

NRF2 and Bip Interconnection Mediates Resistance to the Organometallic Ruthenium-Cymene Bisdemethoxycurcumin Complex Cytotoxicity in Colon Cancer Cells

 , and
, and 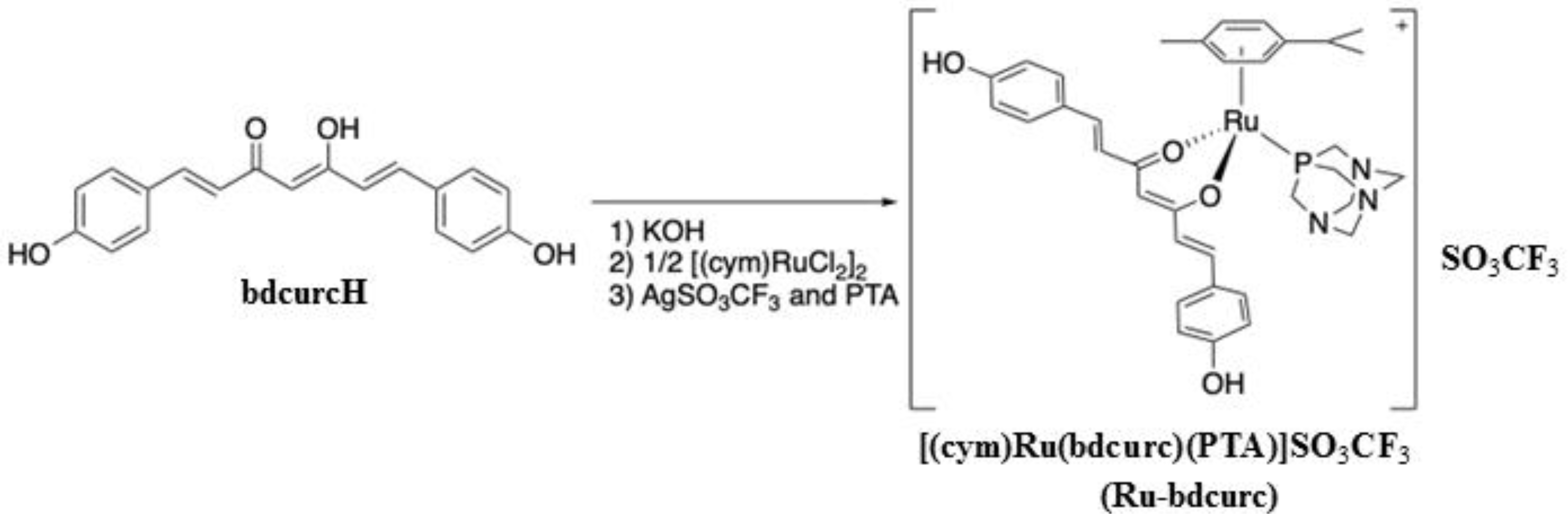{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of [(cym)Ru(bdcurc)(PTA)]SO3CF3 Complex (Ru-bdcurc)
2.2. Cell Culture and Reagents
2.3. Cell Viability Assay
2.4. Proliferation Assay (XTT)
2.5. Western Blotting
2.6. Antibodies
2.7. RNA Extraction and Semiquantitative Reverse Transcription (RT)-Polymerase Chain Reaction (PCR) Analysis
2.8. Statistical Analysis
3. Results
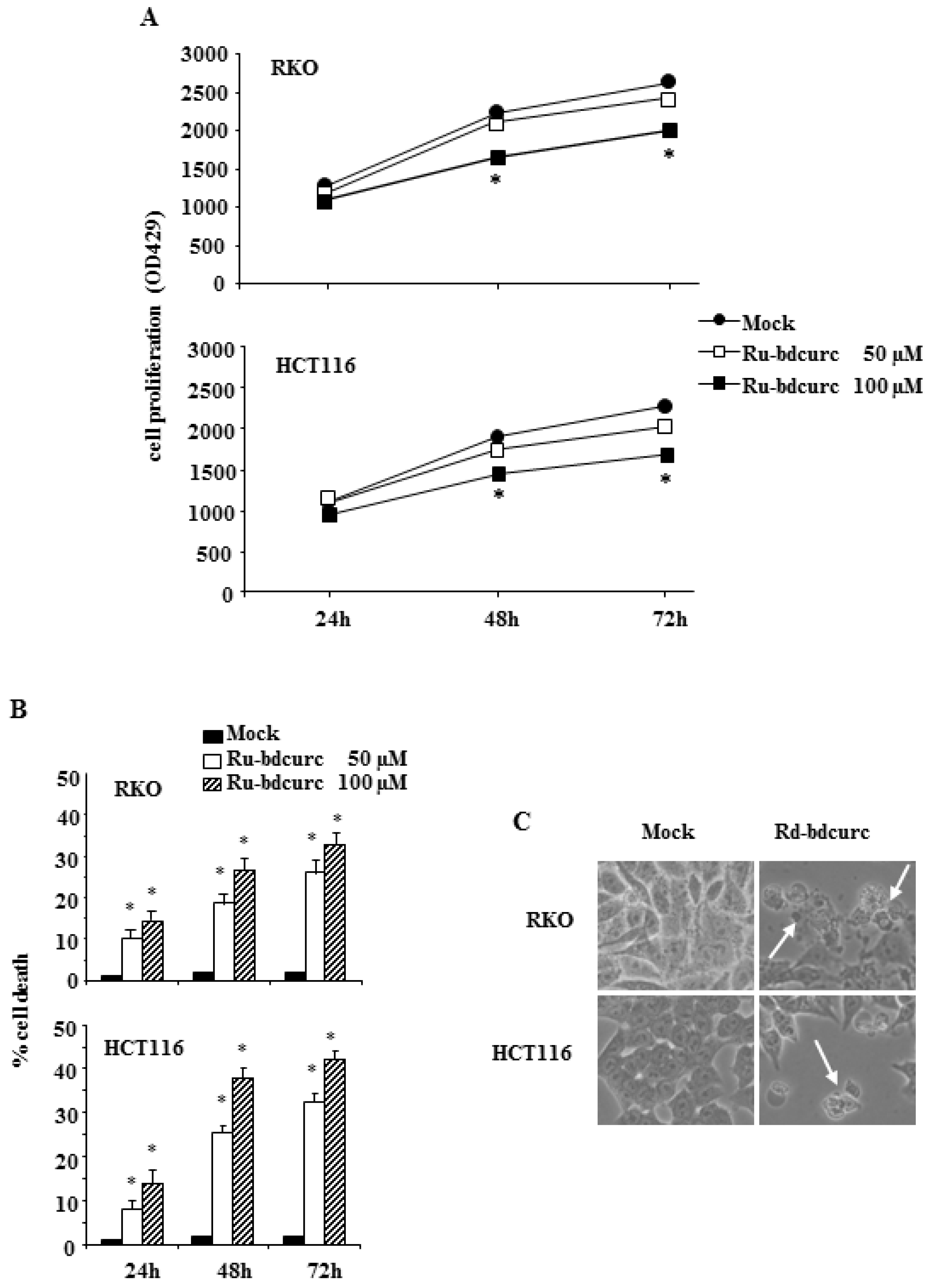
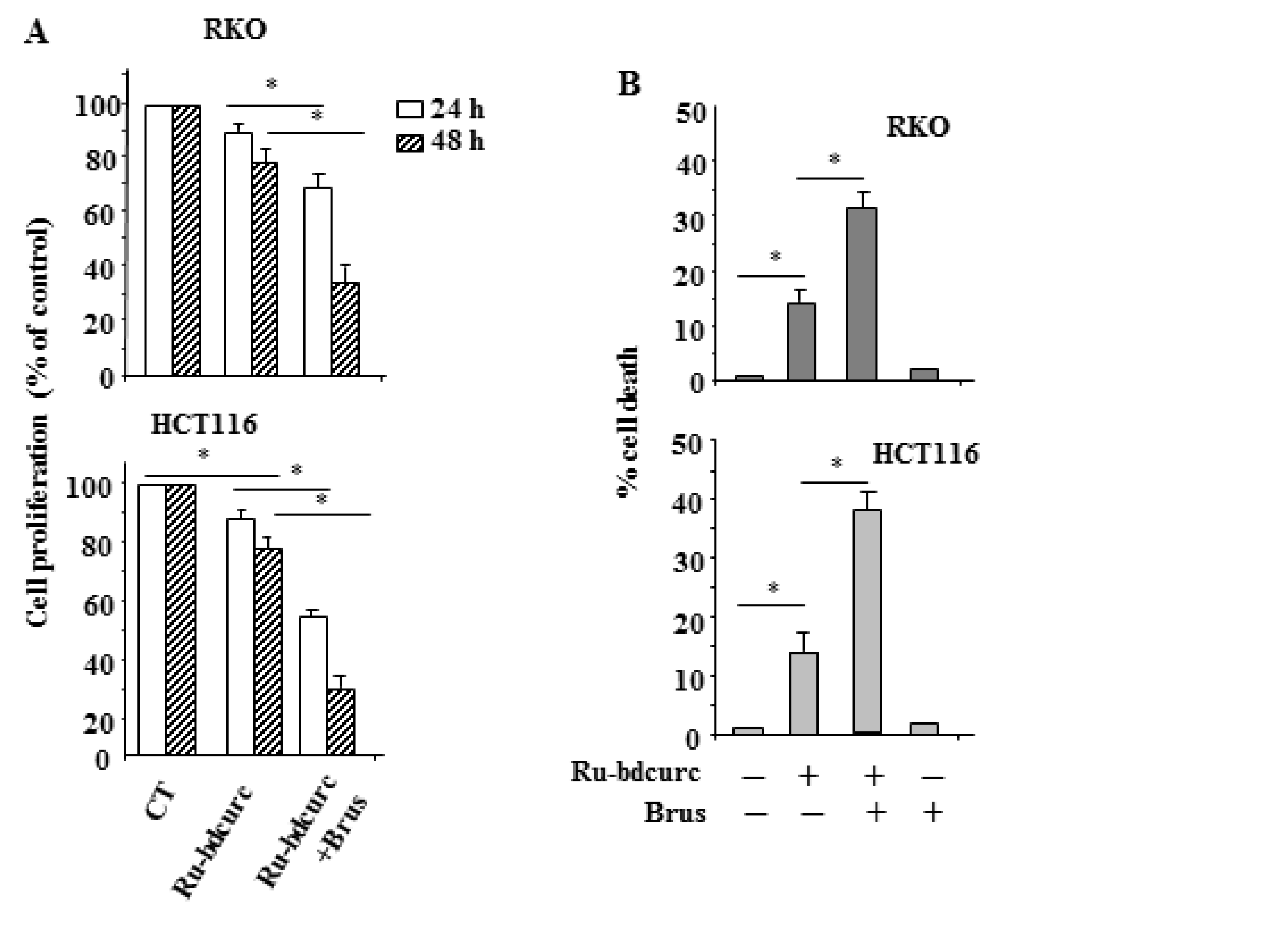
3.1. Ru-bdcurc Compound Induces Cell Death in Colon Cancer Cells
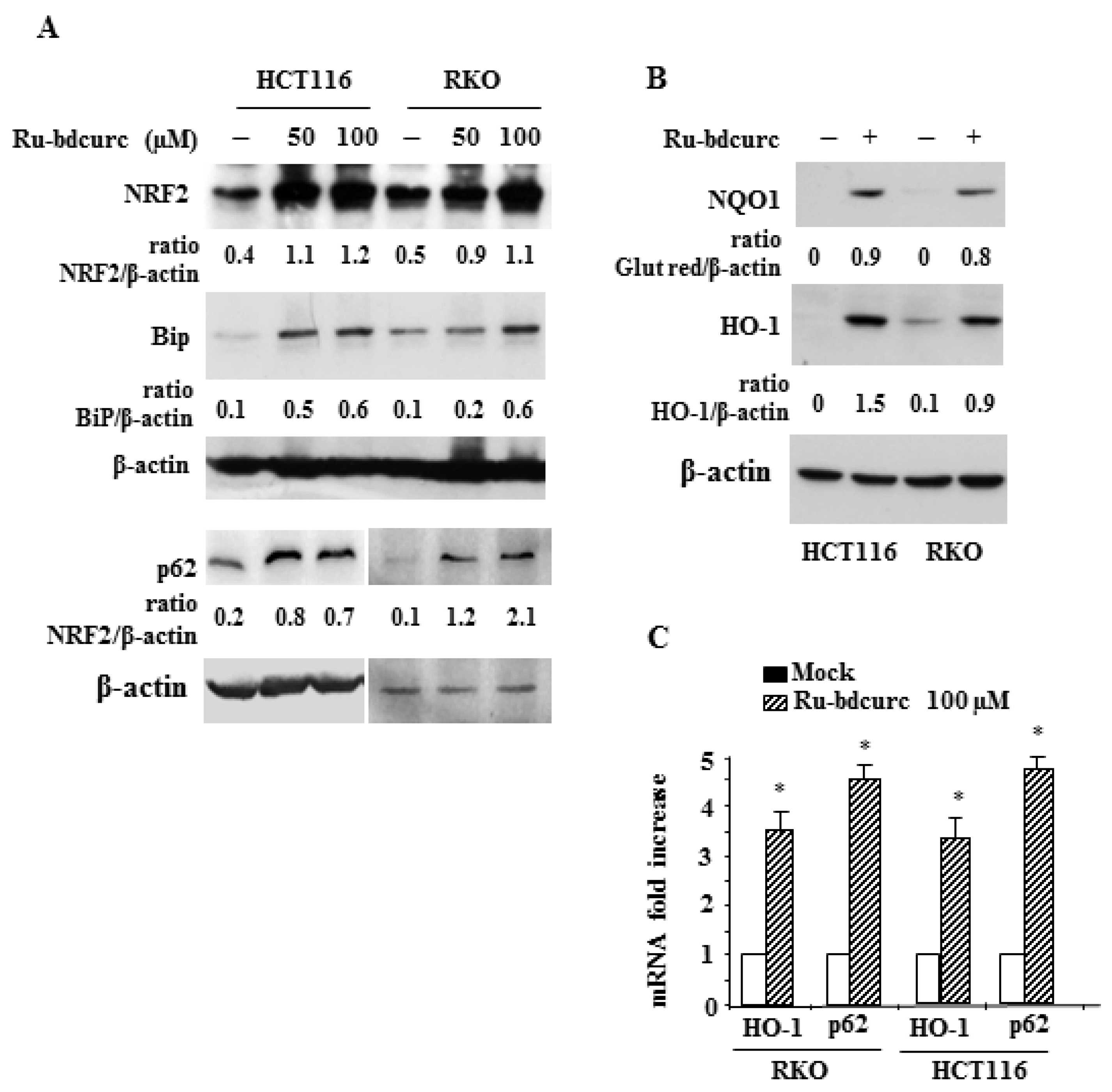
3.2. Induction of NRF2 and BiP Expression in Response to Rubdcurc Treatment
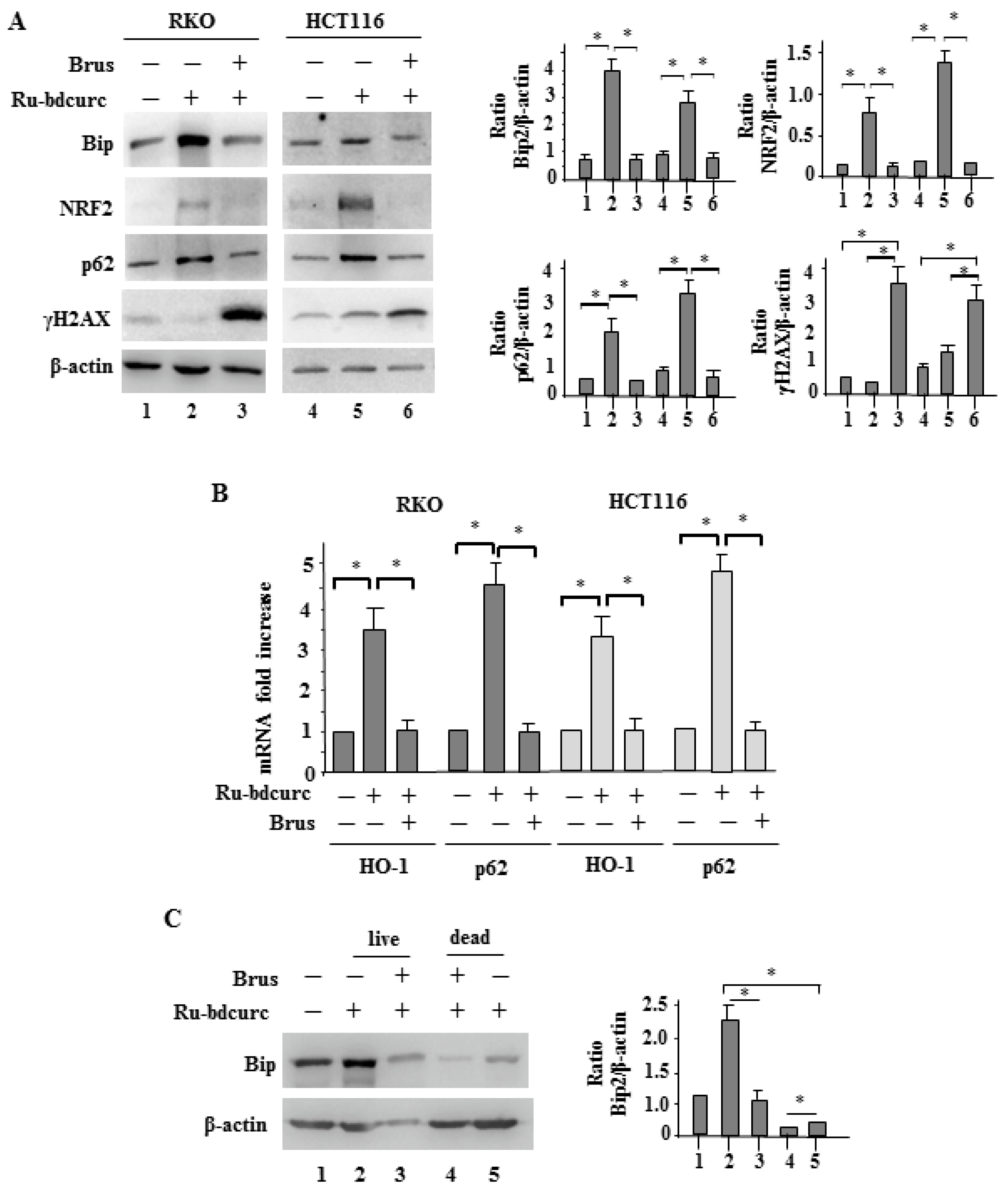
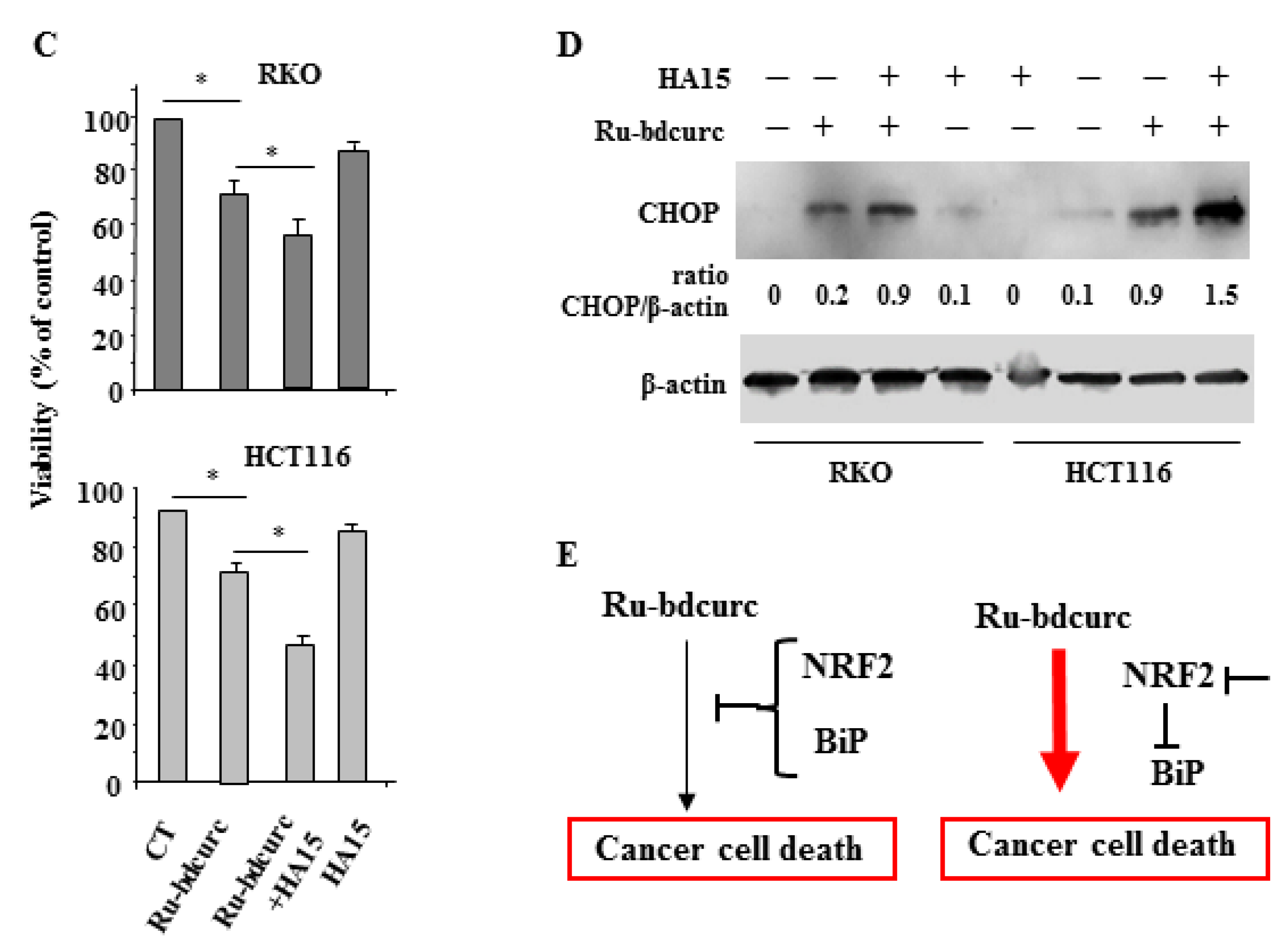
3.3. NRF2 and BiP Pathways as Death Resistant Mechanisms to Ru-bdcurc Cytotocicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Lu, J.J.; Ding, J. Natural Products in Cancer Therapy: Past, Present and Future. Nat. Prod. Bioprospect. 2021, 11, 5–13. [Google Scholar] [CrossRef]
- Gavrilas, L.I.; Cruceriu, D.; Mocan, A.; Loghin, F.; Miere, D.; Balacescu, O. Plat-derived bioactive compounds in colorectal cancer: Insights from combined regimens with conventional chemotherapy to overcome drug-resistance. Biomedicines 2022, 10, 1948. [Google Scholar] [CrossRef]
- Mansouri, K.; Rasoulpoor, S.; Daneshkhah, A.; Abolfatthi, S.; Salari, N.; Mohammadi, M.; Rasoulpoor, S.; Shabani, S. Clinical effects of curcumin in enhancing cancer therapy: A systematic review. BMC Cancer 2020, 20, 791. [Google Scholar] [CrossRef]
- Raduly, F.M.; Raditoiu, V.; Raditoiu, A.; Purcar, V. Curcumin: Modern applications for a versatile additive. Coatings 2021, 11, 519. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin: Miniperspective. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Karthikeyan, A.; Senthil, N.; Min, T. Nanocurcumin: A promising candidate for therapeutic applications. Front. Pharmacol. 2020, 11, 487. [Google Scholar] [CrossRef]
