
The NF-κB Transcriptional Network Is a High-Dose Vitamin C-Targetable Vulnerability in Breast Cancer

 , , ,
, , ,  , , , and
, , , and 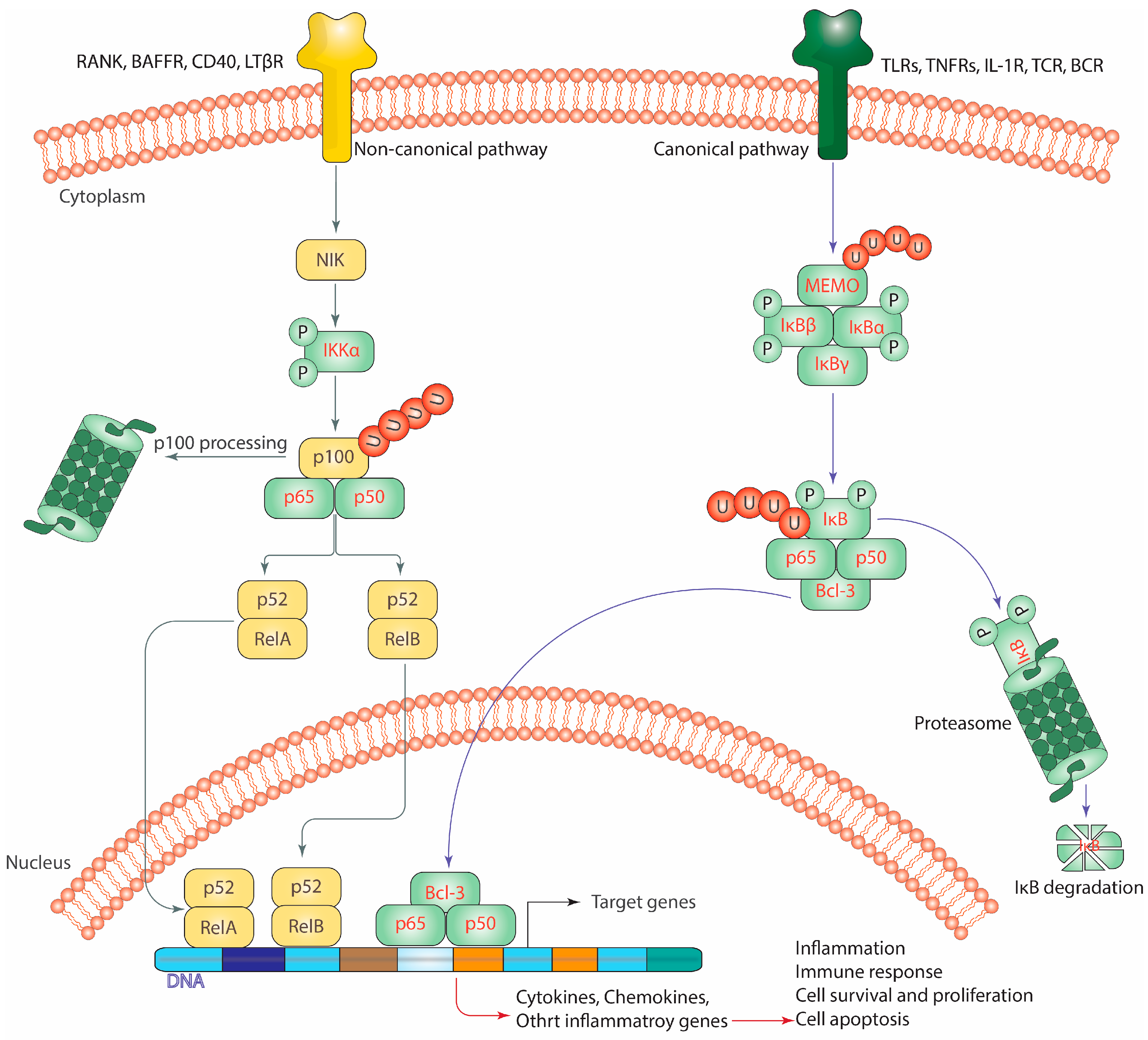{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-κB)
3. The Signaling Pathways of the NF-κB
3.1. The Triggering of the Canonical Pathway of NF-κB
3.2. The Triggering of the Non-Canonical Pathway of NF-κB
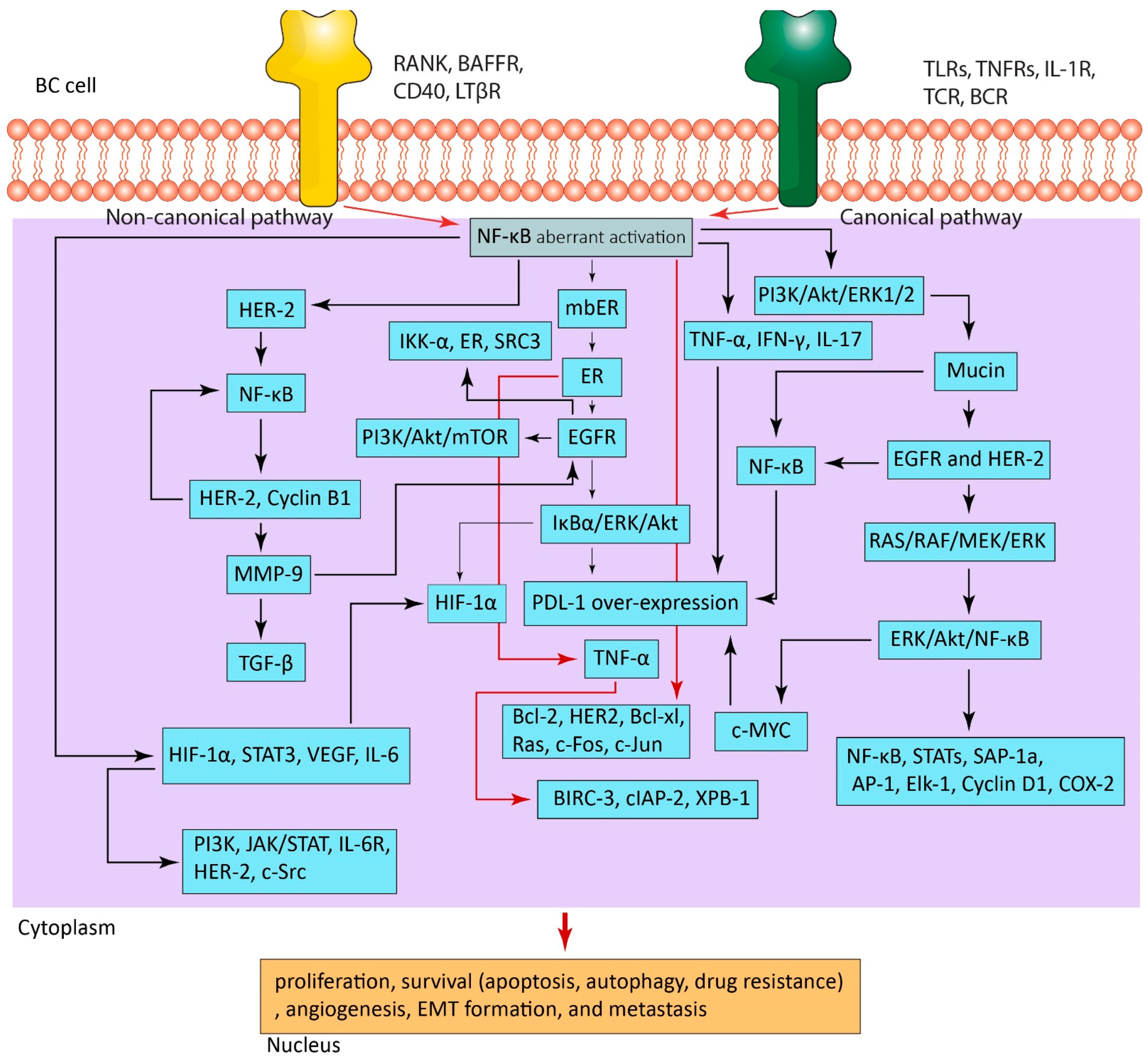
4. NF-κB in BC
4.1. NF-κB Activation in BC
4.2. NF-κB Activates HIF-1α in BC
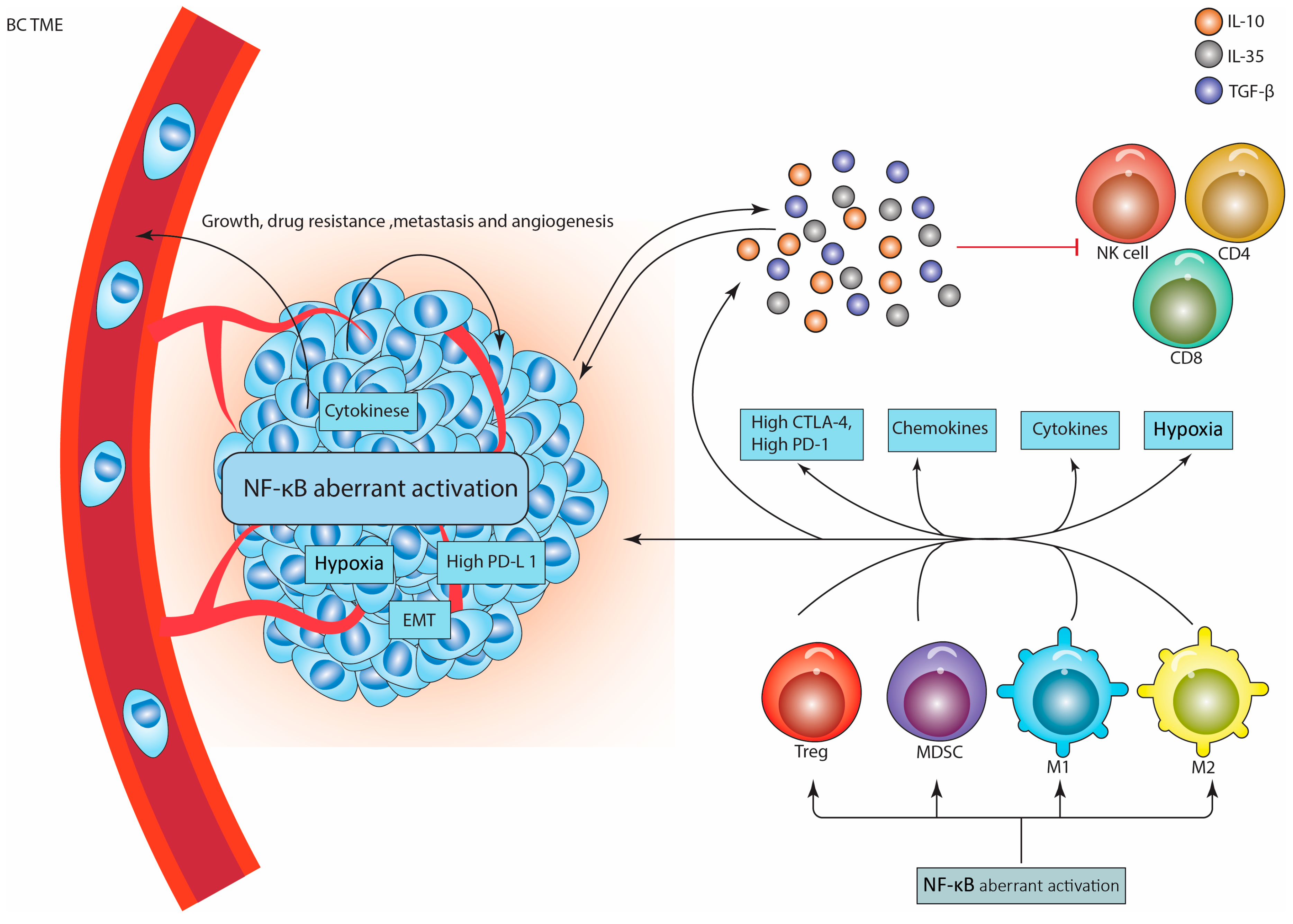
5. NF-κB Recruits and Activates Immunosuppressive Cells in BC Tumor Microenvironment
5.1. Tregs
5.2. MDSCs
5.3. TAMs
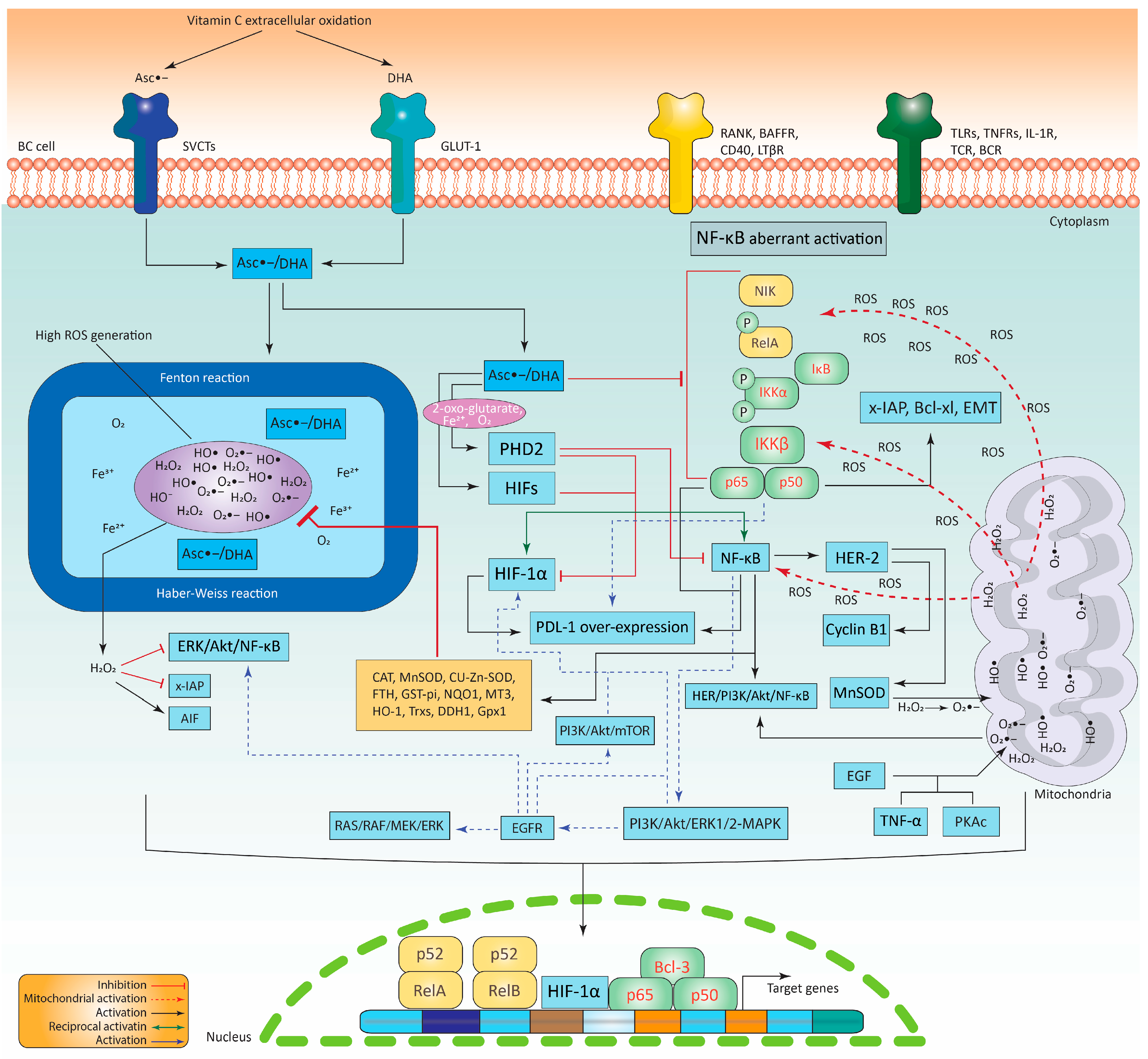
6. High-Dose Vitamin C Targeting the NF-κB Transcriptional Network
The Pro-Oxidant Activity of Vitamin C
7. Conclusions and Feature Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, D.; Sendi, M.A.; Kelly, C.M. Overview of recent advances in metastatic triple negative breast cancer. World J. Clin. Oncol. 2021, 12, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.M.; Hsu, N.C.; Chen, S.C.; Lu, Y.S.; Lin, C.H.; Chang, D.Y.; Li, H.; Lin, Y.C.; Chang, H.K.; Chao, T.C.; et al. Distant metastasis in triple-negative breast cancer. Neoplasma 2013, 60, 290–294. [Google Scholar] [CrossRef]
- Guo, M.; Wang, S.M. Genome Instability-Derived Genes Are Novel Prognostic Biomarkers for Triple-Negative Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 701073. [Google Scholar] [CrossRef]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Wang, Y.; Li, Q.; Cao, L.; Sun, Z.; Jin, J.; Fang, H.; Zhu, A.; Li, Y.; Zhang, W.; et al. Intratumor heterogeneity predicts metastasis of triple-negative breast cancer. Carcinogenesis 2017, 38, 900–909. [Google Scholar] [CrossRef]
- Asleh, K.; Riaz, N.; Nielsen, T.O. Heterogeneity of triple negative breast cancer: Current advances in subtyping and treatment implications. J. Exp. Clin. Cancer Res. 2022, 41, 265. [Google Scholar] [CrossRef]
- Zimmerli, D.; Brambillasca, C.S.; Talens, F.; Bhin, J.; Linstra, R.; Romanens, L.; Bhattacharya, A.; Joosten, S.E.P.; Da Silva, A.M.; Padrao, N.; et al. MYC promotes immune-suppression in triple-negative breast cancer via inhibition of interferon signaling. Nat. Commun. 2022, 13, 6579. [Google Scholar] [CrossRef]
- Su, Y.; Hopfinger, N.R.; Nguyen, T.D.; Pogash, T.J.; Santucci-Pereira, J.; Russo, J. Epigenetic reprogramming of epithelial mesenchymal transition in triple negative breast cancer cells with DNA methyltransferase and histone deacetylase inhibitors. J. Exp. Clin. Cancer Res. 2018, 37, 314. [Google Scholar] [CrossRef]
- Dusenbery, A.C.; Maniaci, J.L.; Hillerson, N.D.; Dill, E.A.; Bullock, T.N.; Mills, A.M. MHC Class I Loss in Triple-negative Breast Cancer: A Potential Barrier to PD-1/PD-L1 Checkpoint Inhibitors. Am. J. Surg. Pathol. 2021, 45, 701–707. [Google Scholar] [CrossRef]
