchell, K.D.; et al. Neuronopathic Gaucher disease: Dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum. Mol. Genet. 2015, 24, 7031–7048. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, W.; Xu, Y.H.; Quinn, B.; Dasgupta, N.; Liou, B.; Setchell, K.D.; Grabowski, G.A. Substrate compositional variation with tissue/region and Gba1 mutations in mouse models--implications for Gaucher disease. PLoS ONE 2013, 8, e57560. [Google Scholar] [CrossRef] [Green Version]
- Liou, B.; Zhang, W.; Fannin, V.; Quinn, B.; Ran, H.; Xu, K.; Setchell, K.D.R.; Witte, D.; Grabowski, G.A.; Sun, Y. Combination of acid β-glucosidase mutation and Saposin C deficiency in mice reveals Gba1 mutation dependent and tissue-specific disease phenotype. Sci. Rep. 2019, 9, 5571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignot, C.; Doummar, D.; Maire, I.; De Villemeur, T.B. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 2006, 28, 39–48. [Google Scholar] [CrossRef]
- Gupta, N.; Oppenheim, I.M.; Kauvar, E.F.; Tayebi, N.; Sidransky, E. Type 2 Gaucher disease: Phenotypic variation and genotypic heterogeneity. Blood Cells Mol. Dis. 2011, 46, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, C.M.; Taylor, D.S.; Vellodi, A. Ocular motor abnormalities in Gaucher disease. Neuropediatrics 1999, 30, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.B.; Kim, E.Y.; Jung, S.C. Upregulation of proinflammatory cytokines in the fetal brain of the Gaucher mouse. J. Korean Med. Sci. 2006, 21, 733–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.Y.; Hong, Y.B.; Go, S.H.; Lee, B.; Jung, S.C. Downregulation of neurotrophic factors in the brain of a mouse model of Gaucher disease; implications for neuronal loss in Gaucher disease. Exp. Mol. Med. 2006, 38, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Tybulewicz, V.L.; Tremblay, M.L.; LaMarca, M.E.; Willemsen, R.; Stubblefield, B.K.; Winfield, S.; Zablocka, B.; Sidransky, E.; Martin, B.M.; Huang, S.P.; et al. Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 1992, 357, 407–410. [Google Scholar] [CrossRef]
- Vitner, E.B.; Salomon, R.; Farfel-Becker, T.; Meshcheriakova, A.; Ali, M.; Klein, A.D.; Platt, F.M.; Cox, T.M.; Futerman, A.H. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 2014, 20, 204–208. [Google Scholar] [CrossRef]
- Vardi, A.; Zigdon, H.; Meshcheriakova, A.; Klein, A.D.; Yaacobi, C.; Eilam, R.; Kenwood, B.M.; Rahim, A.A.; Massaro, G.; Merrill, A.H., Jr.; et al. Delineating pathological pathways in a chemically induced mouse model of Gaucher disease. J. Pathol. 2016, 239, 496–509. [Google Scholar] [CrossRef]
- Kanfer, J.N.; Legler, G.; Sullivan, J.; Raghavan, S.S.; Mumford, R.A. The Gaucher mouse. Biochem. Biophys. Res. Commun. 1975, 67, 85–90. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Vitner, E.B.; Pressey, S.N.; Eilam, R.; Cooper, J.D.; Futerman, A.H. Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum. Mol. Genet. 2011, 20, 1375–1386. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Vitner, E.B.; Kelly, S.L.; Bame, J.R.; Duan, J.; Shinder, V.; Merrill, A.H., Jr.; Dobrenis, K.; Futerman, A.H. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet. 2014, 23, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Vitner, E.B.; Dekel, H.; Zigdon, H.; Shachar, T.; Farfel-Becker, T.; Eilam, R.; Karlsson, S.; Futerman, A.H. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum. Mol. Genet. 2010, 19, 3583–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A.H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain A J. Neurol. 2012, 135, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Enquist, I.B.; Lo Bianco, C.; Ooka, A.; Nilsson, E.; Månsson, J.E.; Ehinger, M.; Richter, J.; Brady, R.O.; Kirik, D.; Karlsson, S. Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17483–17488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.H. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef]
- Koprivica, V.; Stone, D.L.; Park, J.K.; Callahan, M.; Frisch, A.; Cohen, I.J.; Tayebi, N.; Sidransky, E. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am. J. Hum. Genet. 2000, 66, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Orvisky, E.; Park, J.K.; Parker, A.; Walker, J.M.; Martin, B.M.; Stubblefield, B.K.; Uyama, E.; Tayebi, N.; Sidransky, E. The identification of eight novel glucocerebrosidase (GBA) mutations in patients with Gaucher disease. Hum. Mutat. 2002, 19, 458–459. [Google Scholar] [CrossRef]
- Conradi, N.G.; Sourander, P.; Nilsson, O.; Svennerholm, L.; Erikson, A. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta Neuropathol. 1984, 65, 99–109. [Google Scholar] [CrossRef]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H.V. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.N.; Do, H.-S.; Jeong, H.; Kim, T.; Heo, S.H.; Kim, Y.-M.; Cheon, C.K.; Lee, Y.; Choi, Y.; Choi, I.H.; et al. Identification of a novel therapeutic target underlying atypical manifestation of Gaucher disease. Clin. Transl. Med. 2022, 12, e862. [Google Scholar] [CrossRef]
- Serfecz, J.C.; Saadin, A.; Santiago, C.P.; Zhang, Y.; Bentzen, S.M.; Vogel, S.N.; Feldman, R.A. C5a Activates a Pro-Inflammatory Gene Expression Profile in Human Gaucher iPSC-Derived Macrophages. Int. J. Mol. Sci. 2021, 22, 9912. [Google Scholar] [CrossRef] [PubMed]
- Del Pinto, R.; Ferri, C. The role of Immunity in Fabry Disease and Hypertension: A Review of a Novel Common Pathway. High Blood Press Cardiovasc. Prev. 2020, 27, 539–546. [Google Scholar] [CrossRef]
- Ishii, S.; Kase, R.; Sakuraba, H.; Suzuki, Y. Characterization of a Mutant α-Galactosidase Gene Product for the Late-Onset Cardiac Form of Fabry Disease. Biochem. Biophys. Res. Commun. 1993, 197, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Arad, M.; Baron, R.; Burlina, A.; Elliott, P.M.; Feldt-Rasmussen, U.; Fomin, V.V.; Germain, D.P.; Hughes, D.A.; Jovanovic, A.; et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol. Genet. Metab. 2018, 124, 189–203. [Google Scholar] [CrossRef]
- Aguilera-Correa, J.-J.; Madrazo-Clemente, P.; Martínez-Cuesta, M.d.C.; Peláez, C.; Ortiz, A.; Sánchez-Niño, M.D.; Esteban, J.; Requena, T. Lyso-Gb3 modulates the gut microbiota and decreases butyrate production. Sci. Rep. 2019, 9, 12010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-Y.; Park, S.; Lee, S.-W.; Lee, J.-H.; Lee, E.S.; Kim, M.; Kim, Y.; Kang, J.S.; Chung, C.H.; Moon, J.-S.; et al. RIPK3 Contributes to Lyso-Gb3-Induced Podocyte Death. Cells 2021, 10, 245. Available online: https://www.mdpi.com/2073-4409/10/2/245 (accessed on 28 February 2023). [CrossRef] [PubMed]
- Choi, S.; Kim, J.A.; Na, H.Y.; Cho, S.E.; Park, S.; Jung, S.C.; Suh, S.H. Globotriaosylceramide induces lysosomal degradation of endothelial KCa3.1 in fabry disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, H.; Miyata, K.; Mikame, M.; Taguchi, A.; Guili, C.; Shimura, M.; Murayama, K.; Inoue, T.; Yamamoto, S.; Sugimura, K. Effectiveness of plasma lyso-Gb3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis. Genet. Med. 2019, 21, 44–52. [Google Scholar] [CrossRef]
- Choi, L.; Vernon, J.; Kopach, O.; Minett, M.S.; Mills, K.; Clayton, P.T.; Meert, T.; Wood, J.N. The Fabry disease-associated lipid Lyso-Gb3 enhances voltage-gated calcium currents in sensory neurons and causes pain. Neurosci. Lett. 2015, 594, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Heo, S.H.; Kang, E.; Kim, Y.-M.; Go, H.; Kim, K.Y.; Jung, J.Y.; Kang, M.; Kim, G.-H.; Kim, J.-M.; Choi, I.-H. Fabry disease: Characterisation of the plasma proteome pre- and post-enzyme replacement therapy. J. Med. Genet. 2017, 54, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biancini, G.B.; Vanzin, C.S.; Rodrigues, D.B.; Deon, M.; Ribas, G.S.; Barschak, A.G.; Manfredini, V.; Netto, C.B.; Jardim, L.B.; Giugliani, R.; et al. Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2012, 1822, 226–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenfeld, P.A.; Croxatto, O.; Ebner, R.; Fossati, C.A. Immunofluorescence detection of globotriaosylceramide deposits in conjunctival biopsies of Fabry disease patients. Clin. Exp. Ophthalmol. 2006, 34, 689–694. [Google Scholar] [CrossRef]
- Hongo, K.; Harada, T.; Fukuro, E.; Kobayashi, M.; Ohashi, T.; Eto, Y. Massive accumulation of globotriaosylceramide in various tissues from a Fabry patient with a high antibody titer against alpha-galactosidase A after 6 years of enzyme replacement therapy. Mol. Genet. Metab. Rep. 2020, 24, 100623. [Google Scholar] [CrossRef]