- Wanninger, S.; Lorenz, V.; Subhan, A.; Edelmann, F.T. Metal complexes of curcumin–synthetic strategies, structures and medicinal applications. Chem. Soc. Rev. 2015, 44, 4986–5002. [Google Scholar] [CrossRef] [Green Version]
- Amalraj, A.; Pius, A.; Gopi, S.; Gopi, S. Biological activities of curcuminoids, other biomolecules from turmeric and their derivatives. J. Tradit. Complement. Med. 2017, 7, 205–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Khan, A.R.; Fu, M.; Zhai, Y.; Yu, A.; Zhai, G. The progresses in curcuminoids-based metal complexes: Especially in cancer therapy. Future Med. Chem. 2019, 11, 1035–1056. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Pan, H.; Wang, J.; Wang, T.; Huo, X.; Ma, Y.; Lu, Z.; Sun, B.; Jiang, H. Unfolded protein response in colorectal cancer. Cell Biosc. 2021, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Torrente, L.; Maan, G.; Rezig, A.O.; Quinn, J.; Jackson, A.; Grilli, A.; Casares, L.; Zhang, Y.; Kulesskiv, E.; Saarela, J.; et al. High NRF2 levels correlate with poor prognosis in colorectal cancer patients and with sensitivity to the kinase inhibitor AT9283 in vitro. Biomolecules 2020, 10, 1365. [Google Scholar] [CrossRef]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Chadwick, S.R.; Lajoie, P. Endoplasmic reticulum stress coping mechanisms and lifespan regulation in health and diseases. Front. Cell Dev. Biol. 2019, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lee, A.S. Stress induction of GRP78/BiP and its role in cancer. Curr. Mol. Med. 2006, 6, 45–54. [Google Scholar] [CrossRef]
- Benedetti, R.; Gilardini Montani, M.S.; Romeo, M.A.; Arena, A.; Santarelli, R.; D’Orazi, G.; Cirone, M. Role of UPR sensor activation in cell death-survival decision of colon cancer cells stressed by DPE treatment. Biomedicines 2021, 9, 1262. [Google Scholar] [CrossRef]