- Makena, M.R.; Rao, R. Subtype specific targeting of calcium signaling in breast cancer. Cell Calcium 2020, 85, 102109. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.; So, J.Y.; Ahad, A.; Michalowski, A.M.; Son, D.S.; Li, Y. CXCL14 Attenuates Triple-Negative Breast Cancer Progression by Regulating Immune Profiles of the Tumor Microenvironment in a T Cell-Dependent Manner. Int. J. Mol. Sci. 2022, 23, 9314. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Geng, A.; Li, X.; Zhou, Y.; Xu, L.; Zeng, Y.A.; Li, J.; Cai, C. CDK14 inhibition reduces mammary stem cell activity and suppresses triple negative breast cancer progression. Cell Rep. 2022, 40, 111331. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Lyu, Y.L.; Cai, L. NF-κB Affects Proliferation and Invasiveness of Breast Cancer Cells by Regulating CD44 Expression. PLoS ONE 2014, 9, e106966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provance, O.K.; Geanes, E.S.; Lui, A.J.; Roy, A.; Holloran, S.M.; Gunewardena, S.; Hagan, C.R.; Weir, S.; Lewis-Wambi, J. Disrupting interferon-alpha and NF-kappaB crosstalk suppresses IFITM1 expression attenuating triple-negative breast cancer progression. Cancer Lett. 2021, 514, 12–29. [Google Scholar] [CrossRef]
- Zeng, A.; Liang, X.; Zhu, S.; Liu, C.; Luo, X.; Zhang, Q.; Song, L. Baicalin, a Potent Inhibitor of NF-κB Signaling Pathway, Enhances Chemosensitivity of Breast Cancer Cells to Docetaxel and Inhibits Tumor Growth and Metastasis Both In Vitro and In Vivo. Front. Pharmacol. 2020, 11, 879. [Google Scholar] [CrossRef]
- Biswas, D.K.; Martin, K.J.; McAlister, C.; Cruz, A.P.; Graner, E.; Dai, S.C.; Pardee, A.B. Apoptosis caused by chemotherapeutic inhibition of nuclear factor-kappaB activation. Cancer Res. 2003, 63, 290–295. [Google Scholar]
- Murwanti, R.; Kholifah, E.; Sudarmanto, B.S.A.; Hermawan, A. Effect of curcumin on NF-κB P105/50 expression on triple-negative breast cancer (TNBC) and its possible mechanism of action. AIP Conf. Proc. 2020, 2260, 040024. [Google Scholar] [CrossRef]
- Sarkar, D.K.; Jana, D.; Patil, P.S.; Chaudhari, K.S.; Chattopadhyay, B.K.; Chikkala, B.R.; Mandal, S.; Chowdhary, P. Role of NF-κB as a Prognostic Marker in Breast Cancer: A Pilot Study in Indian Patients. Indian J. Surg. Oncol. 2013, 4, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-Y.; Jung, H.H.; Ahn, S.; Bae, S.; Lee, S.K.; Kim, S.W.; Lee, J.E.; Nam, S.J.; Ahn, J.S.; Im, Y.-H.; et al. The relationship between nuclear factor (NF)-κB family gene expression and prognosis in triple-negative breast cancer (TNBC) patients receiving adjuvant doxorubicin treatment. Sci. Rep. 2016, 6, 31804. [Google Scholar] [CrossRef] [Green Version]