- Lang, F.M.; Korner, P.; Harnett, M.; Karunakara, A.; Tifft, C.J. The natural history of Type 1 infantile GM1 gangliosidosis: A literature-based meta-analysis. Mol. Genet. Metab. 2020, 129, 228–235. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.-T.; Ariga, T. Functional Roles of Gangliosides in Neurodevelopment: An Overview of Recent Advances. Neurochem. Res. 2012, 37, 1230–1244. [Google Scholar] [CrossRef] [Green Version]
- Regier, D.S.; Kwon, H.J.; Johnston, J.; Golas, G.; Yang, S.; Wiggs, E.; Latour, Y.; Thomas, S.; Portner, C.; Adams, D.; et al. MRI/MRS as a surrogate marker for clinical progression in GM1 gangliosidosis. Am. J. Med. Genet. Part A 2016, 170, 634–644. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Regier, D.S.; Yoon, R.; Pan, K.S.; Johnston, J.M.; Yang, S.; Spranger, J.W.; Tifft, C.J. The skeletal phenotype of intermediate GM1 gangliosidosis: Clinical, radiographic and densitometric features, and implications for clinical monitoring and intervention. Bone 2020, 131, 115142. [Google Scholar] [CrossRef]
- Lee, J.S.; Choi, J.-M.; Lee, M.; Kim, S.Y.; Lee, S.; Lim, B.C.; Cheon, J.-E.; Kim, I.-O.; Kim, K.J.; Choi, M.; et al. Diagnostic challenge for the rare lysosomal storage disease: Late infantile GM1 gangliosidosis. Brain Dev. 2018, 40, 383–390. [Google Scholar] [CrossRef]
- Sano, R.; Tessitore, A.; Ingrassia, A.; d’Azzo, A. Chemokine-induced recruitment of genetically modified bone marrow cells into the CNS of GM1-gangliosidosis mice corrects neuronal pathology. Blood 2005, 106, 2259–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyakumar, M.; Thomas, R.; Elliot-Smith, E.; Smith, D.A.; van der Spoel, A.C.; d’Azzo, A.; Perry, V.H.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain A J. Neurol. 2003, 126, 974–987. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Feng, Y.; Huang, Y.; Jiang, X.; Tang, C.; Tang, F.; Zeng, C.; Liu, L. A GM1 gangliosidosis mutant mouse model exhibits activated microglia and disturbed autophagy. Exp. Biol. Med. (Maywood NJ) 2021, 246, 1330–1341. [Google Scholar] [CrossRef]
- Tessitore, A.; del PMartin, M.; Sano, R.; Ma, Y.; Mann, L.; Ingrassia, A.; Laywell, E.D.; Steindler, D.A.; Hendershot, L.M.; d’Azzo, A. GM1-Ganglioside-Mediated Activation of the Unfolded Protein Response Causes Neuronal Death in a Neurodegenerative Gangliosidosis. Mol. Cell 2004, 15, 753–766. [Google Scholar] [CrossRef]
- van Doorn, P.A.; Ruts, L.; Jacobs, B.C. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008, 7, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.W.; Li, C.Y.; Ho, T.W.; Tian, M.; Gao, C.Y.; Xue, P.; Mishu, B.; Cornblath, D.R.; Macko, C.; McKhann, G.M.; et al. Pathology of the motor-sensory axonal Guillain-Barré syndrome. Ann. Neurol. 1996, 39, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Yuki, N.; Kuwabara, S.; Koga, M.; Hirata, K. Acute motor axonal neuropathy and acute motor-sensory axonal neuropathy share a common immunological profile. J. Neurol. Sci. 1999, 168, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.L.; Yuki, N.; Koga, M.; Chiang, M.C.; Hsieh, S.T. Acute sensory ataxic neuropathy associated with monospecific anti-GD1b IgG antibody. Neurology 2001, 57, 1316–1318. [Google Scholar] [CrossRef]
- Notturno, F.; Caporale, C.M.; Uncini, A. Acute sensory ataxic neuropathy with antibodies to GD1b and GQ1b gangliosides and prompt recovery. Muscle Nerve 2008, 37, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Matsuno, K.; Sakumoto, Y.; Hirata, K.; Yuki, N. Ataxic Guillain-Barré syndrome and acute sensory ataxic neuropathy form a continuous spectrum. J. Neurol. Neurosurg. Psychiatry 2011, 82, 294–299. [Google Scholar] [CrossRef]
- Susuki, K.; Yuki, N.; Schafer, D.P.; Hirata, K.; Zhang, G.; Funakoshi, K.; Rasband, M.N. Dysfunction of nodes of Ranvier: A mechanism for anti-ganglioside antibody-mediated neuropathies. Exp. Neurol. 2012, 233, 534–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanaka, S.; Yogo, R.; Watanabe, H.; Taniguchi, Y.; Satoh, T.; Komura, N.; Ando, H.; Yagi, H.; Yuki, N.; Uchihashi, T.; et al. On-Membrane Dynamic Interplay between Anti-GM1 IgG Antibodies and Complement Component C1q. Int. J. Mol. Sci. 2019, 21, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.K.; Beutler, E. Hexosaminidase-A and hexosaminidase-B: Studies in Tay-Sachs’ and Sandhoff’s disease. Nature 1973, 241, 463. [Google Scholar] [CrossRef] [PubMed]
- Toro, C.; Zainab, M.; Tifft, C.J. The GM2 gangliosidoses: Unlocking the mysteries of pathogenesis and treatment. Neurosci. Lett. 2021, 764, 136195. [Google Scholar] [CrossRef]
- Blaine, B.; Francesco, S.P.; Martino, C. GM1 and GM2 gangliosides: Recent developments. Biomol. Concepts 2014, 5, 87–93. (In English) [Google Scholar] [CrossRef]
- Novak, A.; Lowden, J.A.; Gravel, Y.L.; Wolfe, L.S. Preparation of radiolabeled GM2 and GA2 gangliosides. J. Lipid Res. 1979, 20, 678–681. Available online: https://www.ncbi.nlm.nih.gov/pubmed/490046 (accessed on 28 February 2023). [CrossRef] [PubMed]
- Leal, A.F.; Benincore-Flórez, E.; Solano-Galarza, D.; Garzón Jaramillo, R.G.; Echeverri-Peña, O.Y.; Suarez, D.A.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. Int. J. Mol. Sci. 2020, 21, 6213. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Katsuyama, K.; Nagahama, K.; Takai, T.; Aoki, I.; Yamanaka, S. Possible role of autoantibodies in the pathophysiology of GM2 gangliosidoses. J. Clin. Investig. 2004, 113, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Quinville, B.M.; Mitchell, M.; Chen, Z.; Walia, J.S. Gene Expression Profile in the Sandhoff Mouse Brain with Progression of Age. Genes 2022, 13, 2020. [Google Scholar] [CrossRef]
- Sargeant, T.J.; Wang, S.; Bradley, J.; Smith, N.J.; Raha, A.A.; McNair, R.; Ziegler, R.J.; Cheng, S.H.; Cox, T.M.; Cachon-Gonzalez, M.B. Adeno-associated virus-mediated expression of beta-hexosaminidase prevents neuronal loss in the Sandhoff mouse brain. Hum. Mol. Genet. 2011, 20, 4371–4380. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Furusawa, E.; Saitoh, T.; Sugimoto, H.; Omori, T.; Shimizu, S.; Kondo, H.; Yamazaki, M.; Sakuraba, H.; Oishi, K. Inhibition of astrocytic adenosine receptor A2A attenuates microglial activation in a mouse model of Sandhoff disease. Neurobiol. Dis. 2018, 118, 142–154. [Google Scholar] [CrossRef]
- Wheeler, S.; Sillence, D.J. Niemann–Pick type C disease: Cellular pathology and pharmacotherapy. J. Neurochem. 2020, 153, 674–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, S.J.; Ward, C.P.; Fensom, A.H. Complementation studies in Niemann-Pick disease type C indicate the existence of a second group. J. Med. Genet. 1994, 31, 317–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanier, M.T.; Duthel, S.; Rodriguez-Lafrasse, C.; Pentchev, P.; Carstea, E.D. Genetic heterogeneity in Niemann-Pick C disease: A study using somatic cell hybridization and linkage analysis. Am. J. Hum. Genet. 1996, 58, 118–125. Available online: http://www.ncbi.nlm.nih.gov/pubmed/8554047 (accessed on 28 February 2023). [PubMed]
- Liscum, L. Niemann-Pick type C mutations cause lipid traffic jam. Traffic 2000, 1, 218–225. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11208105 (accessed on 28 February 2023). [CrossRef] [PubMed]
- Davies, J.P.; Chen, F.W.; Ioannou, Y.A. Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science 2000, 290, 2295–2298. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.C.; Gordon, M.D.; Jin, J.Y.; Scott, M.P. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol. Biol. Cell 2001, 12, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef]
- Babalola, J.O.; Wendeler, M.; Breiden, B.; Arenz, C.; Schwarzmann, G.; Locatelli-Hoops, S.; Sandhoff, K. Development of an assay for the intermembrane transfer of cholesterol by Niemann-Pick C2 protein. Biol. Chem. 2007, 388, 617–626. [Google Scholar] [CrossRef]
- Ioannou, Y.A. Guilty until proven innocent: The case of NPC1 and cholesterol. Trends Biochem. Sci. 2005, 30, 498–505. [Google Scholar] [CrossRef]
- Higgins, M.E.; Davies, J.P.; Chen, F.W.; Ioannou, Y.A. Niemann-Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol. Genet. Metab. 1999, 68, 1–13. [Google Scholar] [CrossRef]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, F.M.; Wassif, C.; Colaco, A.; Dardis, A.; Lloyd-Evans, E.; Bembi, B.; Porter, F.D. Disorders of cholesterol metabolism and their unanticipated convergent mechanisms of disease. Annu. Rev. Genom. Hum. Genet. 2014, 15, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Pinto, C.; Sousa, D.; Ghilas, V.; Dardis, A.; Scarpa, M.; Macedo, M.F. Acid Sphingomyelinase Deficiency: A Clinical and Immunological Perspective. Int. J. Mol. Sci. 2021, 22, 12870. [Google Scholar] [CrossRef]
