
In Silico Identification of Lead Compounds for Pseudomonas Aeruginosa PqsA Enzyme: Computational Study to Block Biofilm Formation
Abstract
:1. Introduction
2. Methodology
2.1. Retrieval of Protein
2.2. Preparation of Quorum Sensing Inhibitor
2.3. Pharmacophore Based Virtual Screening
2.4. Screening of Commercially Available Databases
2.5. Molecular Docking
2.6. Systematic Analysis of the Potent Lead Compound
2.7. Hydrogen Bond Analysis
2.8. Dynamic Cross-Correlation Movement Analysis
2.9. Principal Component Analysis and Free Energy Landscape
2.10. Binding Free Energy Calculations
3. Results and Discussion
3.1. Pharmacophore-based Virtual Screening
3.2. Molecular Docking
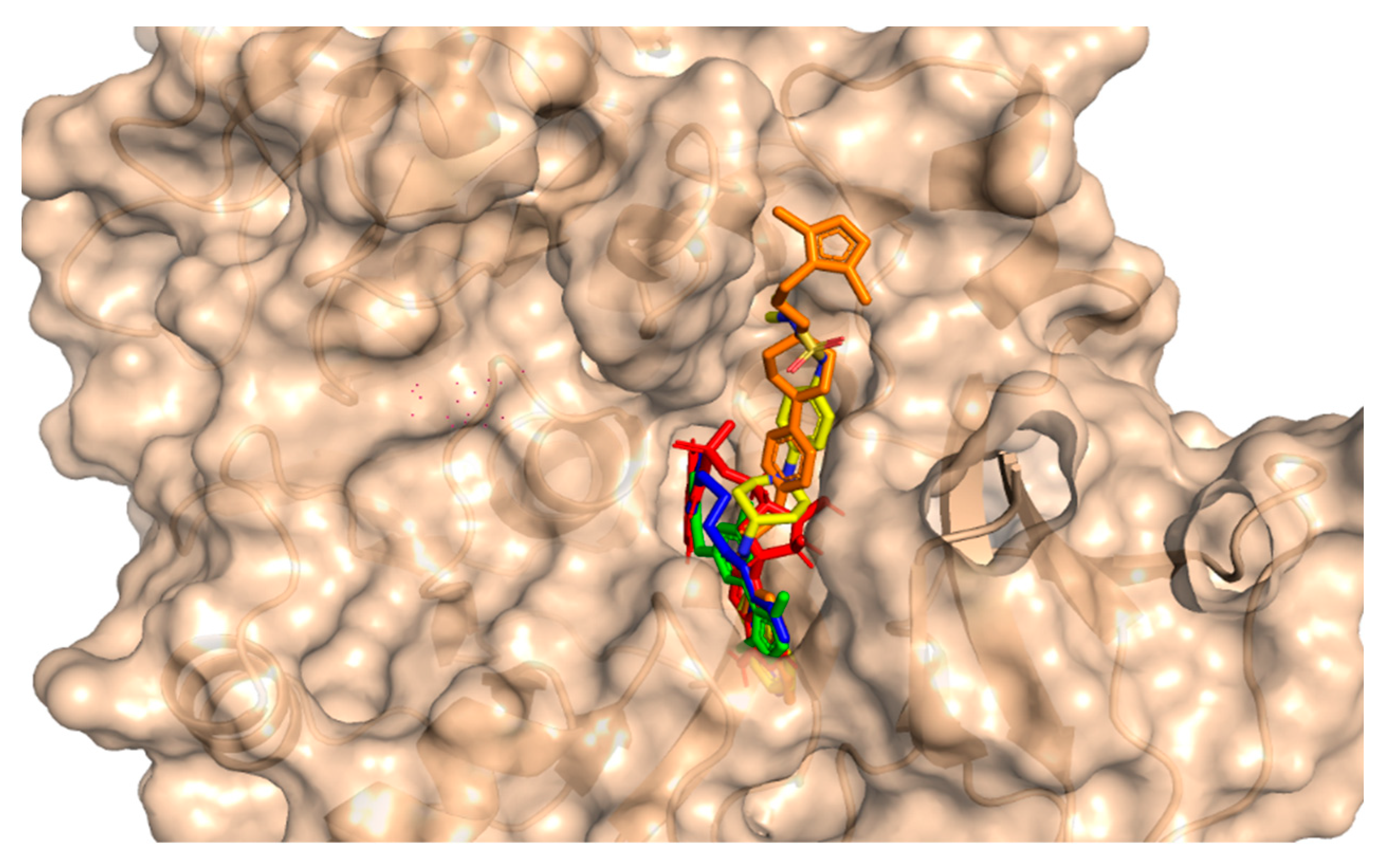
3.3. Binding of Selected Drug-like Compound
3.4. Molecular Dynamics Simulation
3.5. Hydrogen Bond Analysis
3.6. Exploring the Dominant Motions for Lead Compounds
3.7. The Dynamic Cross-Correlation Matrix (DCCM)
3.8. Binding Free Energy Calculation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laborda, P.; Fernando, S.-G.; Sara, H.-A.; José, L.M. Pseudomonas aeruginosa: An antibiotic resilient pathogen with environmental origin. Curr. Opin. Microbiol. 2021, 64, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.R.; Shrivastava, P.S.; Ramasamy, J. World health organization releases global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. J. Med. Soc. 2018, 32, 76. [Google Scholar] [CrossRef]
- Witzgall, F.; Ewert, W.; Blankenfeldt, W. Structures of the N-Terminal Domain of PqsA in Complex with Anthraniloyl-and 6-Fluoroanthraniloyl-AMP: Substrate Activation in Pseudomonas Quinolone Signal (PQS) Biosynthesis. ChemBioChem 2017, 18, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Soukarieh, F.; Vico Oton, E.; Dubern, J.F.; Gomes, J.; Halliday, N.; de Pilar Crespo, M.; Ramírez-Prada, J.; Insuasty, B.; Abonia, R.; Quiroga, J.; et al. In silico and in vitro-guided identification of inhibitors of alkylquinolone-dependent quorum sensing in Pseudomonas aeruginosa. Molecules 2018, 23, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rather, M.A.; Gupta, K.; Bardhan, P.; Borah, M.; Sarkar, A.; Eldiehy, K.S.; Bhuyan, S.; Mandal, M. Microbial biofilm: A matter of grave concern for human health and food industry. J. Basic Microbiol. 2021, 61, 380–395. [Google Scholar] [CrossRef]
- Impey, R.E.; Panjikar, S.; Hall, C.J.; Bock, L.J.; Sutton, J.M.; Perugini, M.A.; Soares da Costa, T.P. Identification of two dihydrodipicolinate synthase isoforms from Pseudomonas aeruginosa that differ in allosteric regulation. FEBS J. 2020, 287, 386–400. [Google Scholar] [CrossRef]
- Shao, X.; Yao, C.; Ding, Y.; Hu, H.; Qian, G.; He, M.; Deng, X. The transcriptional regulators of virulence for Pseudomonas aeruginosa: Therapeutic opportunity and preventive potential of its clinical infections. Genes Dis. 2022. [Google Scholar] [CrossRef]
