

The Effects of the Levosimendan Metabolites OR-1855 and OR-1896 on Endothelial Pro-Inflammatory Responses
 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Chemicals
2.2. Human Umbilical Vein Endothelial Cell Culture
2.3. Western Blotting
2.4. Flow Cytometry
2.5. Quantitative Real-Time PCR
2.6. Measurement of Reactive Oxygen Species
2.7. Measurement of Superoxide Radicals
2.8. Statistics
3. Results
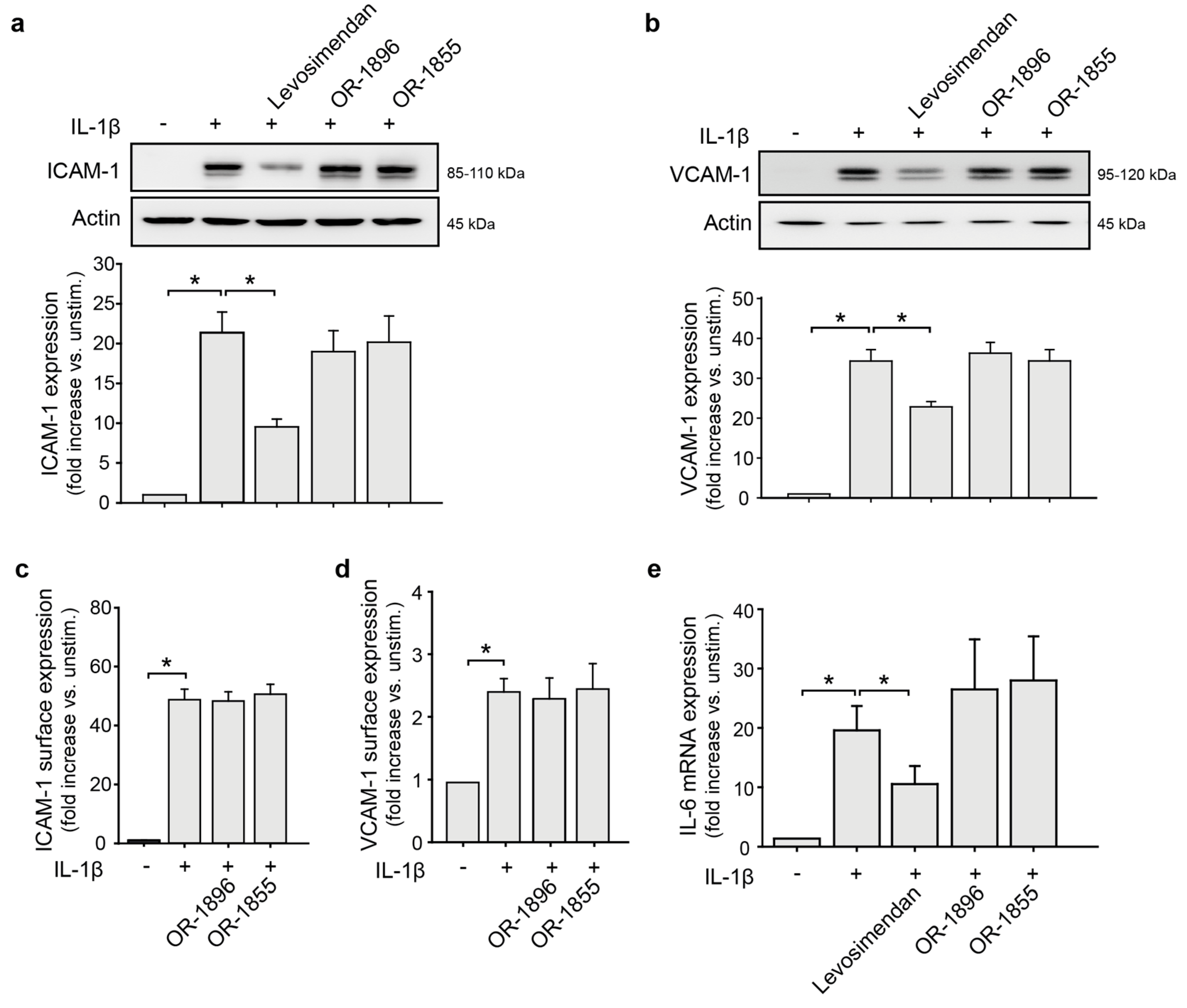
3.1. Levosimendan but Not Its Metabolites Affect the Endothelial Pro-Inflammatory Phenotype
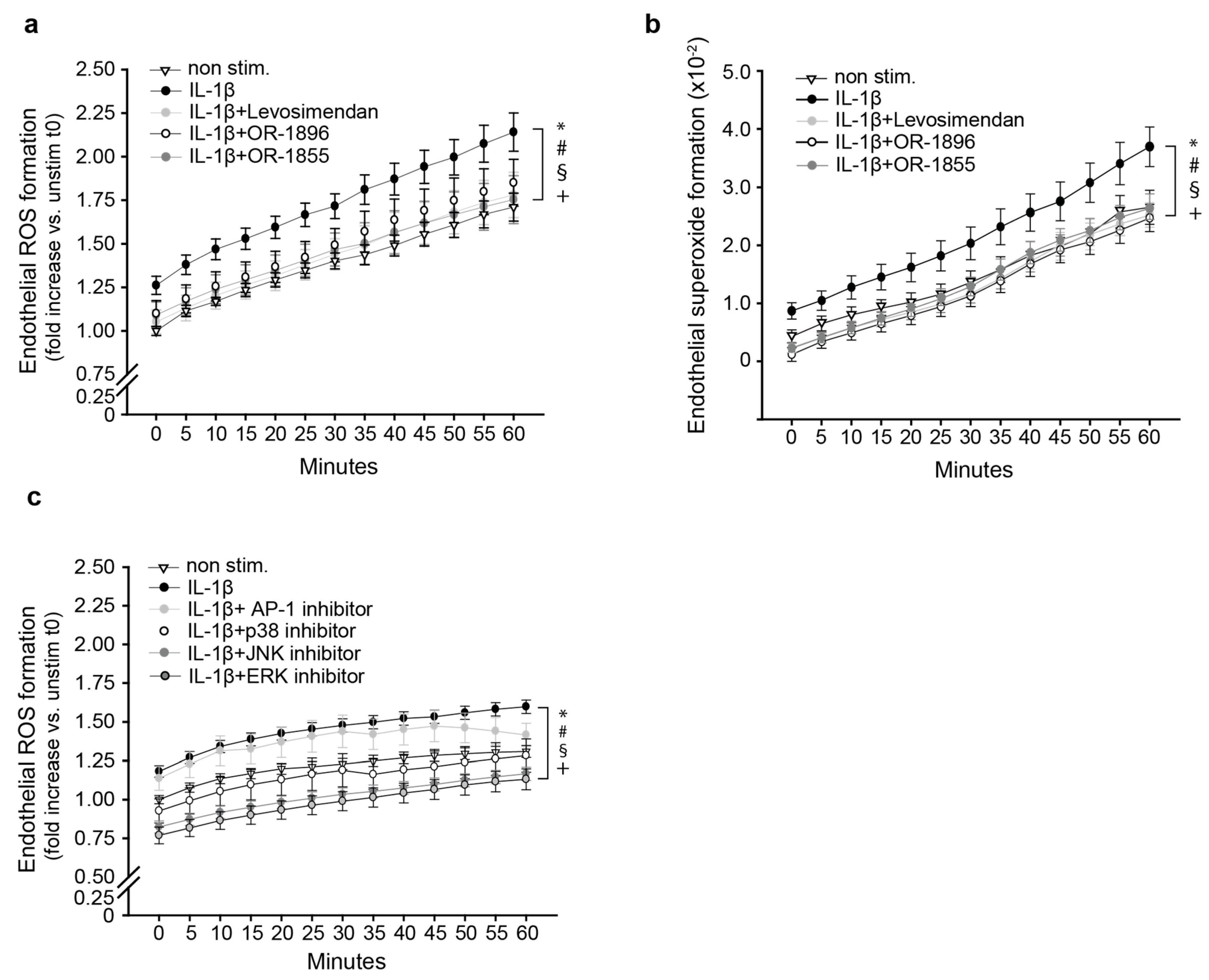
3.2. The Metabolites OR-1896 and OR-1855 Impair Inflammatory ROS Formation
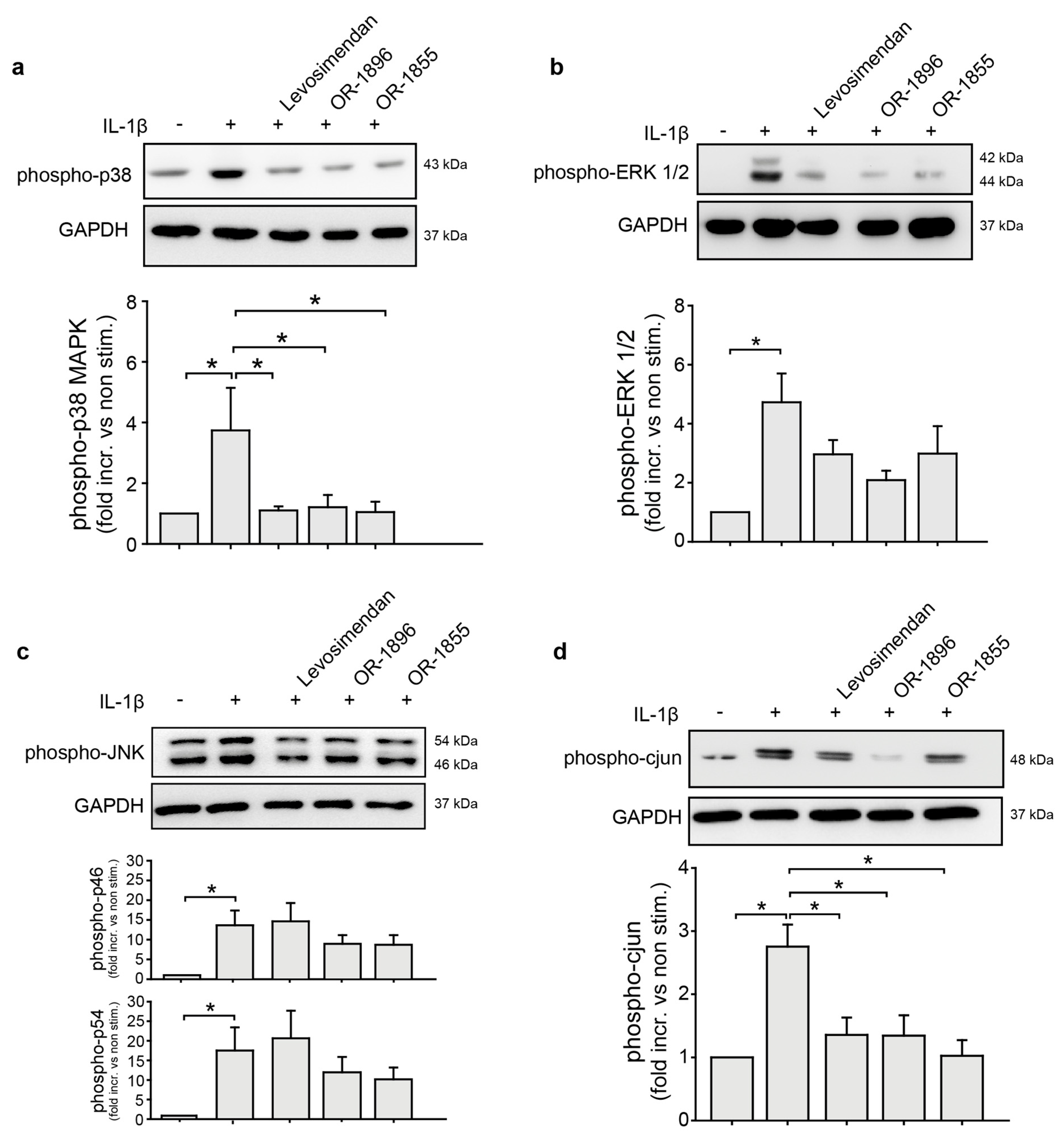
3.3. OR-1855 and OR-1896 Inhibit Pro-Inflammatory MAPK-Signalling in Endothelial Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kasikcioglu, H.A.; Cam, N. A review of levosimendan in the treatment of heart failure. Vasc. Health Risk Manag. 2006, 2, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, L.A.; Antila, S.; Pentikäinen, P.J. Pharmacokinetics and pharmacodynamics of intravenous inotropic agents. Clin. Pharmacokinet. 2004, 43, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Yokoshiki, H.; Katsube, Y.; Sunagawa, M.; Sperelakis, N. Levosimendan, a novel Ca2+ sensitizer, activates the glibenclamide-sensitive K+ channel in rat arterial myocytes. Eur. J. Pharm. 1997, 333, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Usta, C.; Eksert, B.; Gölbasi, I.; Bigat, Z.; Ozdem, S.S. The role of potassium channels in the vasodilatory effect of levosimendan in human internal thoracic arteries. Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2006, 30, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Antila, S.; Sundberg, S.; Lehtonen, L.A. Clinical pharmacology of levosimendan. Clin. Pharmacokinet. 2007, 46, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Antila, S.; Huuskonen, H.; Nevalainen, T.; Kanerva, H.; Vanninen, P.; Lehtonen, L. Site dependent bioavailability and metabolism of levosimendan in dogs. Eur. J. Pharm. Sci. 1999, 9, 85–91. [Google Scholar] [CrossRef]