- Muraro, E.; Comaro, E.; Talamini, R.; Turchet, E.; Miolo, G.; Scalone, S.; Militello, L.; Lombardi, D.; Spazzapan, S.; Perin, T.; et al. Improved Natural Killer cell activity and retained anti-tumor CD8(+) T cell responses contribute to the induction of a pathological complete response in HER2-positive breast cancer patients undergoing neoadjuvant chemotherapy. J. Transl. Med. 2015, 13, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajković-Molek, K.; Mustać, E.; Hadžisejdić, I.; Jonjić, N. The prognostic importance of nuclear factor κB and hypoxia-inducible factor 1α in relation to the breast cancer subtype and the overall survival. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Barbie, T.U.; Alexe, G.; Aref, A.R.; Li, S.; Zhu, Z.; Zhang, X.; Imamura, Y.; Thai, T.C.; Huang, Y.; Bowden, M.; et al. Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth. J. Clin. Investig. 2014, 124, 5411–5423. [Google Scholar] [CrossRef] [PubMed]
- Roseweir, A.K.; Bennett, L.; Dickson, A.; Cheng, K.; Quintayo, M.-A.; Bayani, J.; McMillan, D.C.; Horgan, P.G.; van de Velde, C.J.H.; Seynaeve, C.; et al. Predictive Biomarkers for Endocrine Therapy: Retrospective Study in Tamoxifen and Exemestane Adjuvant Multinational (TEAM) Trial. J. Natl. Cancer Inst. 2017, 110, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Inglés-Esteve, J.; Morales, M.; Dalmases, A.; Garcia-Carbonell, R.; Jené-Sanz, A.; López-Bigas, N.; Iglesias, M.; Ruiz-Herguido, C.; Rovira, A.; Rojo, F.; et al. Inhibition of Specific NF-κB Activity Contributes to the Tumor Suppressor Function of 14-3-3σ in Breast Cancer. PLoS ONE 2012, 7, e38347. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Nag, S.A.; Zhang, R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr. Med. Chem. 2015, 22, 264–289. [Google Scholar] [CrossRef]
- Tegowski, M.; Baldwin, A. Noncanonical NF-κB in Cancer. Biomedicines 2018, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Sun, S.-C.; Chang, J.-H.; Jin, J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Walker, W.; Stein, B.; Ganchi, P.; Hoffman, J.; Kaufman, P.; Ballard, D.; Hannink, M.; Greene, W.C. The v-rel oncogene: Insights into the mechanism of transcriptional activation, repression, and transformation. J. Virol. 1992, 66, 5018–5029. [Google Scholar] [CrossRef] [Green Version]
- Frederiksen, A.L.; Larsen, M.J.; Brusgaard, K.; Novack, D.V.; Knudsen, P.J.T.; Schrøder, H.D.; Qiu, W.; Eckhardt, C.; McAlister, W.H.; Kassem, M.J.J.o.B.; et al. Neonatal high bone mass with first mutation of the NF-κB complex: Heterozygous de novo missense (p. Asp512Ser) RELA (Rela/p65). J. Bone Miner. Res. 2016, 31, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, A.; Leung, T.H.; Baltimore, D.J.T.E.j. Genetic analysis of NF-κB/Rel transcription factors defines functional specificities. EMBO J. 2003, 22, 5530–5539. [Google Scholar] [CrossRef] [PubMed]