- Nathwani, D.; Raman, G.; Sulham, K.; Gavaghan, M.; Menon, V. Clinical and economic consequences of hospital-acquired resistant and multidrug-resistant Pseudomonas aeruginosa infections: A systematic review and meta-analysis. Antimicrob. Resist. Infect. Control 2014, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Tabak, Y.; Merchant, S.; Ye, G.; Vankeepuram, L.; Gupta, V.; Kurtz, S.G.; Puzniak, L.A. Incremental clinical and economic burden of suspected respiratory infections due to multi-drug-resistant Pseudomonas aeruginosa in the United States. J. Hosp. Infect. 2019, 103, 134–141. [Google Scholar] [CrossRef]
- Micoli, F.; Costantino, P.; Adamo, R. Potential targets for next generation antimicrobial glycoconjugate vaccines. FEMS Microbiol. Rev. 2018, 42, 388–423. [Google Scholar] [CrossRef] [Green Version]
- Soukarieh, F.; Williams, P.; Stocks, M.J.; Camara, M. Pseudomonas aeruginosa quorum sensing systems as drug discovery targets: Current position and future perspectives. J. Med. Chem. 2018, 61, 10385–10402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brindhadevi, K.; LewisOscar, F.; Mylonakis, E.; Shanmugam, S.; Verma, T.N.; Pugazhendhi, A. Biofilm and Quorum sensing mediated pathogenicity in Pseudomonas aeruginosa. Process Biochem. 2020, 96, 49–57. [Google Scholar] [CrossRef]
- Subramani, R.; Jayaprakashvel, M. Bacterial quorum sensing: Biofilm formation, survival behaviour and antibiotic resistance. In Implication of Quorum Sensing and Biofilm Formation in Medicine, Agriculture and Food Industry; Springer: Singapore, 2019; pp. 21–37. [Google Scholar]
- Chiang, W.-C.; Nilsson, M.; Jensen, P.Ø.; Høiby, N.; Nielsen, T.E.; Givskov, M.; Tolker-Nielsen, T. Extracellular DNA shields against aminoglycosides in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2013, 57, 2352–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhede, M.; Bjarnsholt, T.; Jensen, P.Ø.; Phipps, R.K.; Moser, C.; Christophersen, L.; Christensen, L.D.; van Gennip, M.; Parsek, M.; Høiby, N.; et al. Pseudomonas aeruginosa recognizes and responds aggressively to the presence of polymorphonuclear leukocytes. Microbiology 2009, 155, 3500–3508. [Google Scholar] [CrossRef] [Green Version]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, W.-L.; Bassler, B.L. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 2009, 43, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühlen, S.; Dersch, P. Anti-virulence strategies to target bacterial infections. In How to Overcome the Antibiotic Crisis: Facts, Challenges, Technologies and Future Perspectives; Springer: Cham, Germany, 2016; pp. 147–183. [Google Scholar]
- Bhardwaj, S.; Gupta, P.S. Virtual Screening of Potential Quorum Sensing Inhibitors of P. aeruginosa. Int. J. Pharm. Investig. 2022, 12, 260–271. [Google Scholar] [CrossRef]
- Kamal, A.A.; Maurer, C.K.; Allegretta, G.; Haupenthal, J.; Empting, M.; Hartmann, R.W. Quorum sensing inhibitors as pathoblockers for Pseudomonas aeruginosa infections: A new concept in anti-infective drug discovery. Antibact. Vol. II 2018, 26, 185–210. [Google Scholar]