- Roller, C.; Maddalo, D. The Molecular Chaperone GRP78/ BiP in the Development of Chemoresistance: Mechanism and Possible Treatment. Front. Pharmacol. 2013, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Mhaidat, N.M.; Alzoubi, K.H.; Khabour, O.F.; Banihani, M.N.; Al-Balas, Q.A. GRP78 regulates sensitivity of human colorectal cancer cells to DNA targeting agents. Cytotechnology 2016, 68, 459–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 axis in cancer cell growth and chemoresistance. Oxid. Med. Cell Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeekpuds, P.; Kukongviriyapan, V.; Senggunprai, L.; Sripa, B.; Prawan, A. Suppression of NAD(P)H-quinone oxidoreductase 1 enhanced the susceptibility of cholangiocarcinoma cells to chemotherapeutic agents. J. Exp. Clin. Cancer Res. 2014, 33, 11. [Google Scholar] [CrossRef] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjottem, E.; Bowitz Larsen, K.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, P.; Saito, T.; Komatsu, M. p62/SQSTM1: “Jack of all trades” in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [Green Version]
- No, J.H.; Kim, Y.B.; Song, Y.S. Targeting Nrf2 signaling to combat chemoresistance. J. Cancer Prev. 2014, 2, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Pettinari, R.; Marchetti, F.; Condello, F.; Pettinari, C.; Lupidi, G.; Scopelliti, R.; Mukhopadhyay, S.; Riedel, T.; Dyson, P.J. Ruthenium(II)−Arene RAPTA type complexes containing curcumin and bisdemethoxycurcumin display potent and selective anticancer activity. Organometallics 2014, 33, 3709–3715. [Google Scholar] [CrossRef]
- Caruso, F.; Pettinari, R.; Rossi, M.; Monti, E.; Gariboldi, M.B.; Marchetti, F.; Pettinari, C.; Caruso, A.; Ramani, M.V.; Subbaraju, G.V.J. The in vitro antitumor activity of arene-ruthenium(II) curcuminoid complexes improves when decreasing curcumin polarity. Inorg. Biochem. 