- Erdei, N.; Papp, Z.; Pollesello, P.; Edes, I.; Bagi, Z. The levosimendan metabolite OR-1896 elicits vasodilation by activating the K(ATP) and BK(Ca) channels in rat isolated arterioles. Br. J. Pharm. 2006, 148, 696–702. [Google Scholar] [CrossRef] [Green Version]
- Gödény, I.; Pollesello, P.; Edes, I.; Papp, Z.; Bagi, Z. Levosimendan and its metabolite OR-1896 elicit KATP channel-dependent dilation in resistance arteries in vivo. Pharmacol. Rep. PR 2013, 65, 1304–1310. [Google Scholar] [CrossRef] [Green Version]
- Ørstavik, Ø.; Manfra, O.; Andressen, K.W.; Andersen, G.; Skomedal, T.; Osnes, J.B.; Levy, F.O.; Krobert, K.A. The inotropic effect of the active metabolite of levosimendan, OR-1896, is mediated through inhibition of PDE3 in rat ventricular myocardium. PLoS ONE 2015, 10, e0115547. [Google Scholar] [CrossRef] [Green Version]
- Krychtiuk, K.A.; Kaun, C.; Hohensinner, P.J.; Stojkovic, S.; Seigner, J.; Kastl, S.P.; Zuckermann, A.; Eppel, W.; Rauscher, S.; de Martin, R.; et al. Anti-thrombotic and pro-fibrinolytic effects of levosimendan in human endothelial cells in vitro. Vasc. Pharmacol. 2017, 90, 44–50. [Google Scholar] [CrossRef]
- Revermann, M.; Schloss, M.; Mieth, A.; Babelova, A.; Schröder, K.; Neofitidou, S.; Buerkl, J.; Kirschning, T.; Schermuly, R.T.; Hofstetter, C.; et al. Levosimendan attenuates pulmonary vascular remodeling. Intensive Care Med. 2011, 37, 1368–1377. [Google Scholar] [CrossRef]
- Grossini, E.; Molinari, C.; Caimmi, P.P.; Uberti, F.; Vacca, G. Levosimendan induces NO production through p38 MAPK, ERK and Akt in porcine coronary endothelial cells: Role for mitochondrial K(ATP) channel. Br. J. Pharm. 