- Tsui, R.; Kearns, J.D.; Lynch, C.; Vu, D.; Ngo, K.A.; Basak, S.; Ghosh, G.; Hoffmann, A. IκBβ enhances the generation of the low-affinity NFκB/RelA homodimer. Nat. Commun. 2015, 6, 7068. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinke, S.; Ley, S.C. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-κB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.C.; Ley, S.C. New insights into NF-kappaB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Kim, H.; Cha, M.Y.; Park, H.G.; Kim, Y.J.; Kim, I.Y.; Kim, J.M. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-kappaB signaling pathway. J. Mol. Med. 2009, 87, 169–180. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujari, R.; Hunte, R.; Khan, W.N.; Shembade, N. A20-mediated negative regulation of canonical NF-κB signaling pathway. Immunol. Res. 2013, 57, 166–171. [Google Scholar] [CrossRef]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell. 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Bonizzi, G.; Bebien, M.; Otero, D.C.; Johnson-Vroom, K.E.; Cao, Y.; Vu, D.; Jegga, A.G.; Aronow, B.J.; Ghosh, G.; Rickert, R.C.; et al. Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers. EMBO J. 2004, 23, 4202–4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O’Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M.J.C. A fourth IκB protein within the NF-κB signaling module. Cell 2007, 128, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, G.; Xiao, G. Essential Role of IκB Kinase α in the Constitutive Processing of NF-κB2 p100*. J. Biol. Chem. 2005, 280, 9765–9768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerlund, R.; Melén, K.; Cao, X.; Julkunen, I. NF-kappaB p52, RelB and c-Rel are transported into the nucleus via a subset of importin alpha molecules. Cell. Signal. 2008, 20, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Belguise, K.; Kersual, N.; Kirsch, K.H.; Mineva, N.D.; Galtier, F.; Chalbos, D.; Sonenshein, G.E. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 2007, 9, 470–478. [Google Scholar] [CrossRef] [Green Version]
- Sas, L.; Lardon, F.; Vermeulen, P.B.; Hauspy, J.; Van Dam, P.; Pauwels, P.; Dirix, L.Y.; Van Laere, S.J. The interaction between ER and NFκB in resistance to endocrine therapy. Breast Cancer Res. 2012, 14, 212. [Google Scholar] [CrossRef] [Green Version]
- Semmler, L.; Reiter-Brennan, C.; Klein, A. BRCA1 and Breast Cancer: A Review of the Underlying Mechanisms Resulting in the Tissue-Specific Tumorigenesis in Mutation Carriers. J. Breast Cancer 2019, 22, 1–14. [Google Scholar] [CrossRef]
- Kufe, D.W. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2013, 32, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.; Herr, D.R. G protein-coupled receptor GPR19 regulates E-cadherin expression and invasion of breast cancer cells. Biochim. Biophys. Acta BBA-Mol. Cell. Res. 2017, 1864, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, L.; Chen, Y.; Wang, X.-y.; Lu, R.-f.; Zhang, S.