2016, 162, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T.J.; Sava, G.; Dyson, P.J. In vitro and in vivo evaluation of ruthenium (II)− arene PTA complexes. J. Med. Chem. 2005, 48, 4161–4171. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.Q.; Lin, Z.X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol Provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity: Implications for therapeutic targeting of Nrf2. Free Rad. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garufi, A.; Traversi, G.; Gilardini Montani, M.S.; D’Orazi, V.; Pistritto, G.; Cirone, M.; D’Orazi, G. Reduced chemotherapeutic sensitivity in high glucose condition: Implication of antioxidant response. Oncotarget 2019, 10, 4691–4702. [Google Scholar] [CrossRef] [Green Version]
- Cerezo, M.; Lehraiki, A.; Millet, A.; Rouaud, F.; Plaisant, M.; Jaune, E.; Botton, T.; Ronco, C.; Abbe, P.; Amdouni, H.; et al. Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer Cell 2016, 30, 183. [Google Scholar] [CrossRef]
- Wu, J.; Wu, Y.; Lian, X. Targeted inhibition of GRP78 by HA15 promotes apoptosis of lung cancer cells accompanied by ER stress and autophagy. Biol. Open 2020, 9, bio053298. [Google Scholar] [CrossRef]
- Garufi, A.; Pistritto, G.; D’Orazi, V.; Cirone, M.; D’Orazi, G. The impact of NRF2 inhibition on drug-induced colon cancer cell death and p53 activity: A pilot study. Biomolecules 2022, 12, 461. [Google Scholar] [CrossRef]
- Garufi, A.; Giorno, E.; Gilardini Montani, M.S.; Pistritto, G.; Crispini, A.; Cirone, M.; D’Orazi, G. P62/SQSTM1/Keap1/NRF2 axis reduces cancer cells death-sensitivity in response to Zn(II)-curcumin complex. Biomolecules 2021, 11, 348. [Google Scholar] [CrossRef]
- Dong, D.; Ni, M.; Li, J.; Xiong, S.; Ye, W.; Virrey, J.J.; Mao, C.; Ye, R.; Wang, M.; Pen, L.; et al. Critical role of the stress chaperone GRP/78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008, 68, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Valente, D.; Gentileschi, M.P.; Guerrisi, A.; Bruzzanti, V.; Morrone, A.; Soddu, S.; Verdina, A. Factors to consider for the correct use of gammaH2AX in the evaluation of DNA double-strand breaks damage caused by ionixing radiation. Cancers 2022, 14, 6204. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Cirone, M. Mutant p53 and cellular stress pathways: A criminal alliance that promotes cancer progression. Cancers 2019, 11, 614. [Google Scholar] [CrossRef] [Green Version]
- Cirone, M.; D’Orazi, G. NRF2 in cancer: Cross-talk with oncogenic pathways and involvement in gammaherpesvirus-driven carcinogenesis. Int. J. Mol. Sci. 2023, 24, 595. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Yao, Y.; Yu, S.; Guo, H.; Han, L.; Wang, W.; Tian, T.; Hao, Y.; Liu, Z.; Nan, K.; et al. Clinicopathologic significance of CXCR4 and Nrf2 in colorectal cancer. J. Biomed. Res. 2013, 27, 283–290. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, W.I.; Bae, J.H.; Cho, M.K.; Lee, S.H.; Nam, H.S.; Choi, I.H.; Cho, S.W. Overexpression of Nrf2 promotes colon cancer progression via ERK and A and AKT signaling pathways. Ann. Sur. Treat. 2020, 98, 159–167. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Cecere, N.; Granato, M.; Romeo, M.A.; Falcinelli, L.; Ciciarelli, U.; D’Orazi, G.; Faggioni, A.; Cirone, M. Mutant p53, stabilized by its interplay with HSP90, activates a positivr feedback loop between NRF2 and p62 that induces chemo-resistance to Apigenin in pancreatic cancer cells. Cancers 2019, 11, 703. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Garufi, A.; Cirone, M. Nuclear factor erythroid (NF-E2) p45-related factor 2 interferes with homeodomain-interacting protein kinase 2/p53 activity to impair solid tumors chemosensitivity. IUBMB Life 2020, 72, 1634–1639. [Google Scholar] [CrossRef]
- Garufi, A.; Baldari, S.; Pettinari, R.; Gilardini Montani, M.S.; D’Orazi, V.; Pistritto, G.; Crispini, E.; Giorno, E.; Toietta, G.; Marchetti, F.; et al. A ruthenium(II) curcumin compound modulates NRF2 expression balancing the cell death/survival outcome in both wild-type and mutant p53-carrying cancer cells. J. Exp. Clin. Cancer Res. 2020, 39, 122. [Google Scholar] [CrossRef]