- Poczobutt, J.M.; Mikosz, A.M.; Poirier, C.; Beatman, E.L.; Serban, K.A.; Gally, F.; Cao, D.; McCubbrey, A.L.; Cornell, C.F.; Schweitzer, K.S.; et al. Altered Macrophage Function Associated with Crystalline Lung Inflammation in Acid Sphingomyelinase Deficiency. Am. J. Respir. Cell Mol. Biol. 2021, 64, 629–640. [Google Scholar] [CrossRef]
- Cluzeau, C.V.; Watkins-Chow, D.E.; Fu, R.; Borate, B.; Yanjanin, N.; Dail, M.K.; Davidson, C.D.; Walkley, S.U.; Ory, D.S.; Wassif, C.A.; et al. Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum. Mol. Genet. 2012, 21, 3632–3646. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.D.; González de la Vega, J.; Zanlungo, S. Complement Component C3 Participates in Early Stages of Niemann–Pick C Mouse Liver Damage. Int. J. Mol. Sci. 2020, 21, 2127. [Google Scholar] [CrossRef] [Green Version]
- Liao, G.; Wen, Z.; Irizarry, K.; Huang, Y.; Mitsouras, K.; Darmani, M.; Leon, T.; Shi, L.; Bi, X. Abnormal gene expression in cerebellum of Npc1-/- mice during postnatal development. Brain Res. 2010, 1325, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, M.E.; Klein, A.D.; Hong, J.; Dimbil, U.J.; Scott, M.P. Neuronal and epithelial cell rescue resolves chronic systemic inflammation in the lipid storage disorder Niemann-Pick C. Hum. Mol. Genet. 2012, 21, 2946–2960. [Google Scholar] [CrossRef]
- Shin, S.D.; Shin, A.; Mayagoitia, K.; Wilson, C.G.; Bellinger, D.L.; Soriano, S. Interferon downstream signaling is activated early in pre-symptomatic Niemann-Pick disease type C. Neurosci. Lett. 2019, 706, 43–50. [Google Scholar] [CrossRef]
- Wu, Y.P.; Mizukami, H.; Matsuda, J.; Saito, Y.; Proia, R.L.; Suzuki, K. Apoptosis accompanied by up-regulation of TNF-alpha death pathway genes in the brain of Niemann-Pick type C disease. Mol. Genet. Metab. 2005, 84, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Cologna, S.M.; Cluzeau, C.V.; Yanjanin, N.M.; Blank, P.S.; Dail, M.K.; Siebel, S.; Toth, C.L.; Wassif, C.A.; Lieberman, A.P.; Porter, F.D. Human and mouse neuroinflammation markers in Niemann-Pick disease, type C1. J. Inherit. Metab. Dis. 2014, 37, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, M.E.; Klein, A.D.; Scott, M.P. Complement is dispensable for neurodegeneration in Niemann-Pick disease type C. J. Neuroinflammation 2012, 9, 216. [Google Scholar] [CrossRef] [Green Version]
- Dimitriou, E.; Paschali, E.; Kanariou, M.; Michelakakis, H. Prevalence of antibodies to ganglioside and Hep 2 in Gaucher, Niemann—Pick type C and Sanfilippo diseases. Mol. Genet. Metab. Rep. 2019, 20, 100477. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Sugimoto, Y.; Ohsaki, Y.; Ueno, M.; Kato, S.; Kitamura, Y.; Hosokawa, H.; Davies, J.P.; Ioannou, Y.A.; Vanier, M.T.; et al. Endosomal Accumulation of Toll-Like Receptor 4 Causes Constitutive Secretion of Cytokines and Activation of Signal Transducers and Activators of Transcription in Niemann–Pick Disease Type C (NPC) Fibroblasts: A Potential Basis for Glial Cell Activation in the NPC Brain. J. Neurosci. 2007, 27, 1879. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.; Tunnage, B.; Yeo, S.T. Drugs for exceptionally rare diseases: Do they deserve special status for funding? QJM 2005, 98, 829–836. [Google Scholar] [CrossRef]
- Sugita, M.; Dulaney, J.T.; Moser, H.W. Ceramidase deficiency in Farber’s disease (lipogranulomatosis). Science 1972, 178, 1100–1102. [Google Scholar] [CrossRef]
- Gatt, S. Enzymic Hydrolysis and Synthesis of Ceramides. J. Biol. Chem. 1963, 238, PC3131–PC3133. [Google Scholar] [CrossRef]
- Coant, N.; Sakamoto, W.; Mao, C.; Hannun, Y.A. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv. Biol. Regul. 2017, 63, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levade, T.; Moser, H.W.; Fensom, A.H.; Harzer, K.; Moser, A.B.; Salvayre, R. Neurodegenerative course in ceramidase deficiency (Farber disease) correlates with the residual lysosomal ceramide turnover in cultured living patient cells. J. Neurol. Sci. 1995, 134, 108–114. [Google Scholar] [CrossRef]
- Meyer, R.C.; Giddens, M.M.; Coleman, B.M.; Hall, R.A. The protective role of prosaposin and its receptors in the nervous system. Brain Res. 2014, 1585, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, S.E.; Valle, D.; Moser, H.W.; Moser, A.; Qualman, S.J.; Zinkham, W.H. Phenotypic variability in siblings with Farber disease. J. Pediatr. 1984, 104, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Qualman, S.J.; Moser, H.W.; Valle, D.; Moser, A.E.; Antonarakis, S.E.; Boitnott, J.K.; Zinkham, W.H.; Opitz, J.M.; Bernstein, J. Farber Disease: Pathologic diagnosis in sibs with phenotypic variability. Am. J. Med. Genet. 1987, 28, 233–241. [Google Scholar] [CrossRef]
- Dworski, S.; Lu, P.; Khan, A.; Maranda, B.; Mitchell, J.J.; Parini, R.; Di Rocco, M.; Hugle, B.; Yoshimitsu, M.; Magnusson, B.; et al. Acid Ceramidase Deficiency is characterized by a unique plasma cytokine and ceramide profile that is altered by therapy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Abenoza, P.; Sibley, R.K. Farber’s disease: A fine structural study. Ultrastruct. Pathol. 1987, 11, 397–403. [Google Scholar] [CrossRef]
- Zappatini-Tommasi, L.; Dumontel, C.; Guibaud, P.; Girod, C. Farber disease: An ultrastructural study. Report of a case and review of the literature. Virchows Arch. A Pathol. Anat. Histopathol. 1992, 420, 281–290. [Google Scholar] [CrossRef]
- Burck, U.; Moser, H.W.; Goebel, H.H.; Grüttner, R.; Held, K.R. A case of lipogranulomatosis Farber: Some clinical and ultrastructural aspects. Eur. J. Pediatr. 1985, 143, 203–208. [Google Scholar] [CrossRef]
- Moser, H.W.; Prensky, A.L.; Wolfe, H.J.; Rosman, N.P. Farber’s lipogranulomatosis: Report of a case and demonstration of an excess of free ceramide and ganglioside. Am. J. Med. 1969, 47, 869–890. [Google Scholar] [CrossRef]
- Zarbin, M.A.; Green, W.R.; Moser, A.B.; Tiffany, C. Increased Levels of Ceramide in the Retina of a Patient with Farber’s Disease. Arch. Ophthalmol. 1988, 106, 1163. [Google Scholar] [CrossRef]
- Molz, G. Farber’s disease. Pathologic anatomical findings. Virchows Arch. A Pathol. Pathol. Anat. 1968, 344, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Barczykowski, A.L.; Foss, A.H.; Duffner, P.K.; Yan, L.; Carter, R.L. Death rates in the U.S. due to Krabbe disease and related leukodystrophy and lysosomal storage diseases. Am. J. Med. Genet. Part A 2012, 158A, 2835–2842. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Rafi, M.A.; de Gala, G.; Wenger, D.A. Cloning and expression cDNA encoding human galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum. Mol. Genet. 1993, 2, 1841–1846. [Google Scholar] [CrossRef]
- Suzuki, K. Twenty five years of the “psychosine hypothesis”: A personal perspective of its history and present status. Neurochem. Res. 1998, 23, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, T.; Suzuki, K. Globoid cell leukodystrophy: Additional deficiency of psychosine galactosidase. Biochem. Biophys. Res. Commun. 1972, 48, 538–543. [Google Scholar] [CrossRef]
- Duffner, P.K.; Jalal, K.; Carter, R.L. The Hunter’s Hope Krabbe family database. Pediatr. Neurol. 2009, 40, 13–18. [Google Scholar] [CrossRef]
- Zaka, M.; Wenger, D.A. Psychosine-induced apoptosis in a mouse oligodendrocyte progenitor cell line is mediated by caspase activation. Neurosci. Lett. 2004, 358, 205–209. [Google Scholar] [CrossRef]
- Haq, E.; Giri, S.; Singh, I.; Singh, A.K. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J. Neurochem. 2003, 86, 1428–1440. [Google Scholar] [CrossRef]
- Jatana, M.; Giri, S.; Singh, A.K. Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci. Lett. 2002, 330, 183–187. [Google Scholar] [CrossRef]
- Castelvetri, L.C.; Givogri, M.I.; Zhu, H.; Smith, B.; Lopez-Rosas, A.; Qiu, X.; van Breemen, R.; Bongarzone, E.R. Axonopathy is a compounding factor in the pathogenesis of Krabbe disease. Acta Neuropathol. 2011, 122, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Escolar, M.L.; Poe, M.D.; Provenzale, J.M.; Richards, K.C.; Allison, J.; Wood, S.; Wenger, D.A.; Pietryga, D.; Wall, D.; Champagne, M.; et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N. Engl. J. Med. 2005, 352, 2069–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snook, E.R.; Fisher-Perkins, J.M.; Sansing, H.A.; Lee, K.M.; Alvarez, X.; MacLean, A.G.; Peterson, K.E.; Lackner, A.A.; Bunnell, B.A. Innate immune activation in the pathogenesis of a murine model of globoid cell leukodystrophy. Am. J. Pathol. 2014, 184, 382–396. [Google Scholar] [CrossRef] [Green Version]