-y.; Tian, M.; Xie, T.; Liu, B.; He, G. Polygonatum odoratum lectin induces apoptosis and autophagy via targeting EGFR-mediated Ras-Raf-MEK-ERK pathway in human MCF-7 breast cancer cells. Phytomedicine 2014, 21, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, P.; Sun, Y.-J.; Wu, Y.-J. Ivermectin reverses the drug resistance in cancer cells through EGFR/ERK/Akt/NF-κB pathway. J. Exp. Clin. Cancer Res. 2019, 38, 265. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996, 74, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Xu, J.; Sun, T. Cyclin-Dependent Kinases 4/6 Inhibitors in Breast Cancer: Current Status, Resistance, and Combination Strategies. J. Cancer 2019, 10, 5504–5517. [Google Scholar] [CrossRef] [PubMed]
- Khongthong, P.; Roseweir, A.K.; Edwards, J. The NF-KB pathway and endocrine therapy resistance in breast cancer. Endocr.-Relat. Cancer 2019, 26, R369–R380. [Google Scholar] [CrossRef]
- Devanaboyina, M.; Kaur, J.; Whiteley, E.; Lin, L.; Einloth, K.; Morand, S.; Stanbery, L.; Hamouda, D.; Nemunaitis, J. NF-κB Signaling in Tumor Pathways Focusing on Breast and Ovarian Cancer. Oncol. Rev. 2022, 16, 10568. [Google Scholar] [CrossRef]
- Butt, A.J.; McNeil, C.M.; Musgrove, E.A.; Sutherland, R.L. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: The potential roles of c-Myc, cyclin D1 and cyclin E. Endocr.-Relat. Cancer 2005, 12, S47–S59. [Google Scholar] [CrossRef]
- Kang, H.; Lee, M.; Choi, K.C.; Shin, D.M.; Ko, J.; Jang, S.W. N-(4-hydroxyphenyl)retinamide inhibits breast cancer cell invasion through suppressing NF-KB activation and inhibiting matrix metalloproteinase-9 expression. J. Cell. Biochem. 2012, 113, 2845–2855. [Google Scholar] [CrossRef]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinases as breast cancer drivers and therapeutic targets. Front. Biosci. 2015, 20, 1144–1163. [Google Scholar] [CrossRef] [PubMed]
- Gomez, B.P.; Riggins, R.B.; Shajahan, A.N.; Klimach, U.; Wang, A.; Crawford, A.C.; Zhu, Y.; Zwart, A.; Wang, M.; Clarke, R. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J. 2007, 21, 4013–4027. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Chen, Z.; Jiang, G.; Zhou, Y.; Liu, Q.; Su, Q.; Wei, W.; Du, J.; Wang, H. Activation of GPER suppresses migration and angiogenesis of triple negative breast cancer via inhibition of NF-κB/IL-6 signals. Cancer Lett. 2017, 386, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- To, S.Q.; Dmello, R.S.; Richards, A.K.; Ernst, M.; Chand, A.L. STAT3 Signaling in Breast Cancer: Multicellular Actions and Therapeutic Potential. Cancers 2022, 14, 429. [Google Scholar] [CrossRef]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Li, L.; Andrew, S.; Allan, D.; Li, X.; Lee, J.; Ji, G.; Yao, Z.; Gadde, S.; Figeys, D.; et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017, 8, e2932. [Google Scholar] [CrossRef] [Green Version]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef]