- Na, H.K.; Surth, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Rad. Biol. Ther. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 induction in cancer progression: A matter of cell adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lu, C.; Cao, Y.; Luo, Y.; Bao, R.; Yan, S.; Xue, M.; Zhu, F.; Wang, Z.; Duan, L. Imatinib induces H2AX phosphorylation and apoptosis in chronic myelogenous leukemia cells in vitro via caspase-3/Mst1 pathway. Acta Pharmacol. Sin. 2012, 33, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Pandita, R.K.; Eskiocak, U.; Ly, P.; Kaisani, A.; Kumar, R.; Cornelius, C.; Wright, W.E.; Pandita, T.K.; Shay, J.W. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc. Natl. Acad. Sci. USA 2012, 109, E2949–E2955. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.S.; Deeni, Y. NRF2 inhibition causes repression of ATM and ATR expression leading to aberrant DNA Damage Response. Biodiscovery 2015, 15, 1. [Google Scholar] [CrossRef] [Green Version]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernandez-Cardenas, L.P.; Blackwell, T.K. Cysteine sulfenylation directs IRE-1 to activate the SKN-1/Nrf2 antioxidant response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Bahar, E.; Kim, J.Y.; Yoon, H. Chemotherapy resistance through endoplasmic reticulum stress-dependent signaling. Cancers 2019, 11, 338. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Zheng, Z.; Hu, S.; Ru, X.; Fan, Z.; Qiu, L.; Zhang, Y. Unification of opposites between two antioxidant transcription factors Nrf1 and Nrf2 in mediating distinct cellular responses to the endoplasmic reticulum stressor Tunicamycin. Antioxidants 2020, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Cirone, M. Interconnected adaptive responses: A way out for cancer cells to avoid cellular demise. Cancers 2022, 14, 2780. [Google Scholar] [CrossRef]
