
TSLP and HMGB1: Inflammatory Targets and Potential Biomarkers for Precision Medicine in Asthma and COPD
 , ,
, ,  , , , , , and
, , , , , and 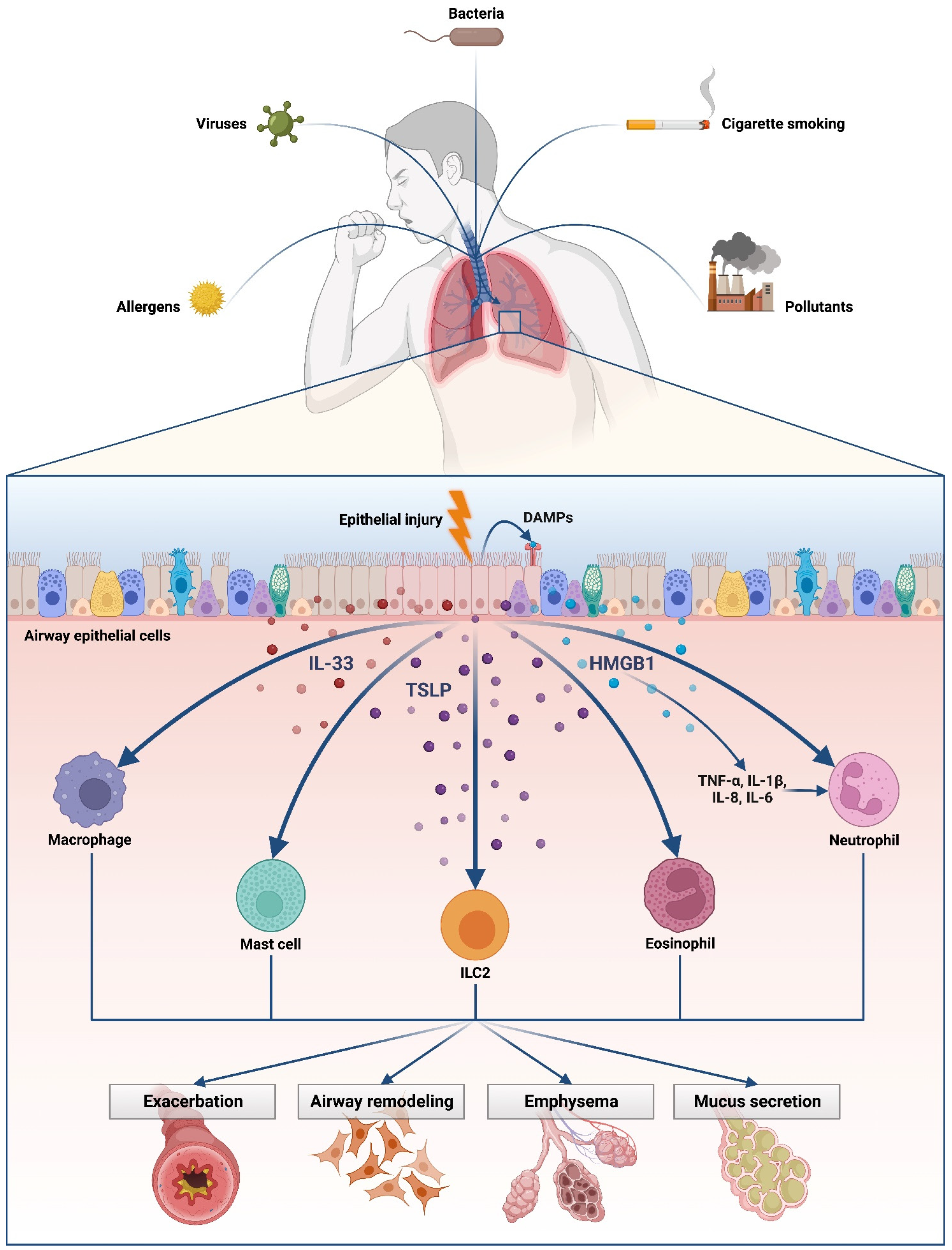{kind=link}
Abstract
:1. Introduction
2. HMGB1: Comparison in Asthma and in COPD
3. Role of Thymic Stromal Lymphopoietin (TSLP) in Asthma and COPD
4. HMGB1: Epigenetics and Omics Approaches
5. Summary and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hernandez Cordero, A.I.; Li, X.; Yang, C.X.; Yang, J.; MacIsaac, J.L.; Dever, K.; Kobor, M.S.; Milne, S.; van Eeden, S.F.; Shaipanich, T.; et al. Systemic and Airway Epigenetic Disruptions Are Associated with Health Status in COPD. Biomedicines 2023, 11, 134. [Google Scholar] [CrossRef]
- Ruaro, B.; Salton, F.; Braga, L.; Wade, B.; Confalonieri, P.; Volpe, M.C.; Baratella, E.; Maiocchi, S.; Confalonieri, M. The History and Mystery of Alveolar Epithelial Type II Cells: Focus on Their Physiologic and Pathologic Role in Lung. Int. J. Mol. Sci. 2021, 22, 2566. [Google Scholar] [CrossRef] [PubMed]
- Confalonieri, M.; Salton, F.; Ruaro, B.; Confaloniero, P.; Volpe, M.C. Alveolar Epithelial Type II Cells. In Encyclopedia of Respiratory Medicine, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 10–17. [Google Scholar] [CrossRef]
- Whetstone, C.E.; Ranjbar, M.; Omer, H.; Cusack, R.P.; Gauvreau, G.M. The Role of Airway Epithelial Cell Alarmins in Asthma. Cells 2022, 11, 1105. [Google Scholar] [CrossRef] [PubMed]
- Gauvreau, G.M.; Bergeron, C.; Boulet, L.P.; Cockcroft, D.W.; Côté, A.; Davis, B.E.; Leigh, R.; Myers, I.; O’Byrne, P.M.; Sehmi, R. Sounding the alarmins—The role of alarmin cytokines in asthma. Allergy 2022. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.A.; Fisher, A.J.; Borthwick, L.A. The Role of Epithelial Damage in the Pulmonary Immune Response. Cells 2021, 10, 2763. [Google Scholar] [CrossRef]
- Crystal, R.G.; Randell, S.H.; Engelhardt, J.; Voynow, J.; Sunday, M.E. Airway Epithelial Cells: Current Concepts and Challenges. Proc. Am. Thorac. Soc. 2008, 5, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, Z.; Huang, H.; Li, J.; Wang, Z.; Yu, Y.; Zhang, C.; Li, J.; Dai, H.; Wang, F.; et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. USA 2018, 115, 2407–2412. [Google Scholar] [CrossRef]
- Leiva-Juárez, M.M.; Kolls, J.K.; Evans, S.E. Lung epithelial cells: Therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2017, 11, 21–34. [Google Scholar] [CrossRef]
- Marchiando, A.M.; Graham, W.; Turner, J.R. Epithelial Barriers in Homeostasis and Disease. Annu. Rev. Pathol. 2010, 5, 119–144. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef]
- Adivitiya; Kaushik, M.S.; Chakraborty, S.; Veleri, S.; Kateriya, S. Mucociliary Respiratory Epithelium Integrity in Molecular Defense and Susceptibility to Pulmonary Viral Infections. Biology 2021, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Uppada, S.; Achkar, I.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2019, 9, 1942. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.; Wadsworth, S.; Dorscheid, D.; Man, S.P.; Sin, D.D. The airway epithelium: More than just a structural barrier. Ther. Adv. Respir. Dis. 2011, 5, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef]
