
The Effects of PP2A Disruption on ER-Mitochondria Contact and Mitochondrial Functions in Neuronal-like Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Immunoblotting
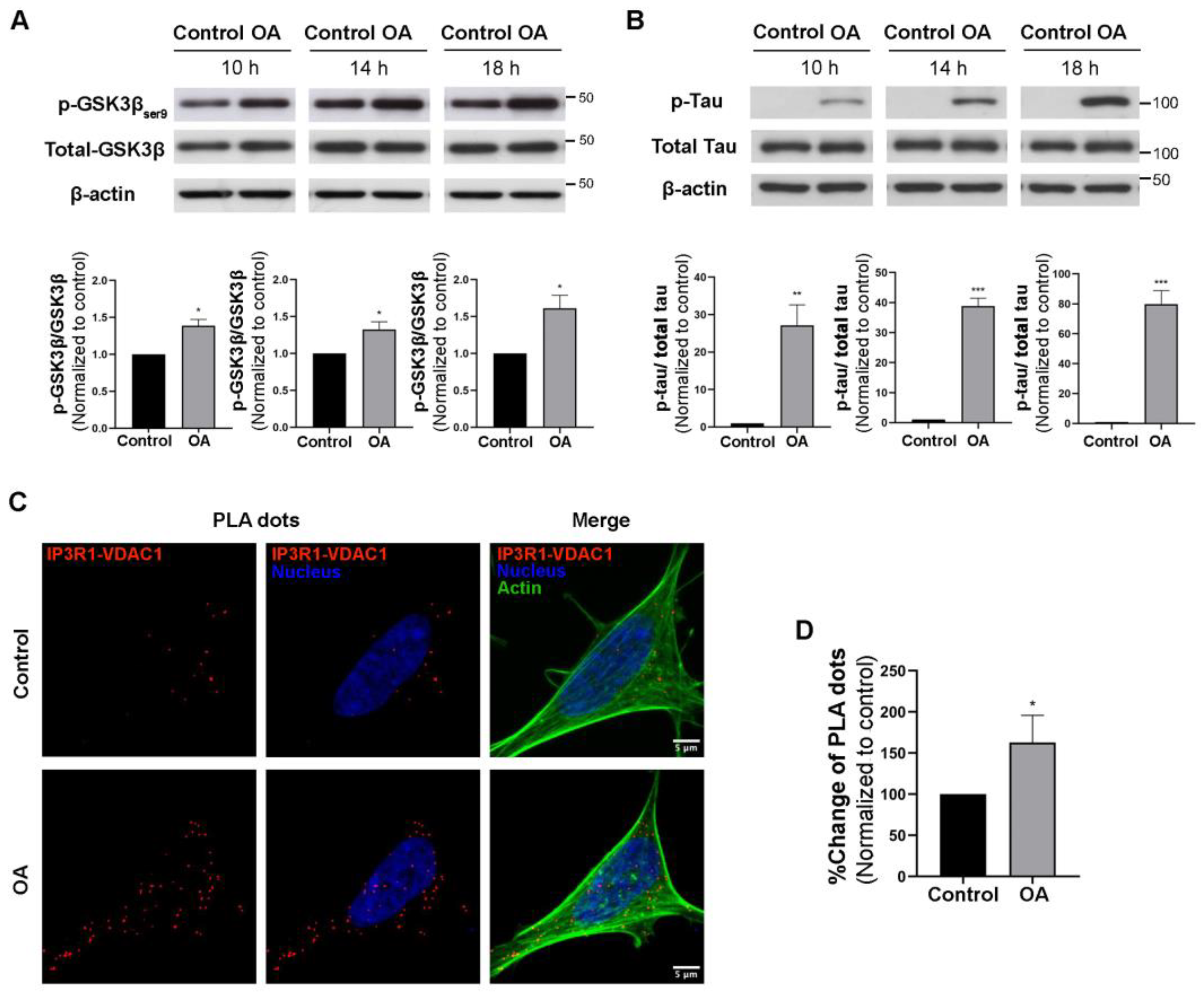
2.3. Proximity Ligation Assay (PLA)
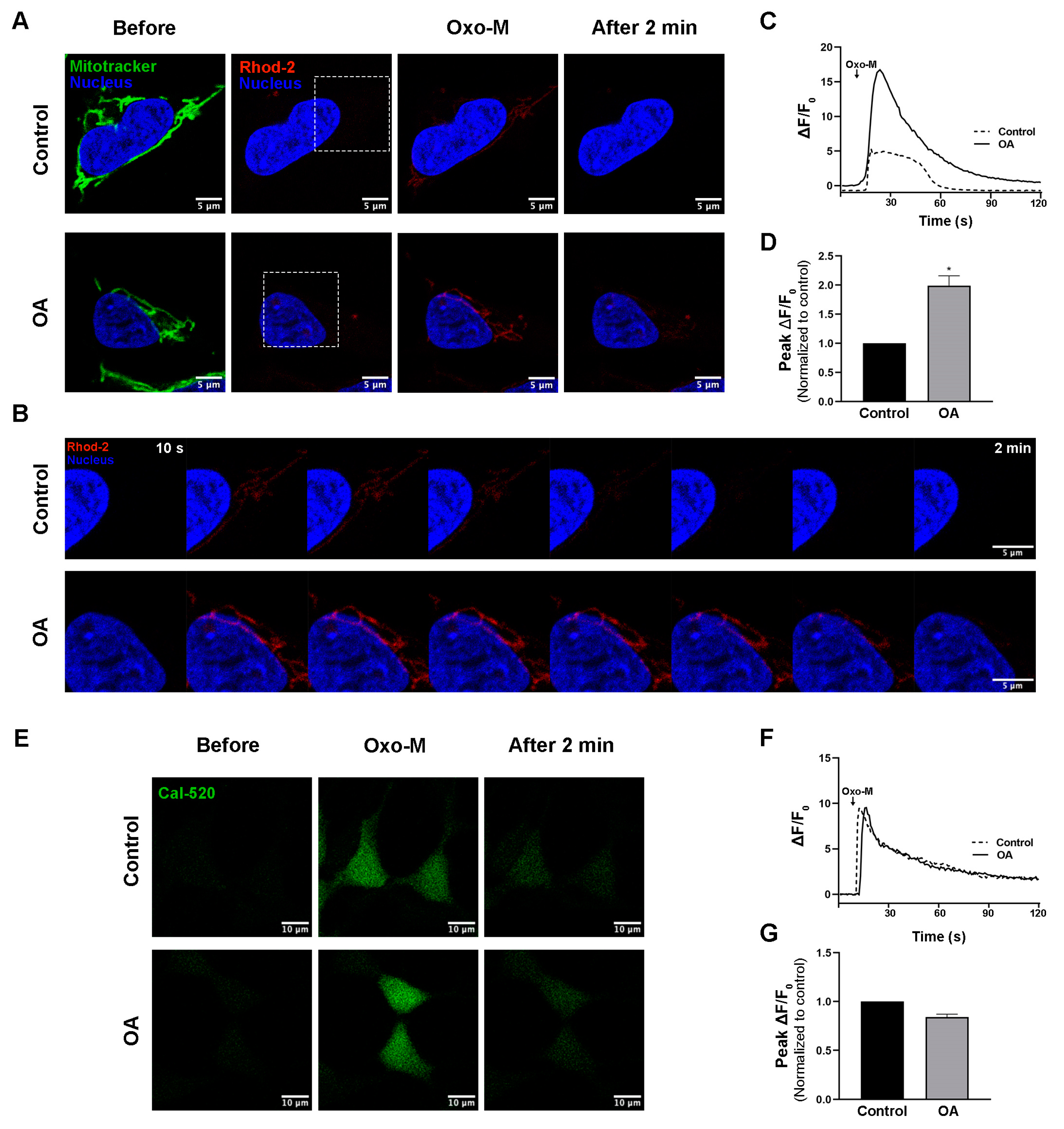
2.4. Ca2+ Measurements
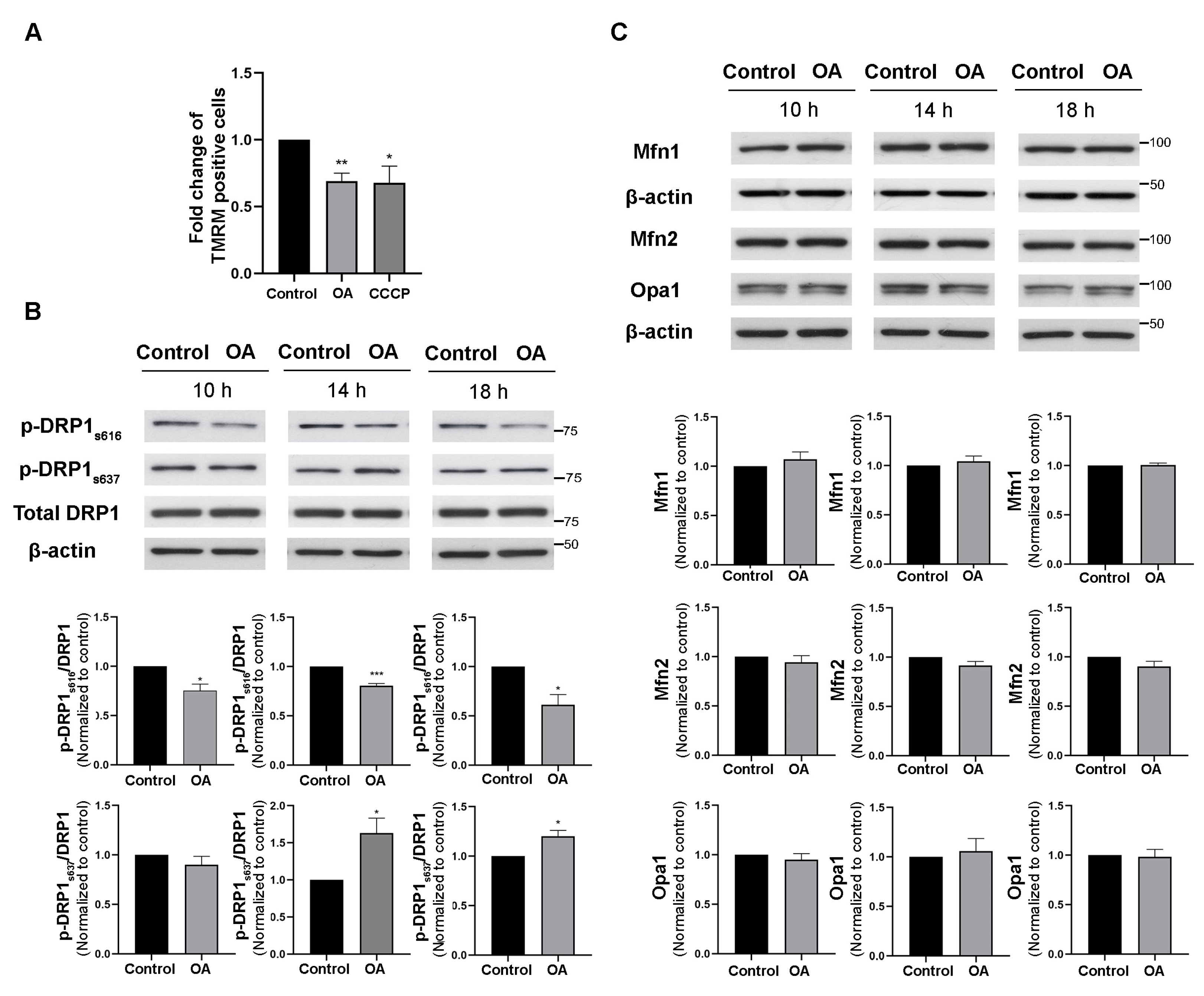
2.5. TMRM Assay
2.6. Statistical Analysis
3. Results
3.1. PP2A Inhibition Increased MAM Formation
3.2. PP2A Inhibition Regulates Mitochondrial Calcium Transfer
3.3. PP2A Inhibition Decreased Mitochondrial Membrane Potential and Mitochondrial Fission
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swerdlow, R.H.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: An update. Exp. Neurol. 2009, 218, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Teo, E.; Ravi, S.; Barardo, D.; Kim, H.-S.; Fong, S.; Cazenave-Gassiot, A.; Tan, T.Y.; Ching, J.; Kovalik, J.-P.; Wenk, M.R. Metabolic stress is a primary pathogenic event in transgenic Caenorhabditis elegans expressing pan-neuronal human amyloid beta. Elife 2019, 8, e50069. [Google Scholar] [CrossRef] [PubMed]
- Sassano, M.L.; Felipe-Abrio, B.; Agostinis, P. ER-mitochondria contact sites; a multifaceted factory for Ca2+ signaling and lipid transport. Front. Cell Develop. Biol. 2022, 10, 988014. [Google Scholar] [CrossRef] [PubMed]
- Loncke, J.; Kaasik, A.; Bezprozvanny, I.; Parys, J.B.; Kerkhofs, M.; Bultynck, G. Balancing ER-mitochondrial Ca2+ fluxes in health and disease. Trends Cell Biol. 2021, 31, 598–612. [Google Scholar] [CrossRef]