2009, 156, 250–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannell, H.K.; Pircher, J.; Chaudhry, D.I.; Alig, S.K.; Koch, E.G.; Mettler, R.; Pohl, U.; Krötz, F. ARNO regulates VEGF-dependent tissue responses by stabilizing endothelial VEGFR-2 surface expression. Cardiovasc. Res. 2012, 93, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heun, Y.; Pogoda, K.; Anton, M.; Pircher, J.; Pfeifer, A.; Woernle, M.; Ribeiro, A.; Kameritsch, P.; Mykhaylyk, O.; Plank, C.; et al. HIF-1alpha Dependent Wound Healing Angiogenesis In Vivo Can Be Controlled by Site-Specific Lentiviral Magnetic Targeting of SHP-2. Mol. Ther. 2017, 25, 1616–1627. [Google Scholar] [CrossRef] [Green Version]
- Krötz, F.; Engelbrecht, B.; Buerkle, M.A.; Bassermann, F.; Bridell, H.; Gloe, T.; Duyster, J.; Pohl, U.; Sohn, H.Y. The tyrosine phosphatase, SHP-1, is a negative regulator of endothelial superoxide formation. J. Am. Coll. Cardiol 2005, 45, 1700–1706. [Google Scholar] [CrossRef] [Green Version]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-González, R.; Pollesello, P.; Baluja, A.; Álvarez, J. Effects of Levosimendan on Inflammation and Oxidative Stress Pathways in a Lipopolysaccharide-Stimulated Human Endothelial Cell Model. Biol. Res. Nurs. 2019, 21, 466–472. [Google Scholar] [CrossRef]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef]
- Alem, M.M. Endothelial Dysfunction in Chronic Heart Failure: Assessment, Findings, Significance, and Potential Therapeutic Targets. Int. J. Mol. Sci. 2019, 20, 3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trepels, T.; Zeiher, A.M.; Fichtlscherer, S. The endothelium and inflammation. Endothel. J. Endothel. Cell Res. 2006, 13, 423–429. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Markousis-Mavrogenis, G.; Tromp, J.; Ouwerkerk, W.; Devalaraja, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.S.; van der Harst, P.; Lang, C.C.; et al. The clinical significance of interleukin-6 in heart failure: Results from the BIOSTAT-CHF study. Eur. J. Heart Fail. 2019, 21, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Munger, M.A.; Johnson, B.; Amber, I.J.; Callahan, K.S.; Gilbert, E.M. Circulating concentrations of proinflammatory cytokines in mild or moderate heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am. J. Cardiol. 