- Pradeu, T.; Cooper, E.L. The danger theory: 20 years later. Front. Immunol. 2012, 3, 287. [Google Scholar] [CrossRef]
- Hiemstra, P.S.; McCray, P.B., Jr.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Lucchini, A.C.; Gachanja, N.N.; Rossi, A.G.; Dorward, D.A.; Lucas, C.D. Epithelial Cells and Inflammation in Pulmonary Wound Repair. Cells 2021, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Imbalzano, E.; Quartuccio, S.; Di Salvo, E.; Crea, T.; Casciaro, M.; Gangemi, S. Association between HMGB1 and asthma: A literature review. Clin. Mol. Allergy. 2017, 15, 12. [Google Scholar] [CrossRef]
- Yayan, J.; Rasche, K. Asthma and COPD: Similarities and Differences in the Pathophysiology, Diagnosis and Therapy. Adv. Exp. Med. Biol. 2016, 910, 31–38. [Google Scholar] [CrossRef]
- Cavone, L.; Cuppari, C.; Manti, S.; Grasso, L.; Arrigo, T.; Calamai, L.; Salpietro, C.; Chiarugi, A. Increase in the Level of Proinflammatory Cytokine HMGB1 in Nasal Fluids of Patients with Rhinitis and its Sequestration by Glycyrrhizin Induces Eosinophil Cell Death. Clin. Exp. Otorhinolaryngol. 2015, 8, 123–128. [Google Scholar] [CrossRef]
- Sukkar, M.B.; Wood, L.G.; Tooze, M.; Simpson, J.L.; McDonald, V.M.; Gibson, P.G.; Wark, P.A. Soluble RAGE is deficient in neutrophilic asthma and COPD. Eur. Respir. J. 2012, 39, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Brightling, C.; Greening, N. Airway inflammation in COPD: Progress to precision medicine. Eur. Respir. J. 2019, 54, 1900651. [Google Scholar] [CrossRef] [PubMed]
- Confalonieri, M.; Braga, L.; Salton, F.; Ruaro, B.; Confalonieri, P. COPD Definition: Is It Time to Incorporate Also the Concept of Lung Regeneration’s Failure? Am. J. Respir. Crit. Care Med. 2022. [Google Scholar] [CrossRef]
- Celli, B.; Fabbri, L.; Criner, G.; Martinez, F.J.; Mannino, D.; Vogelmeier, C.; Montes de Oca, M.; Papi, A.; Sin, D.D.; Han, M.K.; et al. Definition and Nomenclature of Chronic Obstructive Pulmonary Disease: Time for Its Revision. Am. J. Respir. Crit. Care Med. 2022, 206, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Sivapalan, P.; Bikov, A.; Jensen, J.U. Using Blood Eosinophil Count as a Biomarker to Guide Corticosteroid Treatment for Chronic Obstructive Pulmonary Disease. Diagnostics 2021, 11, 236. [Google Scholar] [CrossRef]
- Di Salvo, E.; Di Gioacchino, M.; Tonacci, A.; Casciaro, M.; Gangemi, S. Alarmins, COVID-19 and comorbidities. Ann. Med. 2021, 53, 777–785. [Google Scholar] [CrossRef]
- Gangemi, S.; Casciaro, M.; Trapani, G.; Quartuccio, S.; Navarra, M.; Pioggia, G.; Imbalzano, E. Association between HMGB1 and COPD: A Systematic Review. Mediators Inflamm. 2015, 2015, 164913. [Google Scholar] [CrossRef]
- Romani, M.; Rodman, T.C.; Vidali, G.; Bustin, M. Serological analysis of species specificity in the high mobility group chromosomal proteins. J. Biol. Chem. 1979, 254, 2918–2922. [Google Scholar] [CrossRef]