- Matthes, F.; Andersson, C.; Stein, A.; Eistrup, C.; Fogh, J.; Gieselmann, V.; Wenger, D.A.; Matzner, U. Enzyme replacement therapy of a novel humanized mouse model of globoid cell leukodystrophy. Exp. Neurol. 2015, 271, 36–45. [Google Scholar] [CrossRef]
- Valles-Ayoub, Y.; Esfandiarifard, S.; No, D.; Sinai, P.; Khokher, Z.; Kohan, M.; Kahen, T.; Darvish, D. Wolman Disease (LIPA p.G87V) Genotype Frequency in People of Iranian-Jewish Ancestry. Genet. Test. Mol. Biomark. 2011, 15, 395–398. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Dana, S.E.; Faust, J.R.; Beaudet, A.L.; Brown, M.S. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J. Biol. Chem. 1975, 250, 8487–8495. [Google Scholar] [CrossRef]
- Du, H.; Duanmu, M.; Witte, D.; Grabowski, G.A. Targeted Disruption of the Mouse Lysosomal Acid Lipase Gene: Long-Term Survival with Massive Cholesteryl Ester and Triglyceride Storage. Hum. Mol. Genet. 1998, 7, 1347–1354. [Google Scholar] [CrossRef] [Green Version]
- Castro, B.M.; Prieto, M.; Silva, L.C. Ceramide: A simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 2014, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Villamor, E.; Perez-Vizcaino, F.; Moreno, L. Ceramide and Regulation of Vascular Tone. Int. J. Mol. Sci. 2019, 20, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budoff, M. Triglycerides and Triglyceride-Rich Lipoproteins in the Causal Pathway of Cardiovascular Disease. Am. J. Cardiol. 2016, 118, 138–145. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, B.V.; Dodds, C.M.; Miller, S.R.; Gupta, A.; Lawrence, P.; Bullman, J.; Chen, C.; Dewit, O.; Kumar, S.; Dustagheer, M.; et al. The effects of GSK2981710, a medium-chain triglyceride, on cognitive function in healthy older participants: A randomised, placebo-controlled study. Hum. Psychopharmacol. 2019, 34, e2694. [Google Scholar] [CrossRef]
- Cohen, J.L.; Burfield, J.; Valdez-Gonzalez, K.; Samuels, A.; Stefanatos, A.K.; Yudkoff, M.; Pedro, H.; Ficicioglu, C. Early diagnosis of infantile-onset lysosomal acid lipase deficiency in the advent of available enzyme replacement therapy. Orphanet J. Rare Dis. 2019, 14, 198. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Zhao, T.; Ding, X.; Yan, C. Hepatocyte-Specific Expression of Human Lysosome Acid Lipase Corrects Liver Inflammation and Tumor Metastasis in lal(-/-) Mice. Am. J. Pathol. 2015, 185, 2379–2389. [Google Scholar] [CrossRef] [PubMed]
- Hůlková, H.; Elleder, M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology 2012, 60, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Mohamed, F.E.; Al-Gazali, L.; Al-Jasmi, F.; Ali, B.R. Pharmaceutical Chaperones and Proteostasis Regulators in the Therapy of Lysosomal Storage Disorders: Current Perspective and Future Promises. Front. Pharmacol. 2017, 8, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelmann, M.J.; Maegawa, G.H.B. CNS-Targeting Therapies for Lysosomal Storage Diseases: Current Advances and Challenges. Front. Mol. Biosci. 2020, 7, 559804. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pereira, C.; San Millán-Tejado, B.; Gallardo-Gómez, M.; Pérez-Márquez, T.; Alves-Villar, M.; Melcón-Crespo, C.; Fernández-Martín, J.; Ortolano, S. Therapeutic Approaches in Lysosomal Storage Diseases. Biomolecules 2021, 11, 1775. [Google Scholar] [CrossRef] [PubMed]
- Kido, J.; Sugawara, K.; Nakamura, K. Gene therapy for lysosomal storage diseases: Current clinical trial prospects. Front. Genet. 2023, 14, 1064924. [Google Scholar] [CrossRef]
- Sahasrabudhe, S.A.; Terluk, M.R.; Rudser, K.D.; Cloyd, J.C.; Kartha, R.V. Biological Variation in Peripheral Inflammation and Oxidative Stress Biomarkers in Individuals with Gaucher Disease. Int. J. Mol. Sci. 2022, 23, 9189. [Google Scholar] [CrossRef]
- Katsigianni, E.I.; Petrou, P. A systematic review of economic evaluations of enzyme replacement therapy in Lysosomal storage diseases. Cost Eff. Resour. Alloc. 2022, 20, 51. [Google Scholar] [CrossRef]
- Limgala, R.P.; Goker-Alpan, O. Effect of Substrate Reduction Therapy in Comparison to Enzyme Replacement Therapy on Immune Aspects and Bone Involvement in Gaucher Disease. Biomolecules 2020, 10, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wraith, J.E. Limitations of enzyme replacement therapy: Current and future. J. Inherit. Metab. Dis. 2006, 29, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme replacement therapy: Efficacy and limitations. Italy J. Pediatr. 2018, 44, 120. [Google Scholar] [CrossRef]
- Lowenstein, P.R.; Castro, M.G. Inflammation and adaptive immune responses to adenoviral vectors injected into the brain: Peculiarities, mechanisms, and consequences. Gene Ther. 2003, 10, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Cotter, M.J.; Muruve, D.A. The induction of inflammation by adenovirus vectors used for gene therapy. Front. Biosci.-Landmark 2005, 10, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Yang, K.; Foster, S.J.; Kondo, M.; Kelsoe, G. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J. Exp. Med. 2004, 199, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaoka, H.; Gonzalez-Aseguinolaza, G.; Tsuji, M.; Nussenzweig, M.C. Immunization and infection change the number of recombination activating gene (RAG)-expressing B cells in the periphery by altering immature lymphocyte production. J. Exp. Med. 2000, 191, 2113–2120. Available online: http://www.ncbi.nlm.nih.gov/pubmed/10859336 (accessed on 28 February 2023). [CrossRef] [Green Version]
- Nadazdin, O.; Abrahamian, G.; Boskovic, S.; Smith, R.N.; Schoenfeld, D.A.; Madsen, J.C.; Colvin, R.B.; Sachs, D.H.; Cosimi, A.B.; Kawai, T. Stem cell mobilization and collection for induction of mixed chimerism and renal allograft tolerance in cynomolgus monkeys. J. Surg. Res. 2011, 168, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Ripa, R.S. Granulocyte-colony stimulating factor therapy to induce neovascularization in ischemic heart disease. Dan. Med. J. 2012, 59, B4411. [Google Scholar]
- Zohlnhofer, D.; Ott, I.; Mehilli, J.; Schomig, K.; Michalk, F.; Ibrahim, T.; Meisetschlager, G.; von Wedel, J.; Bollwein, H.; Seyfarth, M.; et al. Stem cell mobilization by granulocyte colony-stimulating factor in patients with acute myocardial infarction: A randomized controlled trial. JAMA 2006, 295, 1003–1010. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bai, H.; Yi, Z.; He, X.; Mo, S. Effect of stem cell factor and granulocyte-macrophage colony-stimulating factor-induced bone marrow stem cell mobilization on recovery from acute tubular necrosis in rats. Ren. Fail. 2012, 34, 350–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, A.P.; Abboud, C.N.; DiPersio, J.F. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF): Receptor biology, signal transduction, and neutrophil activation. Blood Rev. 1992, 6, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, G.J.; Grail, D.; Hodgson, G.; Metcalf, D.; Stanley, E.; Cheers, C.; Fowler, K.J.; Basu, S.; Zhan, Y.F.; Dunn, A.R. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 1994, 84, 1737–1746. Available online: http://www.ncbi.nlm.nih.gov/pubmed/7521686 (accessed on 28 February 2023). [CrossRef]
- Avigan, D.; Wu, Z.; Gong, J.; Joyce, R.; Levine, J.; Elias, A.; Richardson, P.; Milano, J.; Kennedy, L.; Anderson, K.; et al. Selective in vivo mobilization with granulocyte macrophage colony-stimulating factor (GM-CSF)/granulocyte-CSF as compared to G-CSF alone of dendritic cell progenitors from peripheral blood progenitor cells in patients with advanced breast cancer undergoing autologous transplantation. Clin. Cancer Res. 1999, 5, 2735–2741. Available online: http://www.ncbi.nlm.nih.gov/pubmed/10537336 (accessed on 28 February 2023). [PubMed]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Mantovani, A.; Bonecchi, R.; Locati, M. Tuning inflammation and immunity by chemokine sequestration: Decoys and more. Nat. Rev. Immunol. 2006, 6, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, R.; Ho, A.D.; Bredthauer, U.; Cayeux, S.; Egerer, G.; Knauf, W.; Hunstein, W. Successful autologous transplantation of blood stem cells mobilized with recombinant human granulocyte-macrophage colony-stimulating factor. Exp. Hematol. 1990, 18, 94–98. Available online: http://www.ncbi.nlm.nih.gov/pubmed/1968009 (accessed on 28 February 2023).