- Oija, R.; Amaravadi, R.K. Targeting the unfolded protein response in cancer. Pharmacol. Res. 2017, 120, 258–266. [Google Scholar] [CrossRef]
- Walczak, A.; Gradzik, K.; Kabzinski, J.; Przybylowska-Sygut, K.; Majsterek, I. The role of the ER-induced UPR pathway and the efficacy of its inhibitors in the inhibition of tumor progression. Oxid. Med. Cell Longev. 2019, 2019, 5729710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.R.; Chang, C.H.; Hsu, C.F.; Tsai, M.J.; Cheng, H.; Leong, M.K.; Chen, J.C.; Weng, C.F. Natural compounds as potential adjuvants to cancer therapy: Preclinical evidence. Br. J. Pharmacol. 2020, 177, 1409–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stohs, S.S.; Chen, O.; Ray, S.D.; Ji, J.; Bucci, L.R.; Preuss, H.G. Highly Bioavailable Forms of Curcumin and Promising Avenues for Curcumin-Based Research and Application. Molecules 2020, 25, 1397. [Google Scholar] [CrossRef] [Green Version]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure–activity relationships for ruthenium and osmium anticancer agents–towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
- Tremlett, W.D.J.; Goodman, D.M.; Steel, T.R.; Kumar, S.; Wieczorek-Błauz, A.; Walsh, F.P.; Sullivan, M.P.; Hanif, M.; Hartinger, C.G. Design concepts of half-sandwich organoruthenium anticancer agents based on bidentate bioactive ligands. Coord. Chem. Rev. 2021, 445, 213950. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; van Beijnum, J.R.; Casini, A.; Nazarov, A.A.; Wagnieres, G.; van den Bergh, H.; Dyson, P.J.; Griffioen, A.W. Organometallic ruthenium (II) arene compounds with antiangiogenic activity. J. Med. Chem. 2011, 54, 3895–3902. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A.; Hartinger, C.G.; Dyson, P.J. The ruthenium (II)–arene compound RAPTA-C induces apoptosis in EAC cells through mitochondrial and p53–JNK pathways. J. Biol. Inorg. Chem. 2008, 13, 1149–1155. [Google Scholar] [CrossRef]