1996, 77, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Chaar, D.; Dumont, B.; Vulesevic, B.; Neagoe, P.E.; Rakel, A.; Sirois, M.G.; White, M. Neutrophils pro-inflammatory and anti-inflammatory cytokine release in patients with heart failure and reduced ejection fraction. ESC Heart Fail. 2021, 8, 3855–3864. [Google Scholar] [CrossRef]
- Kyrzopoulos, S.; Adamopoulos, S.; Parissis, J.T.; Rassias, J.; Kostakis, G.; Iliodromitis, E.; Degiannis, D.; Kremastinos, D.T. Levosimendan reduces plasma B-type natriuretic peptide and interleukin 6, and improves central hemodynamics in severe heart failure patients. Int. J. Cardiol. 2005, 99, 409–413. [Google Scholar] [CrossRef]
- Navarri, R.; Lunghetti, S.; Cameli, M.; Mondillo, S.; Favilli, R.; Scarpini, F.; Puccetti, L. Neurohumoral improvement and torsional dynamics in patients with heart failure after treatment with levosimendan. Int. J. Cardiol. Heart Vasc. 2015, 7, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Parissis, J.T.; Karavidas, A.; Bistola, V.; Arapi, S.; Paraskevaidis, I.A.; Farmakis, D.; Korres, D.; Filippatos, G.; Matsakas, E.; Kremastinos, D.T. Effects of levosimendan on flow-mediated vasodilation and soluble adhesion molecules in patients with advanced chronic heart failure. Atherosclerosis 2008, 197, 278–282. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Wang, C.; Luo, T.; Lu, B.; Ma, H.; Zhou, Z.; Zhu, D.; Chi, G.; Ge, P.; Luo, Y. JNK Activation Contributes to Oxidative Stress-Induced Parthanatos in Glioma Cells via Increase of Intracellular ROS Production. Mol. Neurobiol. 2017, 54, 3492–3505. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Park, M.T.; Kim, M.J.; Suh, Y.; Kim, R.K.; Kim, H.; Lim, E.J.; Yoo, K.C.; Lee, G.H.; Kim, Y.H.; Hwang, S.G.; et al. Novel signaling axis for ROS generation during K-Ras-induced cellular transformation. Cell Death Differ. 2014, 21, 1185–1197. [Google Scholar] [CrossRef] [Green Version]