- Socinski, M.A.; Cannistra, S.A.; Elias, A.; Antman, K.H.; Schnipper, L.; Griffin, J.D. Granulocyte-macrophage colony stimulating factor expands the circulating haemopoietic progenitor cell compartment in man. Lancet 1988, 1, 1194–1198. Available online: http://www.ncbi.nlm.nih.gov/pubmed/2897009 (accessed on 28 February 2023). [CrossRef]
- Brugger, W.; Bross, K.; Frisch, J.; Dern, P.; Weber, B.; Mertelsmann, R.; Kanz, L. Mobilization of peripheral blood progenitor cells by sequential administration of interleukin-3 and granulocyte-macrophage colony-stimulating factor following polychemotherapy with etoposide, ifosfamide, and cisplatin. Blood 1992, 79, 1193–1200. Available online: http://www.ncbi.nlm.nih.gov/pubmed/1371415 (accessed on 28 February 2023). [CrossRef] [Green Version]
- Henon, P.R.; Becker, M. Cytokine enhancement of peripheral blood stem cells. Stem Cells 1993, 11, 65–71. [Google Scholar] [CrossRef]
- Luyckx, A.; Schouppe, E.; Rutgeerts, O.; Lenaerts, C.; Fevery, S.; Devos, T.; Dierickx, D.; Waer, M.; Van Ginderachter, J.A.; Billiau, A.D. G-CSF stem cell mobilization in human donors induces polymorphonuclear and mononuclear myeloid-derived suppressor cells. Clin. Immunol. 2012, 143, 83–87. [Google Scholar] [CrossRef]
- Yamamoto, J.; Adachi, Y.; Onoue, Y.; Adachi, Y.S.; Okabe, Y.; Itazawa, T.; Toyoda, M.; Seki, T.; Morohashi, M.; Matsushima, K.; et al. Differential expression of the chemokine receptors by the Th1- and Th2-type effector populations within circulating CD4+ T cells. J. Leukoc. Biol. 2000, 68, 568–574. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11037980 (accessed on 28 February 2023). [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Montoya, M.C.; Mellado, M.; Frade, J.M.; del Pozo, M.A.; Nieto, M.; de Landazuri, M.O.; Martinez, A.C.; Sanchez-Madrid, F. The chemokine SDF-1alpha triggers a chemotactic response and induces cell polarization in human B lymphocytes. Eur. J. Immunol. 1998, 28, 2197–2207. [Google Scholar] [CrossRef]
- Rainey-Barger, E.K.; Rumble, J.M.; Lalor, S.J.; Esen, N.; Segal, B.M.; Irani, D.N. The lymphoid chemokine, CXCL13, is dispensable for the initial recruitment of B cells to the acutely inflamed central nervous system. Brain Behav. Immun. 2011, 25, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Pranzatelli, M.R.; Tate, E.D.; McGee, N.R.; Travelstead, A.L.; Ransohoff, R.M.; Ness, J.M.; Colliver, J.A. Key role of CXCL13/CXCR5 axis for cerebrospinal fluid B cell recruitment in pediatric OMS. J. Neuroimmunol. 2012, 243, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Schall, T.J.; Bacon, K.; Toy, K.J.; Goeddel, D.V. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 1990, 347, 669–671. [Google Scholar] [CrossRef]
- Neote, K.; DiGregorio, D.; Mak, J.Y.; Horuk, R.; Schall, T.J. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell 1993, 72, 415–425. [Google Scholar] [CrossRef]
- Murphy, W.J.; Tian, Z.G.; Asai, O.; Funakoshi, S.; Rotter, P.; Henry, M.; Strieter, R.M.; Kunkel, S.L.; Longo, D.L.; Taub, D.D. Chemokines and T lymphocyte activation: II. Facilitation of human T cell trafficking in severe combined immunodeficiency mice. J. Immunol. 1996, 156, 2104–2111. Available online: http://www.ncbi.nlm.nih.gov/pubmed/8690898 (accessed on 28 February 2023). [CrossRef]
- Sozzani, S.; Luini, W.; Borsatti, A.; Polentarutti, N.; Zhou, D.; Piemonti, L.; D’Amico, G.; Power, C.A.; Wells, T.N.; Gobbi, M. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J. Immunol. 1997, 159, 1993–2000. Available online: http://www.ncbi.nlm.nih.gov/pubmed/9257866 (accessed on 28 February 2023). [CrossRef]
- Heine, S.J.; Olive, D.; Gao, J.L.; Murphy, P.M.; Bukrinsky, M.I.; Constant, S.L. Cyclophilin A cooperates with MIP-2 to augment neutrophil migration. J. Inflamm. Res. 2011, 4, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarafi, M.N.; Garcia-Zepeda, E.A.; MacLean, J.A.; Charo, I.F.; Luster, A.D. Murine monocyte chemoattractant protein (MCP)-5: A novel CC chemokine that is a structural and functional homologue of human MCP-1. J. Exp. Med. 1997, 185, 99–109. Available online: http://www.ncbi.nlm.nih.gov/pubmed/8996246 (accessed on 28 February 2023). [CrossRef] [PubMed] [Green Version]
- Young, E.P.; Patrick, A.D. Deficiency of acid esterase activity in Wolman’s disease. Arch. Dis. Child. 1970, 45, 664–668. [Google Scholar] [CrossRef] [Green Version]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5–deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Wolburg-Buchholz, K.; Kraus, J.; Rascher-Eggstein, G.; Liebner, S.; Hamm, S.; Duffner, F.; Grote, E.-H.; Risau, W.; Engelhardt, B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003, 105, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Lal-Nag, M.; Morin, P.J. The claudins. Genome Biol. 2009, 10, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhold, A.; Rittner, H. Barrier function in the peripheral and central nervous system—A review. Pflügers Arch.-Eur. J. Physiol. 2017, 469, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Møllgård, K.; Saunders, N.R. The Development of the Human Blood-Brain and Blood-Csf Barriers. Neuropathol. Appl. Neurobiol. 1986, 12, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Runkle, E.A.; Mu, D. Tight junction proteins: From barrier to tumorigenesis. Cancer Lett. 2013, 337, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pummi, K.P.; Heape, A.M.; Grénman, R.A.; Peltonen, J.T.; Peltonen, S.A. Tight junction proteins ZO-1, occludin, and claudins in developing and adult human perineurium. J. Histochem. Cytochem. 2004, 52, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H.; Rosenberg, G.A. The Neurovascular Unit in Health and Disease. Stroke 2009, 40, S2–S3. [Google Scholar] [CrossRef] [Green Version]
- Segura, I.; De Smet, F.; Hohensinner, P.J.; Almodovar CRd Carmeliet, P. The neurovascular link in health and disease: An update. Trends Mol. Med. 2009, 15, 439–451. [Google Scholar] [CrossRef]
- Daneman, R. The blood–brain barrier in health and disease. Ann. Neurol. 2012, 72, 648–672. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Roe, K.; Kumar, M.; Lum, S.; Orillo, B.; Nerurkar, V.R.; Verma, S. West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J. Gen. Virol. 2012, 93, 1193–1203. [Google Scholar] [CrossRef]
- Chang, C.Y.; Li, J.R.; Chen, W.Y.; Ou, Y.C.; Lai, C.Y.; Hu, Y.H.; Wu, C.C.; Chang, C.J.; Chen, C.J. Disruption of in vitro endothelial barrier integrity by Japanese encephalitis virus-Infected astrocytes. Glia 2015, 63, 1915–1932. [Google Scholar] [CrossRef]
- Lischper, M.; Beuck, S.; Thanabalasundaram, G.; Pieper, C.; Galla, H.J. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res. 2010, 1326, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Nishikawa, T. Oxidative stress: A cause and therapeutic target of diabetic complications. J. Diabetes Investig. 2010, 1, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Q.; He, W.Q.; Zhou, M.; Lu, H.; Fu, Z.F. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J. Virol. 2014, 88, 4698–4710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowska, G.B.; Chen, X.; Zhang, J.; Lim, Y.P.; Cummings, E.E.; Makeyev, O.; Besio, W.G.; Gaitanis, J.; Padbury, J.F.; Banks, W.A.; et al. Interleukin-1β transfer across the blood-brain barrier in the ovine fetus. J. Cereb. Blood Flow Metab. 2015, 35, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Wang, L.; Zhang, L.; Qin, C.; Song, Y.; Ma, Y.; Chen, Y.; Chen, S.; Wang, Y.; Zhang, Z.; et al. Blood-Brain Barrier Disruption Induced Cognitive Impairment Is Associated with Increase of Inflammatory Cytokine. Front. Aging Neurosci. 2018, 10, 129. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Kastin, A.J. TNFalpha transport across the blood-brain barrier is abolished in receptor knockout mice. Exp. Neurol. 