- Cuppari, C.; Manti, S.; Chirico, V.; Caruso, R.; Salpietro, V.; Giacchi, V.; Laganà, F.; Arrigo, T.; Salpietro, C.; Leonardi, S. Sputum high mobility group box-1 in asthmatic children: A noninvasive sensitive biomarker reflecting disease status. Ann. Allergy Asthma Immunol. 2015, 115, 103–107. [Google Scholar] [CrossRef]
- Goodwin, G.H.; Johns, E.W. Are the high mobility group non-histone chromosomal proteins associated with ‘active’ chromatin? Biochim. Biophys. Acta. 1978, 519, 279–284. [Google Scholar] [CrossRef]
- Sohun, M.; Shen, H. The implication and potential applications of high-mobility group box 1 protein in breast cancer. Ann. Transl. Med. 2016, 4, 217. [Google Scholar] [CrossRef] [PubMed]
- Landsman, D.; Bustin, M. A signature for the HMG-1 box DNA-binding proteins. Bioessays 1993, 15, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Asai, K.; Fujimoto, H.; Tanaka, H.; Kanazawa, H.; Hirata, K. Increased levels of HMGB-1 and endogenous secretory RAGE in induced sputum from asthmatic patients. Respir. Med. 2011, 105, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Ferhani, N.; Letuve, S.; Kozhich, A.; Thibaudeau, O.; Grandsaigne, M.; Maret, M.; Dombret, M.C.; Sims, G.P.; Kolbeck, R.; Coyle, A.J.; et al. Expression of high-mobility group box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 181, 917–927. [Google Scholar] [CrossRef]
- Straub, C.; Midoro-Horiuti, T.; Goldblum, R.; Pazdrak, K.; Kurosky, A. Elucidating the role of high mobility group box 1 (HMGB1) cytokine in a murine model of allergic asthma. J. Allergy Clin. Immunol. 2010, 125, AB108. [Google Scholar] [CrossRef]
- Hou, C.; Zhao, H.; Liu, L.; Li, W.; Zhou, X.; Lv, Y.; Shen, X.; Liang, Z.; Cai, S.; Zou, F. High mobility group protein B1 (HMGB1) in Asthma: Comparison of patients with chronic obstructive pulmonary disease and healthy controls. Mol. Med. 2011, 17, 807–815. [Google Scholar] [CrossRef]
- Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2008, 8, 183–192. [Google Scholar] [CrossRef]
- Straub, C.; Pazdrak, K.; Kurosky, A. The novel inflammatory cytokine high mobility group box 1 protein (HMGB1) is actively released from human eosinophils upon stimulation with proinflammatory cytokines. J. Allergy Clin. Immunol. 2008, 121, S4. [Google Scholar] [CrossRef]
- Pace, E.; Di Sano, C.; Sciarrino, S.; Scafidi, V.; Ferraro, M.; Chiappara, G.; Siena, L.; Gangemi, S.; Vitulo, P.; Giarratano, A.; et al. Cigarette smoke alters IL-33 expression and release in airway epithelial cells. Biochim. Biophys. Acta 2014, 1842, 1630–1637. [Google Scholar] [CrossRef] [Green Version]
- Rowe, S.M.; Jackson, P.L.; Liu, G.; Hardison, M.; Livraghi, A.; Solomon, G.M.; McQuaid, D.B.; Noerager, B.D.; Gaggar, A.; Clancy, J.P.; et al. Potential role of high-mobility group box 1 in cystic fibrosis airway disease. Am. J. Respir. Crit. Care Med. 2008, 178, 822–831. [Google Scholar] [CrossRef]