- Williams, P.; Cámara, M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: A tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 2009, 12, 182–191. [Google Scholar] [CrossRef]
- Nadal Jimenez, P.; Koch, G.; Thompson, J.A.; Xavier, K.B.; Cool, R.H.; Quax, W.J. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2012, 76, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Mudduluru, G.; Walther, W.; Kobelt, D.; Dahlmann, M.; Treese, C.; Assaraf, Y.G.; Stein, U. Repositioning of drugs for intervention in tumor progression and metastasis: Old drugs for new targets. Drug Resist. Updates 2016, 26, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Oprea, T.I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laghari, S.; Niazi, M.A. Modeling the internet of things, self-organizing and other complex adaptive communication networks: A cognitive agent-based computing approach. PLoS ONE 2016, 11, e0146760. [Google Scholar] [CrossRef]
- Zhou, T.; Wu, Z.; Das, S.; Eslami, H.; Muller-Plathe, F. How Ethanolic Disinfectants Disintegrate Coronavirus Model Membranes: A Dissipative Particle Dynamics Simulation Study. J. Chem. Theory Comput. 2022, 18, 2597–2615. [Google Scholar] [CrossRef]
- Flydal, M.I.; Alcorlo-Pagés, M.; Johannessen, F.G.; Martínez-Caballero, S.; Skjærven, L.; Fernandez-Leiro, R.; Martinez, A.; Hermoso, J.A. Structure of full-length human phenylalanine hydroxylase in complex with tetrahydrobiopterin. Proc. Natl. Acad. Sci. USA 2019, 116, 11229–11234. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Sekine, S.-i.; Yokoyama, S. Crystal structure of the full-length bacterial selenocysteine-specific elongation factor SelB. Nucleic Acids Res. 2015, 43, 9028–9038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandeel, M.; Yamamoto, M.; Al-Taher, A.; Watanabe, A.; Oh-Hashi, K.; Park, B.K.; Kwon, H.J.; Inoue, J.I.; Al-Nazawi, M. Small molecule inhibitors of Middle East respiratory syndrome coronavirus fusion by targeting cavities on heptad repeat trimers. Biomol. Ther. 2020, 28, 311. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Wadood, A.; Wadood, A.; Riaz, M.; Uddin, R.; Ul-Haq, Z. In silico identification and evaluation of leads for the simultaneous inhibition of protease and helicase activities of HCV NS3/4A protease using complex based pharmacophore mapping and virtual screening. PLoS ONE 2014, 9, e89109. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Kashefolgheta, S.; Verde, A.V. Developing force fields when experimental data is sparse: AMBER/GAFF-compatible parameters for inorganic and alkyl oxoanions. Phys. Chem. Chem. Phys. 2017, 19, 20593–20607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, A.; Avashthi, H.; Jha, R.; Srivastava, A.; Garg, V.K.; Ramteke, P.W.; Kumar, A. Insights using the molecular model of Lipoxygenase from Finger millet (Eleusine coracana (L.)). Bioinformation 2016, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.A.; Liu, R.; Martins de Oliveira, V.; Vázquez-Montelongo, E.A.; Henderson, J.A.; Shen, J. GPU-Accelerated All-atom Particle-Mesh Ewald Continuous Constant pH Molecular Dynamics in Amber. J. Chem. Theory Comput. 2022, 18, 7510–7527. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Rehman, A.U.; Ali, S.; Rafiq, H.; Rasheed, S.; Nouroz, F.; Wadood, A. Computational Insight into the Binding Mechanism of Pyrazinoic Acid to RpsA Protein. Curr. Chin. Sci. 2021, 1, 207–215. [Google Scholar] [CrossRef]