- Yong, L.; Tang, S.; Yu, H.; Zhang, H.; Zhang, Y.; Wan, Y.; Cai, F. The role of hypoxia-inducible factor-1 alpha in multidrug-resistant breast cancer. Front. Oncol. 2022, 12, 964934. [Google Scholar] [CrossRef]
- Rani, S.; Roy, S.; Singh, M.; Kaithwas, G. Regulation of Transactivation at C-TAD Domain of HIF-1α by Factor-Inhibiting HIF-1α (FIH-1): A Potential Target for Therapeutic Intervention in Cancer. Oxid. Med. Cell. Longev. 2022, 2022, 2407223. [Google Scholar] [CrossRef]
- Choi, J.Y.; Jang, Y.S.; Min, S.Y.; Song, J.Y. Overexpression of MMP-9 and HIF-1α in Breast Cancer Cells under Hypoxic Conditions. J. Breast Cancer 2011, 14, 88–95. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.L.; Joyce, M.R.; Shannon, A.M.; Harmey, J.; Geraghty, J.; Bouchier-Hayes, D. Oncological implications of hypoxia inducible factor-1α (HIF-1α) expression. Cancer Treat. Rev. 2006, 32, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bonello, S. The cross-talk between NF-κB and HIF-1: Further evidence for a significant liaison. Biochem. J. 2008, 412, e17–e19. [Google Scholar] [CrossRef] [PubMed]
- Khattri, R.; Cox, T.; Yasayko, S.-A.; Ramsdell, F. An essential role for Scurfin in CD4+ CD25+ T regulatory cells. Nat. Immunol. 2003, 4, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. FoxP3+ T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett. 2020, 490, 174–185. [Google Scholar] [CrossRef]
- Cinier, J.; Hubert, M.; Besson, L.; Di Roio, A.; Rodriguez, C.; Lombardi, V.; Caux, C.; Ménétrier-Caux, C. Recruitment and Expansion of Tregs Cells in the Tumor Environment—How to Target Them? Cancers 2021, 13, 1850. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Deng, Y.; Yu, X.; Wang, H.; Li, Z. Research progress on the role of regulatory T cell in tumor microenvironment in the treatment of breast cancer. Front. Oncol. 2021, 11, 766248. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Allaoui, R.; Hagerling, C.; Desmond, E.; Warfvinge, C.-F.; Jirström, K.; Leandersson, K. Infiltration of γ × δ T cells, IL-17+ T cells and FoxP3+ T cells in human breast cancer. Cancer Biomark. 2017, 20, 395–409. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Bleyer, Y.; Caron, R.; Seeley, J.J.; De Silva, N.S.; Schindler, C.W.; Hayden, M.S.; Klein, U.; Ghosh, S. The alternative NF-κB pathway in regulatory T cell homeostasis and suppressive function. J. Immunol. 2018, 200, 2362–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Hawke, N.; Baldwin, A.S. NF-κB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006, 13, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Tabana, Y.; Okoye, I.S.; Siraki, A.; Elahi, S.; Barakat, K.H. Tackling Immune Targets for Breast Cancer: Beyond PD-1/PD-L1 Axis. Front. Oncol. 2021, 11, 628138. [Google Scholar] [CrossRef]