- Besirli, C.G.; Johnson, E.M. JNK-independent activation of c-Jun during neuronal apoptosis induced by multiple DNA-damaging agents. J. Biol. Chem. 2003, 278, 22357–22366. [Google Scholar] [CrossRef] [Green Version]
- Naumann, M.; Bech-Otschir, D.; Huang, X.; Ferrell, K.; Dubiel, W. COP9 signalosome-directed c-Jun activation/stabilization is independent of JNK. J. Biol. Chem. 1999, 274, 35297–35300. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, J.M. Activation of the AP-1 transcription factor by inflammatory cytokines of the TNF family. Gene Expr. 1999, 7, 217–231. [Google Scholar] [PubMed]
- Yan, W.; Zhao, K.; Jiang, Y.; Huang, Q.; Wang, J.; Kan, W.; Wang, S. Role of p38 MAPK in ICAM-1 expression of vascular endothelial cells induced by lipopolysaccharide. Shock 2002, 17, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.M.; Luo, S.F.; Hsieh, H.L.; Chi, P.L.; Lin, C.C.; Wu, C.C.; Hsiao, L.D. Interleukin-1beta induces ICAM-1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: Involvement of ERK, JNK, AP-1, and NF-kappaB. J. Cell. Physiol. 2010, 224, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Heun, Y.; Pircher, J.; Czermak, T.; Bluem, P.; Hupel, G.; Bohmer, M.; Kraemer, B.F.; Pogoda, K.; Pfeifer, A.; Woernle, M.; et al. Inactivation of the tyrosine phosphatase SHP-2 drives vascular dysfunction in Sepsis. EBioMedicine 2019, 42, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of Inflammation in Heart Failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef]
- Yang, O.; Li, J.; Kong, J. The Endothelium as a Target for the Treatment of Heart Failure. Cell Biochem. Biophys. 2015, 72, 751–756. [Google Scholar] [CrossRef]
- Leppikangas, H.; Järvelä, K.; Sisto, T.; Maaranen, P.; Virtanen, M.; Lehto, P.; Karlsson, S.; Kööbi, T.; Lindgren, L. Preoperative levosimendan infusion in combined aortic valve and coronary bypass surgery. Br. J. Anaesth. 2011, 106, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.H.; Leimberger, J.D.; van Diepen, S.; Meza, J.; Wang, A.; Jankowich, R.; Harrison, R.W.; Hay, D.; Fremes, S.; Duncan, A.; et al. Levosimendan in Patients with Left Ventricular Dysfunction Undergoing Cardiac Surgery. N. Engl. J. Med. 2017, 376, 2032–2042. [Google Scholar] [CrossRef] [PubMed]