- Krols, M.; Bultynck, G.; Janssens, S. ER–Mitochondria contact sites: A new regulator of cellular calcium flux comes into play. J. Cell Biol. 2016, 214, 367. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F. Increased localization of APP–C99 in mitochondria–associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)–mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M. Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proc. Nat. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef]
- Sarasija, S.; Laboy, J.T.; Ashkavand, Z.; Bonner, J.; Tang, Y.; Norman, K.R. Presenilin mutations deregulate mitochondrial Ca2+ homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. Elife 2018, 7, e33052. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S. FUNDC 1 regulates mitochondrial dynamics at the ER–mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-H.; Lin, S.-C.A.; Kuo, C.-H.; Li, C.-J. Molecular machinery and pathophysiology of mitochondrial dynamics. Front. Cell Dev. Biol. 2021, 9, 743892. [Google Scholar] [CrossRef] [PubMed]
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 2020, 219, e201911122. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Human Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef]
- Vogelsberg-Ragaglia, V.; Schuck, T.; Trojanowski, J.Q.; Lee, V.M.-Y. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp. Neurol. 2001, 168, 402–412. [Google Scholar] [CrossRef]
- Sontag, E.; Hladik, C.; Montgomery, L.; Luangpirom, A.; Mudrak, I.; Ogris, E.; White III, C.L. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropapathol. Exp. Neurol. 2004, 63, 1080–1091. [Google Scholar] [CrossRef]