2002, 174, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tang, X.; Li, J.; Hu, B.; Yang, W.; Zhan, M.; Ma, T.; Xu, S. IL-17 crosses the blood–brain barrier to trigger neuroinflammation: A novel mechanism in nitroglycerin-induced chronic migraine. J. Headache Pain 2022, 23, 1. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.F.; Abbas, A.R.; Jeet, S.; Wong, K.; Bischof, A.; Peng, I.; Lee, J.; Bremer, M.; Eggers, E.L.; DeVoss, J.; et al. IL-17A is associated with the breakdown of the blood-brain barrier in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2019, 332, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.K.; Eugenin, E.A.; Lopez, L.; Romero, I.A.; Weksler, B.B.; Couraud, P.-O.; Berman, J.W. CCL2 disrupts the adherens junction: Implications for neuroinflammation. Lab. Investig. 2012, 92, 1213–1233. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Xu, D.; Lin, Y.; Wang, G.; Wang, F.; Gao, Q.; Wei, Q.; Lei, S. Chemokine CCL2 contributes to BBB disruption via the p38 MAPK signaling pathway following acute intracerebral hemorrhage. FASEB J. 2020, 34, 1872–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estevao, C.; Bowers, C.E.; Luo, D.; Sarker, M.; Hoeh, A.E.; Frudd, K.; Turowski, P.; Greenwood, J. CCL4 induces inflammatory signalling and barrier disruption in the neurovascular endothelium. Brain Behav. Immun. Health 2021, 18, 100370. [Google Scholar] [CrossRef] [PubMed]
- Ubogu, E.E.; Callahan, M.K.; Tucky, B.H.; Ransohoff, R.M. Determinants of CCL5-driven mononuclear cell migration across the blood-brain barrier. Implications for therapeutically modulating neuroinflammation. J. Neuroimmunol. 2006, 179, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, H.; Lou, W.; Ma, L.; Li, Y.; Zhang, N.; Wang, C.; Li, F.; Awais, M.; Cao, S. IP-10 Promotes Blood-Brain Barrier Damage by Inducing Tumor Necrosis Factor Alpha Production in Japanese Encephalitis. Front. Immunol. 2018, 9, 1148. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.D.; Parikh, N.U.; Woodruff, T.M.; Jarvis, J.N.; Lopez, M.; Hennon, T.; Cunningham, P.; Quigg, R.J.; Schwartz, S.A.; Alexander, J.J. C5a alters blood-brain barrier integrity in a human in vitro model of systemic lupus erythematosus. Immunology 2015, 146, 130–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, A.; Hack, B.; Chen, P.; Quigg, R.J.; Alexander, J.J. C5a/CD88 signaling alters blood-brain barrier integrity in lupus through nuclear factor-kappaB. J. Neurochem. 2011, 119, 1041–1051. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.; Hack, B.; Chiang, E.; Garcia, J.G.; Quigg, R.J.; Alexander, J.J. C5a alters blood-brain barrier integrity in experimental lupus. FASEB J. 2010, 24, 1682–1688. [Google Scholar] [CrossRef] [Green Version]
- Flierl, M.A.; Stahel, P.F.; Rittirsch, D.; Huber-Lang, M.; Niederbichler, A.D.; Hoesel, L.M.; Touban, B.M.; Morgan, S.J.; Smith, W.R.; Ward, P.A.; et al. Inhibition of complement C5a prevents breakdown of the blood-brain barrier and pituitary dysfunction in experimental sepsis. Crit. Care 2009, 13, R12. [Google Scholar] [CrossRef] [Green Version]
- Landlinger, C.; Oberleitner, L.; Gruber, P.; Noiges, B.; Yatsyk, K.; Santic, R.; Mandler, M.; Staffler, G. Active immunization against complement factor C5a: A new therapeutic approach for Alzheimer’s disease. J. Neuroinflammation 2015, 12, 150. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, T.M.; Crane, J.W.; Proctor, L.M.; Buller, K.M.; Shek, A.B.; de Vos, K.; Pollitt, S.; Williams, H.M.; Shiels, I.A.; Monk, P.N. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006, 20, 1407–1417. [Google Scholar] [CrossRef] [Green Version]
- Farkas, I.; Takahashi, M.; Fukuda, A.; Yamamoto, N.; Akatsu, H.; Baranyi, L.; Tateyama, H.; Yamamoto, T.; Okada, N.; Okada, H. Complement C5a receptor-mediated signaling may be involved in neurodegeneration in Alzheimer’s disease. J. Immunol. 2003, 170, 5764–5771. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Fu, F.; Huang, S.; Lin, C.; Yang, G.; Ma, K.; Shi, H.; Yang, Z. C5a/C5aR Pathway Plays a Vital Role in Brain Inflammatory Injury via Initiating Fgl-2 in Intracerebral Hemorrhage. Mol. Neurobiol. 2017, 54, 6187–6197. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Mocco, J.; Hahn, D.K.; Kellner, C.P.; Komotar, R.J.; Ducruet, A.F.; Mack, W.J.; Connolly, E.S., Jr. Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery 2008, 63, 122–125; discussion 125–126. [Google Scholar] [CrossRef]
- Lee, J.D.; Kumar, V.; Fung, J.N.; Ruitenberg, M.J.; Noakes, P.G.; Woodruff, T.M. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2017, 174, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piatek, P.; Domowicz, M.; Lewkowicz, N.; Przygodzka, P.; Matysiak, M.; Dzitko, K.; Lewkowicz, P. C5a-Preactivated Neutrophils Are Critical for Autoimmune-Induced Astrocyte Dysregulation in Neuromyelitis Optica Spectrum Disorder. Front. Immunol. 2018, 9, 1694. [Google Scholar] [CrossRef]
- Dhillon, S. Eculizumab: A Review in Generalized Myasthenia Gravis. Drugs 2018, 78, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Leal, E.C.; Martins, J.; Voabil, P.; Liberal, J.; Chiavaroli, C.; Bauer, J.; Cunha-Vaz, J.; Ambrósio, A.F. Calcium Dobesilate Inhibits the Alterations in Tight Junction Proteins and Leukocyte Adhesion to Retinal Endothelial Cells Induced by Diabetes. Diabetes 2010, 59, 2637–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, G.; Wang, Z.; Zhang, X.; Yao, L.; Wang, F.; Liu, S.; Yin, J.; Ling, E.A.; Wang, L.; et al. High glucose-induced expression of inflammatory cytokines and reactive oxygen species in cultured astrocytes. Neuroscience 2012, 202, 58–68. [Google Scholar] [CrossRef]
- van Vliet, E.A.; da Costa Araújo, S.; Redeker, S.; van Schaik, R.; Aronica, E.; Gorter, J.A. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain A J. Neurol. 2007, 130, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Raabe, A.; Schmitz, A.K.; Pernhorst, K.; Grote, A.; von der Brelie, C.; Urbach, H.; Friedman, A.; Becker, A.J.; Elger, C.E.; Niehusmann, P. Cliniconeuropathologic correlations show astroglial albumin storage as a common factor in epileptogenic vascular lesions. Epilepsia 2012, 53, 539–548. [Google Scholar] [CrossRef]
- Baeten, K.M.; Akassoglou, K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev. Neurobiol. 2011, 71, 1018–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Buckwalter, M.; Soreq, H.; Vezzani, A.; Kaufer, D. Blood–brain barrier dysfunction–induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia 2012, 53, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Réus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Allan, S.M.; Rothwell, N.J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001, 2, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Hassan, W.; Noreen, H.; Castro-Gomes, V.; Mohammadzai, I.; da Rocha, J.B.; Landeira-Fernandez, J. Association of Oxidative Stress with Psychiatric Disorders. Curr. Pharm. Des. 2016, 22, 2960–2974. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Duman, R.; Sanacora, G. Serum Brain-Derived Neurotrophic Factor, Depression, and Antidepressant Medications: Meta-Analyses and Implications. Biol. Psychiatry 2008, 64, 527–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronfol, Z.; Remick, D.G. Cytokines and the brain: Implications for clinical psychiatry. Am. J. Psychiatry 2000, 157, 683–694. [Google Scholar] [CrossRef]
- Pollak, T.A.; Drndarski, S.; Stone, J.M.; David, A.S.; McGuire, P.; Abbott, N.J. The blood–brain barrier in psychosis. Lancet Psychiatry 2018, 5, 79–92. [Google Scholar] [CrossRef]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, B.; Adams, R.J. Induction of refractoriness to thyrotropin stimulation in cultured thyroid cells. Dependence on new protein synthesis. J. Biol. Chem. 1976, 251, 6653–6661. [Google Scholar] [CrossRef]