- Squiccimarro, E.; Labriola, C.; Malvindi, P.G.; Margari, V.; Guida, P.; Visicchio, G.; Kounakis, G.; Favale, A.; Dambruoso, P.; Mastrototaro, G.; et al. Prevalence and Clinical Impact of Systemic Inflammatory Reaction After Cardiac Surgery. J. Cardiothorac. Vasc. Anesth. 2019, 33, 1682–1690. [Google Scholar] [CrossRef]
- Day, J.R.; Taylor, K.M. The systemic inflammatory response syndrome and cardiopulmonary bypass. Int. J. Surg. 2005, 3, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paparella, D.; Yau, T.M.; Young, E. Cardiopulmonary bypass induced inflammation: Pathophysiology and treatment. An update. Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2002, 21, 232–244. [Google Scholar] [CrossRef] [Green Version]
- Laffey, J.G.; Boylan, J.F.; Cheng, D.C. The systemic inflammatory response to cardiac surgery: Implications for the anesthesiologist. Anesthesiology 2002, 97, 215–252. [Google Scholar] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kipka, H.; Schaflinger, R.; Tomasi, R.; Pogoda, K.; Mannell, H. The Effects of the Levosimendan Metabolites OR-1855 and OR-1896 on Endothelial Pro-Inflammatory Responses. Biomedicines 2023, 11, 918. https://doi.org/10.3390/biomedicines11030918
Kipka H, Schaflinger R, Tomasi R, Pogoda K, Mannell H. The Effects of the Levosimendan Metabolites OR-1855 and OR-1896 on Endothelial Pro-Inflammatory Responses. Biomedicines. 2023; 11(3):918. https://doi.org/10.3390/biomedicines11030918
Chicago/Turabian StyleKipka, Hannah, Rebecca Schaflinger, Roland Tomasi, Kristin Pogoda, and Hanna Mannell. 2023. "The Effects of the Levosimendan Metabolites OR-1855 and OR-1896 on Endothelial Pro-Inflammatory Responses" Biomedicines 11, no. 3: 918. https://doi.org/10.3390/biomedicines11030918