- Ito, I.; Fukazawa, J.; Yoshida, M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J. Biol. Chem. 2007, 282, 16336–16344. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hou, C.; Kong, J.; Wen, H.; Zheng, X.; Wu, L.; Huang, H.; Chen, Y. HMGB1 binding to receptor for advanced glycation end products enhances inflammatory responses of human bronchial epithelial cells by activating p38 MAPK and ERK1/2. Mol. Cell. Biochem. 2015, 405, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.O.; Ryu, M.H.; Jha, A.; Unruh, H.; Halayko, A.J. High-mobility group box 1 promotes extracellular matrix synthesis and wound repair in human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1354–L1366. [Google Scholar] [CrossRef]
- Shang, G.H.; Jia, C.Q.; Tian, H.; Xiao, W.; Li, Y.; Wang, A.H.; Dong, L.; Lin, D.J. Serum high mobility group box protein 1 as a clinical marker for non-small cell lung cancer. Respir. Med. 2009, 103, 1949–1953. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.D.; Nawijn, M.C.; Bathoorn, E.; Riezebos-Brilman, A.; van Oosterhout, A.J.; Kerstjens, H.A.; Heijink, I.H. Increased serum levels of LL37, HMGB1 and S100A9 during exacerbation in COPD patients. Eur. Respir. J. 2015, 45, 1482–1485. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, D.; Wang, B.; Li, H.; Xiong, J.; Xu, S.; Chen, Q.; Tao, K.; Yang, X.; Zhu, Y.; et al. HMGB1 translocation and release mediate cigarette smoke-induced pulmonary inflammation in mice through a TLR4/MyD88-dependent signaling pathway. Mol. Biol. Cell. 2017, 28, 201–209. [Google Scholar] [CrossRef]
- Rochman, Y.; Spolski, R.; Leonard, W.J. New insights into the regulation of T cells by gamma(c) family cytokines. Nat. Rev. Immunol. 2009, 9, 480–490. [Google Scholar] [CrossRef]
- West, E.E.; Spolski, R.; Kazemian, M.; Yu, Z.X.; Kemper, C.; Leonard, W.J. A TSLP-complement axis mediates neutrophil killing of methicillin-resistant Staphylococcus aureus. Sci. Immunol. 2016, 1, eaaf8471. [Google Scholar] [CrossRef]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Bals, R.; Hiemstra, P.S. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef]
- Stier, M.T.; Bloodworth, M.H.; Toki, S.; Newcomb, D.C.; Goleniewska, K.; Boyd, K.L.; Quitalig, M.; Hotard, A.L.; Moore, M.L.; Hartert, T.V.; et al. Respiratory syncytial virus infection activates IL-13-producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2016, 138, 814–824.e11. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Headley, M.B.; Loo, Y.M.; Berlin, A.; Gale, M., Jr.; Debley, J.S.; Lukacs, N.W.; Ziegler, S.F. Thymic stromal lymphopoietin is induced by respiratory syncytial virus-infected airway epithelial cells and promotes a type 2 response to infection. J. Allergy Clin. Immunol. 2012, 130, 1187–1196.e5. [Google Scholar] [CrossRef] [PubMed]
- Ebina-Shibuya, R.; Leonard, W.J. Role of thymic stromal lymphopoietin in allergy and beyond. Nat. Rev. Immunol. 2022, 1, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Bjerkan, L.; Schreurs, O.; Engen, S.A.; Jahnsen, F.L.; Baekkevold, E.S.; Blix, I.J.; Schenck, K. The short form of TSLP is constitutively translated in human keratinocytes and has characteristics of an antimicrobial peptide. Mucosal. Immunol. 2015, 8, 49–56. [Google Scholar] [CrossRef]