- Bonfili, L.; Pettinari, R.; Cuccioloni, M.; Cecarini, V.; Mozzicafreddo, M.; Angeletti, M.; Lupidi, G.; Marchetti, F.; Pettinari, C.; Eleuteri, A.M. Arene–RuII Complexes of Curcumin Exert Antitumor Activity via Proteasome Inhibition and Apoptosis Induction. ChemMedChem 2012, 7, 2010–2020. [Google Scholar] [CrossRef]
- Caruso, F.; Rossi, M.; Benson, A.; Opazo, C.; Freedman, D.; Monti, E.; Gariboldi, M.B.; Shaulky, J.; Marchetti, F.; Pettinari, R.; et al. Ruthenium−Arene Complexes of Curcumin: X-Ray and Density Functional Theory Structure, Synthesis, and Spectroscopic Characterization, in Vitro Antitumor Activity, and DNA Docking Studies of (p-Cymene)Ru(curcuminato)chloro. J. Med. Chem. 2012, 55, 1072–1081. [Google Scholar] [CrossRef]
- Pagliaricci, N.; Pettinari, R.; Marchetti, F.; Pettinari, C.; Cappellacci, L.; Tombesi, A.; Cuccioloni, M.; Hadijid, M.; Dyson, P.J. Potent and selective anticancer activity of half-sandwich ruthenium and osmium complexes with modified curcuminoid ligands. Dalton Trans. 2022, 51, 13311–13321. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garufi, A.; Pettinari, R.; Marchetti, F.; Cirone, M.; D’Orazi, G. NRF2 and Bip Interconnection Mediates Resistance to the Organometallic Ruthenium-Cymene Bisdemethoxycurcumin Complex Cytotoxicity in Colon Cancer Cells. Biomedicines 2023, 11, 593. https://doi.org/10.3390/biomedicines11020593
Garufi A, Pettinari R, Marchetti F, Cirone M, D’Orazi G. NRF2 and Bip Interconnection Mediates Resistance to the Organometallic Ruthenium-Cymene Bisdemethoxycurcumin Complex Cytotoxicity in Colon Cancer Cells. Biomedicines. 2023; 11(2):593. https://doi.org/10.3390/biomedicines11020593
Chicago/Turabian StyleGarufi, Alessia, Riccardo Pettinari, Fabio Marchetti, Mara Cirone, and Gabriella D’Orazi. 2023. "NRF2 and Bip Interconnection Mediates Resistance to the Organometallic Ruthenium-Cymene Bisdemethoxycurcumin Complex Cytotoxicity in Colon Cancer Cells" Biomedicines 11, no. 2: 593. https://doi.org/10.3390/biomedicines11020593