- Grimm, A.; Friedland, K.; Eckert, A. Mitochondrial dysfunction: The missing link between aging and sporadic Alzheimer’s disease. Biogerontology 2016, 17, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Kruszon-Moran, D.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA 2016, 315, 2284–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantzer, R.; Kelley, K.W. Twenty years of research on cytokine-induced sickness behavior. Brain Behav. Immun. 2007, 21, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, E.G.; Banks, W.A.; Kastin, A.J. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J. Neuroimmunol. 1993, 47, 169–176. [Google Scholar] [CrossRef]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef]
- Devanney, N.A.; Stewart, A.N.; Gensel, J.C. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp. Neurol. 2020, 329, 113310. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Ruckh, J.M.; Zhao, J.W.; Shadrach, J.L.; van Wijngaarden, P.; Rao, T.N.; Wagers, A.J.; Franklin, R.J. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 2012, 10, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Kotter, M.R.; Li, W.W.; Zhao, C.; Franklin, R.J. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. 2006, 26, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Kotter, M.R.; Zhao, C.; van Rooijen, N.; Franklin, R.J. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol. Dis. 2005, 18, 166–175. [Google Scholar] [CrossRef]
- Persidsky, Y.; Ghorpade, A.; Rasmussen, J.; Limoges, J.; Liu, X.J.; Stins, M.; Fiala, M.; Way, D.; Kim, K.S.; Witte, M.H. Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 1999, 155, 1599–1611. [Google Scholar] [CrossRef] [Green Version]
- De Bock, M.; Wang, N.; Decrock, E.; Bol, M.; Gadicherla, A.K.; Culot, M.; Cecchelli, R.; Bultynck, G.; Leybaert, L. Endothelial calcium dynamics, connexin channels and blood-brain barrier function. Prog. Neurobiol. 2013, 108, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, S.; Bennett, D.L. Pathogenic mechanisms in inflammatory and paraproteinaemic peripheral neuropathies. Curr. Opin. Neurol. 2014, 27, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Anghelescu, D.L.; Goldberg, J.L.; Faughnan, L.G.; Wu, J.; Mao, S.; Furman, W.L.; Santana, V.M.; Navid, F. Comparison of pain outcomes between two anti-GD2 antibodies in patients with neuroblastoma. Pediatr. Blood Cancer 2015, 62, 224–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, P.C.; Graham, M.A.; Triano, J.J.; Hondras, M.A.; Anderson, R.J. Lymphocyte profiles in patients with chronic low back pain enrolled in a clinical trial. J. Manip. Physiol. Ther. 1994, 17, 219–227. [Google Scholar]
- Brück, W.; Friede, R. The role of complement in myelin phagocytosis during PNS wallerian degeneration. J. Neurol. Sci. 1991, 103, 182–187. [Google Scholar] [CrossRef]
- Ren, K.; Dubner, R. Interactions between the immune and nervous systems in pain. Nat. Med. 2010, 16, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Calvo, M.; Dawes, J.M.; Bennett, D.L. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 2012, 11, 629–642. [Google Scholar] [CrossRef]
- Fregnan, F.; Muratori, L.; Simões, A.R.; Giacobini-Robecchi, M.G.; Raimondo, S. Role of inflammatory cytokines in peripheral nerve injury. Neural. Regen. Res. 2012, 7, 2259–2266. [Google Scholar] [CrossRef]
- Hung, A.L.; Lim, M.; Doshi, T.L. Targeting cytokines for treatment of neuropathic pain. Scand J. Pain 2017, 17, 287–293. [Google Scholar] [CrossRef]
- Magrinelli, F.; Briani, C.; Romano, M.; Ruggero, S.; Toffanin, E.; Triolo, G.; Peter, G.C.; Praitano, M.; Lauriola, M.F.; Zanette, G.; et al. The Association between Serum Cytokines and Damage to Large and Small Nerve Fibers in Diabetic Peripheral Neuropathy. J. Diabetes Res. 2015, 2015, 547834. [Google Scholar] [CrossRef] [Green Version]
- Palladino, S.P.; Helton, E.S.; Jain, P.; Dong, C.; Crowley, M.R.; Crossman, D.K.; Ubogu, E.E. The human blood-nerve barrier transcriptome. Sci. Rep. 2017, 7, 17477. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Sarkar, C.; Singh, S.P.; Zhang, Z.; Munasinghe, J.; Peng, S.; Chandra, G.; Kong, E.; Mukherjee, A.B. The blood-brain barrier is disrupted in a mouse model of infantile neuronal ceroid lipofuscinosis: Amelioration by resveratrol. Hum. Mol. Genet. 2012, 21, 2233–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.C.; Lee, I.S.; Seo, H.; Ju, A.; Youn, D.; Kim, Y.; Choun, J.; Cho, S. Methanol extracts of Euphorbia cooperi inhibit the production of inflammatory mediators by inhibiting the activation of c-Jun N-terminal kinase and p38 in murine macrophages. Mol. Med. Rep. 2014, 10, 2663–2668. [Google Scholar] [CrossRef] [PubMed]
- Gajanayaka, N.; Dong, S.X.M.; Ali, H.; Iqbal, S.; Mookerjee, A.; Lawton, D.A.; Caballero, R.E.; Cassol, E.; Cameron, D.W.; Angel, J.B.; et al. TLR-4 Agonist Induces IFN-γ Production Selectively in Proinflammatory Human M1 Macrophages through the PI3K-mTOR- and JNK-MAPK-Activated p70S6K Pathway. J. Immunol. 2021, 207, 2310–2324. [Google Scholar] [CrossRef]
- Wang, X.; Li, G.-J.; Hu, H.-X.; Ma, C.; Ma, D.-H.; Liu, X.-L.; Jiang, X.-M. Cerebral mTOR signal and pro-inflammatory cytokines in Alzheimer’s disease rats. Transl. Neurosci. 2016, 7, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fan, H.; Han, X.; Sun, J.; Ni, M.; Zhang, L.; Fang, F.; Zhang, W.; Ma, P. PR-957 Suppresses Th1 and Th17 Cell Differentiation via Inactivating PI3K/AKT Pathway in Alzheimer’s Disease. Neuroscience 2023, 510, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Li, F.H.; Zhang, Y.; Zhang, X.F.; Yang, J. Tangeretin has anti-asthmatic effects via regulating PI3K and Notch signaling and modulating Th1/Th2/Th17 cytokine balance in neonatal asthmatic mice. Braz. J. Med. Biol. Res. 2017, 50, e5991. [Google Scholar] [CrossRef] [Green Version]
- Nasso, M.; Fedele, G.; Spensieri, F.; Palazzo, R.; Costantino, P.; Rappuoli, R.; Ausiello, C.M. Genetically detoxified pertussis toxin induces Th1/Th17 immune response through MAPKs and IL-10-dependent mechanisms. J. Immunol. 2009, 183, 1892–1899. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Guo, Y.; Han, R.; Liu, H.; Ding, Y.; Shi, Y.; Kong, D.; Ma, X. Class I PI3K inhibitor ZSTK474 attenuates experimental autoimmune neuritis by decreasing the frequency of Th1/Th17 cells and reducing the production of proinflammatory cytokines. Cell Immunol. 2018, 329, 41–49. [Google Scholar] [CrossRef]
- Koga, T.; Hedrich, C.M.; Mizui, M.; Yoshida, N.; Otomo, K.; Lieberman, L.A.; Rauen, T.; Crispín, J.C.; Tsokos, G.C. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J. Clin. Investig. 2014, 124, 2234–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orozco, S.L.; Daniels, B.P.; Yatim, N.; Messmer, M.N.; Quarato, G.; Chen-Harris, H.; Cullen, S.P.; Snyder, A.G.; Ralli-Jain, P.; Frase, S.; et al. RIPK3 Activation Leads to Cytokine Synthesis that Continues after Loss of Cell Membrane Integrity. Cell Rep. 2019, 28, 2275–2287.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winzen, R.; Kracht, M.; Ritter, B.; Wilhelm, A.; Chen, C.-Y.A.; Shyu, A.-B.; Müller, M.; Gaestel, M.; Resch, K.; Holtmann, H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999, 18, 4969–4980. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.N.; Konrad, S.; Wiege, K.; Nieswandt, B.; Nimmerjahn, F.; Schmidt, R.E.; Gessner, J.E. Both FcgammaRIV and FcgammaRIII are essential receptors mediating type II and type III autoimmune responses via FcRgamma-LAT-dependent generation of C5a. Eur. J. Immunol. 2009, 39, 3343–3356. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Meng, L.-Q.; Xu, P.-C.; Chen, M.; Zhao, M.-H. p38MAPK, ERK and PI3K signaling pathways are involved in C5a-primed neutrophils for ANCA-mediated activation. PLoS ONE 2012, 7, e38317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, S.; Ali, S.; Wiege, K.; Syed, S.N.; Mueller-Engling, L.; Piekorz, R.; Hirsch, E.; Nürnberg, B.; Schmidt, R.; Gessner, J. Phosphoinositide 3-Kinases γ and δ, Linkers of Coordinate C5a Receptor-Fcγ Receptor Activation and Immune Complex-induced Inflammation. J. Biol. Chem. 2008, 283, 33296–33303. [Google Scholar] [CrossRef] [Green Version]