- Dawod, B.; Liu, J.; Gebremeskel, S.; Yan, C.; Sappong, A.; Johnston, B.; Hoskin, D.W.; Marshall, J.S.; Wang, J. Myeloid-derived suppressor cell depletion therapy targets IL-17A-expressing mammary carcinomas. Sci. Rep. 2020, 10, 13343. [Google Scholar] [CrossRef]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Zhao, T.; Luo, R.; Qiu, R.; Li, Z. Tumor-associated macrophages: Key players in triple-negative breast cancer. Front. Oncol. 2022, 12, 772615. [Google Scholar] [CrossRef]
- Singh, K.; Bhori, M.; Kasu, Y.A.; Bhat, G.; Marar, T. Antioxidants as precision weapons in war against cancer chemotherapy induced toxicity—Exploring the armoury of obscurity. Saudi Pharm. J. 2018, 26, 177–190. [Google Scholar] [CrossRef]
- Seifried, H.E.; McDonald, S.S.; Anderson, D.E.; Greenwald, P.; Milner, J.A. The antioxidant conundrum in cancer. Cancer Res. 2003, 63, 4295–4298. [Google Scholar]
- Wondrak, G.T. Redox-Directed Cancer Therapeutics: Molecular Mechanisms and Opportunities. Antioxid. Redox. Signal. 2009, 11, 3013–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, C.; Buc Calderon, P. Vitamin C (Ascorbate) and Redox Topics in Cancer. Antioxid. Redox. Signal. 2021, 35, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Mohd Idris, R.A.; Ahmed, N.; Ahmad, S.; Murtadha, A.H.; Tengku Din, T.; Yean, C.Y.; Wan Abdul Rahman, W.F.; Mat Lazim, N.; Uskoković, V.; et al. High-Dose Vitamin C for Cancer Therapy. Pharmaceuticals 2022, 15, 711. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Calfee-Mason, K.G.; Spear, B.T.; Glauert, H.P. Vitamin E Inhibits Hepatic NF-κB Activation in Rats Administered the Hepatic Tumor Promoter, Phenobarbital. J. Nutr. 2002, 132, 3178–3185. [Google Scholar] [CrossRef] [Green Version]
- Vlahopoulos, S.; Boldogh, I.; Casola, A.; Brasier, A.R. Nuclear Factor-κB–Dependent Induction of Interleukin-8 Gene Expression by Tumor Necrosis Factor α: Evidence for an Antioxidant Sensitive Activating Pathway Distinct From Nuclear Translocation. Blood 1999, 94, 1878–1889. [Google Scholar] [CrossRef]
- Jamaluddin, M.; Wang, S.; Boldogh, I.; Tian, B.; Brasier, A.R. TNF-α-induced NF-κB/RelA Ser276 phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell. Signal. 2007, 19, 1419–1433. [Google Scholar] [CrossRef]
- Nowak, D.E.; Tian, B.; Jamaluddin, M.; Boldogh, I.; Vergara, L.A.; Choudhary, S.; Brasier, A.R. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol. Cell. Biol. 2008, 28, 3623–3638. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.M.; Cao, N.; Li, J.J. HER-2 and NF-kappaB as the targets for therapy-resistant breast cancer. Anticancer Res. 2006, 26, 4235–4243. [Google Scholar]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, W.L.; Wang, B.F.; Liang, B.B.; Zhang, L.; Pan, J.Y.; Huang, Y.; Zhao, Y.; Lin, S.; Zhao, Y.H.; Zhang, S.Q.; et al. A ROS/Akt/NF-κB Signaling Cascade Mediates Epidermal Growth Factor-Induced Epithelial-Mesenchymal Transition and Invasion in Human Breast Cancer Cells. World J. Oncol. 