- Swaminathan, S.; Ichiye, T.; Van Gunsteren, W.; Karplus, M. Time dependence of atomic fluctuations in proteins: Analysis of local and collective motions in bovine pancreatic trypsin inhibitor. Biochemistry 1982, 21, 5230–5241. [Google Scholar] [CrossRef]
- Henderson, J.A.; Liu, R.; Harris, J.A.; Huang, Y.; de Oliveira, V.M.; Shen, J. A Guide to the Continuous Constant pH Molecular Dynamics Methods in Amber and CHARMM [Article v1. 0]. Living J. Comput. Mol. Sci. 2022, 4, 1563. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Liu, Q.; Chen, A.; Janssens, I.A.; Fu, Y.; Dai, J.; Liu, L.; Lian, X.U.; Shen, M.; Zhu, X. Plant phenology and global climate change: Current progresses and challenges. Glob. Change Biol. 2019, 25, 1922–1940. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Hoang, T.X.; Trovato, A.; Seno, F.; Banavar, J.R.; Maritan, A. Geometry and symmetry presculpt the free-energy landscape of proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 7960–7964. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Luo, W.; Wang, Y. PARP-1 and its associated nucleases in DNA damage response. DNA Repair 2019, 81, 102651. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the generalized Born model suitable for macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Paschek, D. Temperature dependence of the hydrophobic hydration and interaction of simple solutes: An examination of five popular water models. J. Chem. Phys. 2004, 120, 6674–6690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surabhi, S.; Singh, B.K. Computer aided drug design: An overview. J. Drug Deliv. Ther. 2018, 8, 504–509. [Google Scholar] [CrossRef] [Green Version]
- Leach, A.R.; Shoichet, B.K.; Peishoff, C.E. Prediction of protein−ligand interactions. Docking and scoring: Successes and gaps. J. Med. Chem. 2006, 49, 5851–5855. [Google Scholar] [CrossRef] [PubMed]
- Wei-Ya, L.; u-Qing, D.; Yang-Chun, M.; Xin-Hua, L.; Ying, M.; Wang, R.L. Exploring the cause of the inhibitor 4AX attaching to binding site disrupting protein tyrosine phosphatase 4A1 trimerization by molecular dynamic simulation. J. Biomol. Struct. Dyn. 2019, 37, 4840–4851. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Fu, H.; Fryar, K.L.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of hydrogen bonds to protein stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef]
- Islam, M.S.; Al-Majid, A.M.; Sholkamy, E.N.; Yousuf, S.; Ayaz, M.; Nawaz, A.; Wadood, A.; Rehman, A.U.; Verma, V.P.; Bari, A.; et al. Synthesis, molecular docking and enzyme inhibitory approaches of some new chalcones engrafted pyrazole as potential antialzheimer, antidiabetic and antioxidant agents. J. Mol. Struct. 2022, 1269, 133843. [Google Scholar] [CrossRef]