- Shentu, Y.-P.; Huo, Y.; Feng, X.-L.; Gilbert, J.; Zhang, Q.; Liuyang, Z.-Y.; Wang, X.-L.; Wang, G.; Zhou, H.; Wang, X.-C. CIP2A causes Tau/APP phosphorylation, synaptopathy, and memory deficits in Alzheimer’s disease. Cell Rep. 2018, 24, 713–723. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Terro, F. Tau protein phosphatases in Alzheimer’s disease: The leading role of PP2A. Ageing Res. Rev. 2013, 12, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.-A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Rai, S.; Nath, C. Okadaic acid induced neurotoxicity: An emerging tool to study Alzheimer’s disease pathology. Neurotoxicology 2013, 37, 163–172. [Google Scholar] [CrossRef]
- Zhang, Z.; Simpkins, J.W. Okadaic acid induces tau phosphorylation in SH-SY5Y cells in an estrogen-preventable manner. Brain Res. 2010, 1345, 176–181. [Google Scholar] [CrossRef]
- Boban, M.; Leko, M.B.; Miškić, T.; Hof, P.R.; Šimić, G. Human neuroblastoma SH-SY5Y cells treated with okadaic acid express phosphorylated high molecular weight tau-immunoreactive protein species. J. Neurosci. Methods 2019, 319, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Hayashi, T. New insights into the role of mitochondria-associated endoplasmic reticulum membrane. Int. Rev. Cell Mol. Biol. 2011, 292, 73–117. [Google Scholar]
- Hayashi, T.; Su, T.-P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Plácido, A.I.; Pereira, C.M.; Correira, S.C.; Carvalho, C.; Oliveira, C.R.; Moreira, P.I. Phosphatase 2A inhibition affects endoplasmic reticulum and mitochondria homeostasis via cytoskeletal alterations in brain endothelial cells. Mol. Neurobiol. 2017, 54, 154–168. [Google Scholar] [CrossRef]