- Davis, O.B.; Shin, H.R.; Lim, C.-Y.; Wu, E.Y.; Kukurugya, M.; Maher, C.F.; Perera, R.M.; Ordonez, M.P.; Zoncu, R. NPC1-mTORC1 Signaling Couples Cholesterol Sensing to Organelle Homeostasis and Is a Targetable Pathway in Niemann-Pick Type C. Dev. Cell 2021, 56, 260–276.e7. [Google Scholar] [CrossRef]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9–Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.A.; Voit, A.; Srikanth, M.P.; Thayer, J.A.; Kingsbury, T.J.; Jacobson, M.A.; Lipinski, M.M.; Feldman, R.A.; Awad, O. mTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. Dis. Model Mech. 2019, 12, dmm038596. [Google Scholar] [CrossRef] [Green Version]
- Bartolomeo, R.; Cinque, L.; De Leonibus, C.; Forrester, A.; Salzano, A.C.; Monfregola, J.; De Gennaro, E.; Nusco, E.; Azario, I.; Lanzara, C. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J. Clin. Investig. 2017, 127, 3717–3729. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, X.; Lin, Y.; Pan, D. A multifaceted evaluation of microgliosis and differential cellular dysregulation of mammalian target of rapamycin signaling in neuronopathic Gaucher disease. Front. Mol. Neurosci. 2022, 15, 944883. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Vardi, A.; Cox, T.M.; Futerman, A.H. Emerging therapeutic targets for Gaucher disease. Expert Opin. Ther. Targets 2015, 19, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Sural-Fehr, T.; Singh, H.; Cantuti-Catelvetri, L.; Zhu, H.; Marshall, M.S.; Rebiai, R.; Jastrzebski, M.J.; Givogri, M.I.; Rasenick, M.M.; Bongarzone, E.R. Inhibition of the IGF-1-PI3K-Akt-mTORC2 pathway in lipid rafts increases neuronal vulnerability in a genetic lysosomal glycosphingolipidosis. Dis. Model Mech. 2019, 12, dmm036590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitatani, K.; Wada, M.; Perry, D.; Usui, T.; Sun, Y.; Obeid, L.M.; Yaegashi, N.; Grabowski, G.A.; Hannun, Y.A. Activation of p38 Mitogen-Activated Protein Kinase in Gaucher’s Disease. PLoS ONE 2015, 10, e0136633. [Google Scholar] [CrossRef]
- Ługowska, A.; Hetmańczyk-Sawicka, K.; Iwanicka-Nowicka, R.; Fogtman, A.; Cieśla, J.; Purzycka-Olewiecka, J.K.; Sitarska, D.; Płoski, R.; Filocamo, M.; Lualdi, S.; et al. Gene expression profile in patients with Gaucher disease indicates activation of inflammatory processes. Sci. Rep. 2019, 9, 6060. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, M.P.; Jones, J.W.; Kane, M.; Awad, O.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Elevated glucosylsphingosine in Gaucher disease induced pluripotent stem cell neurons deregulates lysosomal compartment through mammalian target of rapamycin complex 1. STEM CELLS Transl. Med. 2021, 10, 1081–1094. [Google Scholar] [CrossRef]
- Kang, J.J.; Treadwell, T.A.; Bodary, P.F.; Shayman, J.A. Voluntary wheel running activates Akt/AMPK/eNOS signaling cascades without improving profound endothelial dysfunction in mice deficient in α-galactosidase A. PLoS ONE 2019, 14, e0217214. [Google Scholar] [CrossRef]
- Kawashima, N.; Tsuji, D.; Okuda, T.; Itoh, K.; Nakayama, K. Mechanism of abnormal growth in astrocytes derived from a mouse model of GM2 gangliosidosis. J. Neurochem. 2009, 111, 1031–1041. [Google Scholar] [CrossRef]


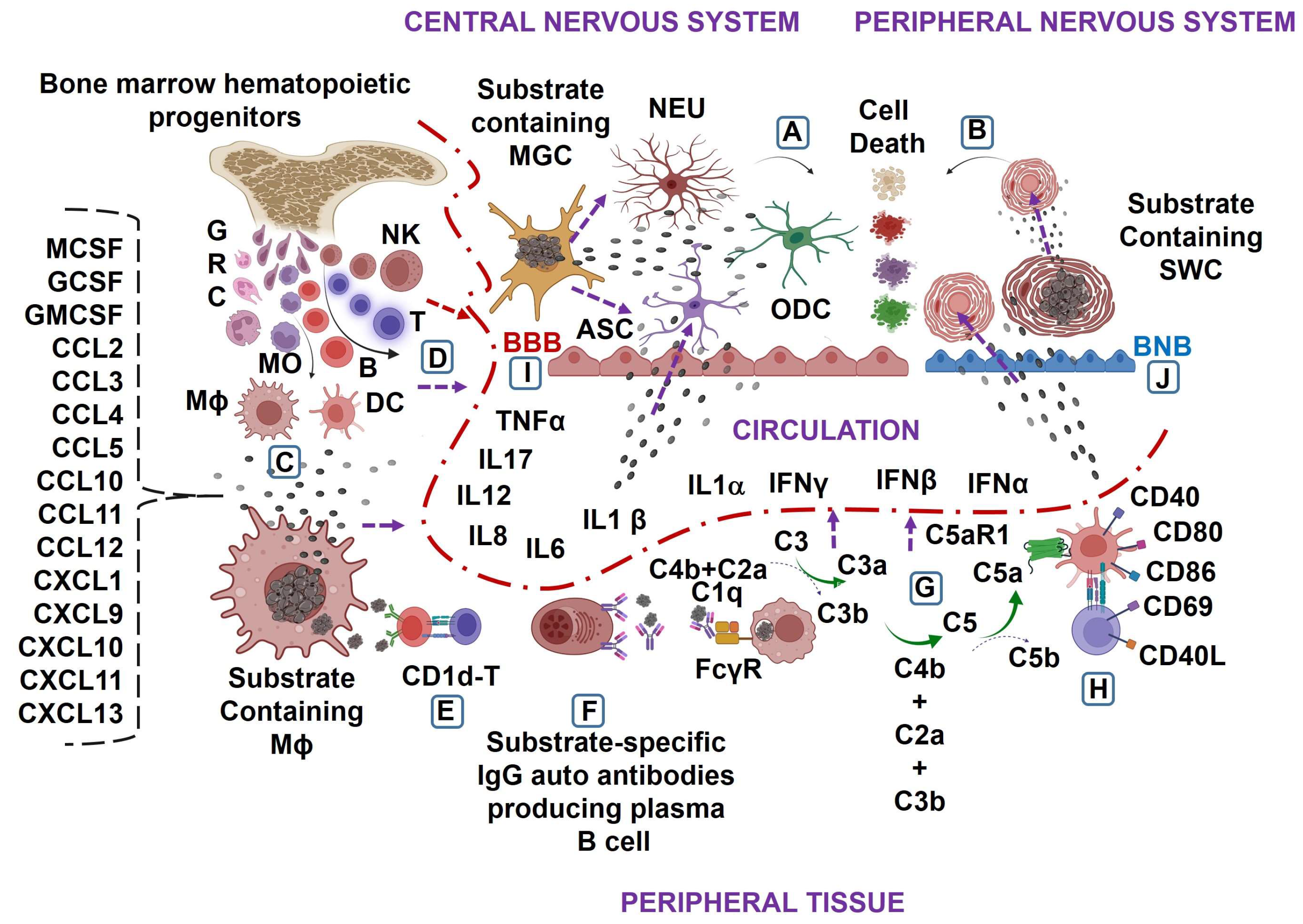{kind=link}
| Lysosomal Storage Disease | Gene Defects | Enzyme or Protein Deficiency | Substrate Accumulation | Affected Organs and Cells | Clinical Symptoms | Reference |
|---|---|---|---|---|---|---|
| Gaucher disease | GBA1(in human)/ Gba1(in mouse) | GCase | GC, GS | Central nervous system tissues, microglial cells, neurons, liver, spleen, lung, kidney MO, Mϕ, DC | Seizure, abnormal eye movements, developmental delay, hepatosplenomegaly, pulmonary inflammation, skeletal weakness, hypergamma globulinemia, B-cell malignancies, anemia, and thrombocytopenia | [5,6,7] |
| Fabry disease | GLA (in human)/Gla (in mouse) | α Gal A | Gb3 and lyso-Gb3 | Central nervous system tissue, and their cells, liver, spleen, kidney, blood vessel walls, renal epithelial cell, endothelial cell, pericyte podocytes, tubular cell of the loops of Henle and the thick ascending limb of the distal tubule, vascular smooth muscle cell, and cardiomyocyte | Stroke, burning pain in the arms and legs, cardiomegaly, renal failure, gastrointestinal difficulties, decreased sweating, fever, and angiokeratomas | [8,9,10,11,12,13,14,15,16] |
| GM1 gangliosidosis | GLB1 (in Human)/Glb1(in mouse) | β gal | GM1 | Central and peripheral nervous system tissue, spleen, bone, and muscle | Dementia, seizures, hyperekplexia, abnormal gait, ataxia, stuttering, apraxia, dysarthria, coarse facies, muscle atrophy, dystonia, angiokeratoma, skeletal irregularities, joint stiffness, abdominal distension, muscle weakness, and hepatosplenomegaly | [17,18,19] |
| GM2 gangliosidosis: Tay–Sachs disease | HEXA (in human)/Hexa (in mouse) | α subunit of the β hex | GM2 | Central nervous system tissue | Dementia, paralysis, seizures decreased eye contact, increased startle response to noise, deafness, dysphagia, blindness, cherry-red spots in the retina | [20,21,22,23] |
| GM2 gangliosidosis: Sandhoff disease | HEXB (in human)/Hexb (in mouse) | β subunit of the β hex | GM2 | Central nervous system tissues, liver, spleen, lung, and heart | Motor weakness, early blindness, startle response to sound, spasticity, myoclonus, seizures, macrocephaly, cherry red spots in the eye, doll-like facies, frequent respiratory infections, heart murmurs, and hepatosplenomegaly | [20,21,22,23,24,25] |
| Niemann–Pick type C disease | NPC1/NPC2(in human)/Npc1/Npc2(in mouse) | NPC1 and NPC2 | Sph, GlycSph, Sm, and Ch | Peripheral nervous system tissue, nerve cell, liver, spleen, lymph node, hard lump under the skin | Progressive dementia, difficulty in walking, dysphagia, progressive loss of hearing, hepatosplenomegaly, anemia, and susceptible to recurring infection | [26] |
| Farber disease | ASAH1 (in human/Asah1(in mouse) | ACDase | Cer | Central nervous system tissue, liver, heart, kidney, lymph node, and joints | Increased sleep and tiredness, dysphagia, dysphonia, arthrogryposis, vomiting, and arthritis | [27,28] |
| Krabbe disease | GALC (in human/Galc (in mouse) | GALCERase | GalCer and GalSph or psychosine | Myelin sheath | Dyspraxia, myoclonic seizures, deafness, blindness, paralysis, dysphagia, muscle weakness, hypertonia, spasticity, and weight loss | [29,30] |
| Wolman disease | LIPA (in human)/Lipa (in mouse) | LAL | CEs and TGs | Liver, spleen, blood, lymph, adrenal gland, MO, and Mϕ | Mental deterioration, Hepatomegaly, abdominal distension, gastrointestinal problems, jaundice, anemia, vomiting, and calcium deposits in the adrenal glands | [31,32] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, M.K. Exploring Pro-Inflammatory Immunological Mediators: Unraveling the Mechanisms of Neuroinflammation in Lysosomal Storage Diseases. Biomedicines 2023, 11, 1067. https://doi.org/10.3390/biomedicines11041067
Pandey MK. Exploring Pro-Inflammatory Immunological Mediators: Unraveling the Mechanisms of Neuroinflammation in Lysosomal Storage Diseases. Biomedicines. 2023; 11(4):1067. https://doi.org/10.3390/biomedicines11041067
Chicago/Turabian StylePandey, Manoj Kumar. 2023. "Exploring Pro-Inflammatory Immunological Mediators: Unraveling the Mechanisms of Neuroinflammation in Lysosomal Storage Diseases" Biomedicines 11, no. 4: 1067. https://doi.org/10.3390/biomedicines11041067