2022, 13, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive Oxygen Species Activate the HIF-1α Promoter Via a Functional NFκB Site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, Y.H.; Chong, S.J.F.; Kong, L.R.; Goh, B.C.; Pervaiz, S. Sustained IKKβ phosphorylation and NF-κB activation by superoxide-induced peroxynitrite-mediated nitrotyrosine modification of B56γ3 and PP2A inactivation. Redox. Biol. 2021, 41, 101834. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.G.; O’Neill, L.A. Vitamin C inhibits NF-kappa B activation by TNF via the activation of p38 mitogen-activated protein kinase. J. Immunol. 2000, 165, 7180–7188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.T.; Long, Y.; Tang, W.; Liu, X.F.; Dai, F.; Zhou, B. Prooxidative inhibition against NF-κB-mediated inflammation by pharmacological vitamin C. Free Radic. Biol. Med. 2022, 180, 85–94. [Google Scholar] [CrossRef]
- Chun, K.S.; Joo, S.H. Modulation of Reactive Oxygen Species to Overcome 5-Fluorouracil Resistance. Biomol. Ther. 2022, 30, 479–489. [Google Scholar] [CrossRef]
- Cárcamo, J.M.; Pedraza, A.; Bórquez-Ojeda, O.; Golde, D.W. Vitamin C Suppresses TNFα-Induced NFκB Activation by Inhibiting IκBα Phosphorylation. Biochemistry 2002, 41, 12995–13002. [Google Scholar] [CrossRef]
- McCarty, M.F. Expression and/or activity of the SVCT2 ascorbate transporter may be decreased in many aggressive cancers, suggesting potential utility for sodium bicarbonate and dehydroascorbic acid in cancer therapy. Med. Hypotheses 2013, 81, 664–670. [Google Scholar] [CrossRef]
- Evans, M.K.; Tovmasyan, A.; Batinic-Haberle, I.; Devi, G.R. Mn porphyrin in combination with ascorbate acts as a pro-oxidant and mediates caspase-independent cancer cell death. Free Radic. Biol. Med. 2014, 68, 302–314. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mussa, A.; Afolabi, H.A.; Syed, N.H.; Talib, M.; Murtadha, A.H.; Hajissa, K.; Mokhtar, N.F.; Mohamud, R.; Hassan, R. The NF-κB Transcriptional Network Is a High-Dose Vitamin C-Targetable Vulnerability in Breast Cancer. Biomedicines 2023, 11, 1060. https://doi.org/10.3390/biomedicines11041060
Mussa A, Afolabi HA, Syed NH, Talib M, Murtadha AH, Hajissa K, Mokhtar NF, Mohamud R, Hassan R. The NF-κB Transcriptional Network Is a High-Dose Vitamin C-Targetable Vulnerability in Breast Cancer. Biomedicines. 2023; 11(4):1060. https://doi.org/10.3390/biomedicines11041060
Chicago/Turabian StyleMussa, Ali, Hafeez Abiola Afolabi, Nazmul Huda Syed, Mustafa Talib, Ahmad Hafiz Murtadha, Khalid Hajissa, Noor Fatmawati Mokhtar, Rohimah Mohamud, and Rosline Hassan. 2023. "The NF-κB Transcriptional Network Is a High-Dose Vitamin C-Targetable Vulnerability in Breast Cancer" Biomedicines 11, no. 4: 1060. https://doi.org/10.3390/biomedicines11041060