- Jin, J.; Hulette, C.; Wang, Y.; Zhang, T.; Pan, C.; Wadhwa, R.; Zhang, J. Proteomic Identification of a Stress Protein, Mortalin/mthsp70/GRP75: Relevance To Parkinson Disease* S. Mol. Cell. Proteomics 2006, 5, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Honrath, B.; Metz, I.; Bendridi, N.; Rieusset, J.; Culmsee, C.; Dolga, A.M. Glucose-regulated protein 75 determines ER–mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 2017, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yu, Q.; Chen, J.; Deng, B.; Qian, L.; Le, Y. PP2A mediated AMPK inhibition promotes HSP70 expression in heat shock response. PLoS ONE 2010, 5, e13096. [Google Scholar] [CrossRef]
- Csordás, G.R.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G.R. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic reticulum–mitochondrial contactology: Structure and signaling functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C. The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.-P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef]
- Weng, T.-Y.; Tsai, S.-Y.A.; Su, T.-P. Roles of sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases. J. Biomed. Sci. 2017, 24, 1–14. [Google Scholar] [CrossRef]
- Moltedo, O.; Remondelli, P.; Amodio, G. The Mitochondria–Endoplasmic Reticulum Contacts and Their Critical Role in Aging and Age-Associated Diseases. Front. Cell Develop. Biol. 2019, 7. [Google Scholar] [CrossRef]
- Kiryu-Seo, S.; Tamada, H.; Kato, Y.; Yasuda, K.; Ishihara, N.; Nomura, M.; Mihara, K.; Kiyama, H. Mitochondrial fission is an acute and adaptive response in injured motor neurons. Sci. Rep. 2016, 6, 28331. [Google Scholar] [CrossRef]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C. α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca 2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.E.; Oh, J.Y.; Lee, H.J.; Chae, C.W.; Kim, J.S.; Jung, Y.H.; Han, H.J. Glucocorticoid-mediated ER-mitochondria contacts reduce AMPA receptor and mitochondria trafficking into cell terminus via microtubule destabilization. Cell Death Dis. 2018, 9, 1137. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhang, H.-L.; Xie, J.-Z.; Meng, D.-L.; Wang, X.-C.; Ke, D.; Zeng, J.; Liu, R. Protein phosphatase 2A as a drug target in the treatment of cancer and Alzheimer’s disease. Curr. Med. Sci. 2020, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Voronkov, M.; Braithwaite, S.P.; Stock, J.B. Phosphoprotein phosphatase 2A: A novel druggable target for Alzheimer’s disease. Future Med. Chem. 2011, 3, 821–833. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaiwijit, P.; Uppakara, K.; Asavapanumas, N.; Saengsawang, W. The Effects of PP2A Disruption on ER-Mitochondria Contact and Mitochondrial Functions in Neuronal-like Cells. Biomedicines 2023, 11, 1011. https://doi.org/10.3390/biomedicines11041011
Chaiwijit P, Uppakara K, Asavapanumas N, Saengsawang W. The Effects of PP2A Disruption on ER-Mitochondria Contact and Mitochondrial Functions in Neuronal-like Cells. Biomedicines. 2023; 11(4):1011. https://doi.org/10.3390/biomedicines11041011
Chicago/Turabian StyleChaiwijit, Phaewa, Kwanchanok Uppakara, Nithi Asavapanumas, and Witchuda Saengsawang. 2023. "The Effects of PP2A Disruption on ER-Mitochondria Contact and Mitochondrial Functions in Neuronal-like Cells" Biomedicines 11, no. 4: 1011. https://doi.org/10.3390/biomedicines11041011