- Kumari, M.; Singh, R.; Subbarao, N. Exploring the interaction mechanism between potential inhibitor and multi-target Mur enzymes of mycobacterium tuberculosis using molecular docking, molecular dynamics simulation, principal component analysis, free energy landscape, dynamic cross-correlation matrices, vector movements, and binding free energy calculation. J. Biomol. Struct. Dyn. 2021, 24, 13497–13526. [Google Scholar]
- Khan, A.; Umbreen, S.; Hameed, A.; Fatima, R.; Zahoor, U.; Babar, Z.; Waseem, M.; Hussain, Z.; Rizwan, M.; Zaman, N.; et al. In silico mutagenesis-based remodelling of SARS-CoV-1 peptide (ATLQAIAS) to inhibit SARS-CoV-2: Structural-dynamics and free energy calculations. Interdiscip. Sci. Comput. Life Sci. 2021, 13, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Ghufran, M.; Rehman, A.U.; Shah, M.; Ayaz, M.; Ng, H.L.; Wadood, A. In-silico design of peptide inhibitors of K-Ras target in cancer disease. J. Biomol. Struct. Dyn. 2020, 38, 5488–5499. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.N | ZINC and ChemBridge ID | MW | LogP | Don | Acc | Docking Score | TPSA (Angstrom) | Binding Energy (kcal/moL) |
|---|---|---|---|---|---|---|---|---|
| 1 | ZINC79107864 | 341.45 | 3.52 | 2 | 4 | −7.76 | 52.93 | −52.75 |
| 2 | ZINC32573386 | 370.49 | 3.12 | 2 | 5 | −8.33 | 53.96 | −43.76 |
| 3 | ChemBridge54245649 | 338.43 | 0.47 | 3 | 5 | −8.27 | 44.73 | −46.95 |
| 4 | ChemBridge53910279 | 366.50 | 3.45 | 2 | 4 | −8.90 | 96.37 | −50.48 |
| 5 | Reference compound | 444.21 | 2.27 | 4 | 5 | −6.65 | 143.72 | −40.14 |
| No | Compound ID | vdW | EEL | ESURF | EGB | TOTAL |
|---|---|---|---|---|---|---|
| 1 | ZINC32573386 | −38.8733 | −0.9948 | −3.9755 | 10.3074 | −33.5365 |
| 2 | ZINC79107864 | −51.8814 | 1.0207 | −4.6930 | 10.7405 | −44.8132 |
| 3 | Ch53910279 | −46.5828 | −43.0180 | −5.8361 | 47.5489 | −47.8880 |
| 4 | Ch54245649 | −65.0533 | −3.2224 | −7.5232 | −18.6792 | −57.1197 |
| 5 | Reference | −40.5049 | −9.0757 | −3.9914 | −30.2265 | −23.3445 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahab, M.; Danial, M.; Khan, T.; Liang, C.; Duan, X.; Wang, D.; Gao, H.; Zheng, G. In Silico Identification of Lead Compounds for Pseudomonas Aeruginosa PqsA Enzyme: Computational Study to Block Biofilm Formation. Biomedicines 2023, 11, 961. https://doi.org/10.3390/biomedicines11030961
Shahab M, Danial M, Khan T, Liang C, Duan X, Wang D, Gao H, Zheng G. In Silico Identification of Lead Compounds for Pseudomonas Aeruginosa PqsA Enzyme: Computational Study to Block Biofilm Formation. Biomedicines. 2023; 11(3):961. https://doi.org/10.3390/biomedicines11030961
Chicago/Turabian StyleShahab, Muhammad, Muhammad Danial, Taimur Khan, Chaoqun Liang, Xiuyuan Duan, Daixi Wang, Hanzi Gao, and Guojun Zheng. 2023. "In Silico Identification of Lead Compounds for Pseudomonas Aeruginosa PqsA Enzyme: Computational Study to Block Biofilm Formation" Biomedicines 11, no. 3: 961. https://doi.org/10.3390/biomedicines11030961