- Varricchi, G.; Pecoraro, A.; Marone, G.; Criscuolo, G.; Spadaro, G.; Genovese, A.; Marone, G. Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front. Immunol. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Dong, H.; Hu, Y.; Liu, L.; Zou, M.; Huang, C.; Luo, L.; Yu, C.; Wan, X.; Zhao, H.; Chen, J.; et al. Distinct roles of short and long thymic stromal lymphopoietin isoforms in house dust mite-induced asthmatic airway epithelial barrier disruption. Sci. Rep. 2016, 6, 39559. [Google Scholar] [CrossRef]
- Harada, M.; Hirota, T.; Jodo, A.I.; Hitomi, Y.; Sakashita, M.; Tsunoda, T.; Miyagawa, T.; Doi, S.; Kameda, M.; Fujita, K.; et al. Thymic stromal lymphopoietin gene promoter polymorphisms are associated with susceptibility to bronchial asthma. Am. J. Respir. Cell Mol. Biol. 2011, 44, 787–793. [Google Scholar] [CrossRef]
- Torgerson, D.G.; Ampleford, E.J.; Chiu, G.Y.; Gauderman, W.J.; Gignoux, C.R.; Graves, P.E.; Himes, B.E.; Levin, A.M.; Mathias, R.A.; Hancock, D.B.; et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet. 2011, 43, 887–892. [Google Scholar] [CrossRef]
- Hirota, T.; Takahashi, A.; Kubo, M.; Tsunoda, T.; Tomita, K.; Doi, S.; Fujita, K.; Miyatake, A.; Enomoto, T.; Miyagawa, T.; et al. Genome-wide association study identifies three new susceptibility loci for adult asthma in the Japanese population. Nat. Genet. 2011, 43, 893–896. [Google Scholar] [CrossRef]
- Murrison, L.B.; Ren, X.; Preusse, K.; He, H.; Kroner, J.; Chen, X.; Jenkins, S.; Johansson, E.; Biagini, J.M.; Weirauch, M.T.; et al. TSLP disease-associated genetic variants combined with airway TSLP expression influence asthma risk. J. Allergy Clin. Immunol. 2022, 149, 79–88. [Google Scholar] [CrossRef]
- Ko, H.K.; Cheng, S.L.; Lin, C.H.; Lin, S.H.; Hsiao, Y.H.; Su, K.C.; Yu, C.J.; Wang, H.C.; Sheu, C.C.; Chiu, K.C.; et al. Blood tryptase and thymic stromal lymphopoietin levels predict the risk of exacerbation in severe asthma. Sci. Rep. 2021, 11, 8425. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Lv, Z.; Chen, Y.; Li, Y.; Huang, K.; Corrigan, C.J.; Ying, S. Bronchial Allergen Challenge of Patients with Atopic Asthma Triggers an Alarmin (IL-33, TSLP, and IL-25) Response in the Airways Epithelium and Submucosa. J. Immunol. 2018, 201, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Gong, J.; Mu, M.; Zhu, Y.; Wang, W.; Chen, W.; Han, G.; Hu, H.; Bao, P. The Pathogenesis of Eosinophilic Asthma: A Positive Feedback Mechanism That Promotes Th2 Immune Response via Filaggrin Deficiency. Front. Immunol. 2021, 12, 672312. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; O’Connor, B.; Ratoff, J.; Meng, Q.; Fang, C.; Cousins, D.; Zhang, G.; Gu, S.; Gao, Z.; Shamji, B.; et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J. Immunol. 2008, 181, 2790–2798. [Google Scholar] [CrossRef]
- Smelter, D.F.; Sathish, V.; Thompson, M.A.; Pabelick, C.M.; Vassallo, R.; Prakash, Y.S. Thymic stromal lymphopoietin in cigarette smoke-exposed human airway smooth muscle. J. Immunol. 2010, 185, 3035–3040. [Google Scholar] [CrossRef]
- Xue, J.; Suarez, J.S.; Minaai, M.; Li, S.; Gaudino, G.; Pass, H.I.; Carbone, M.; Yang, H. HMGB1 as a therapeutic target in disease. J. Cell. Physiol. 2021, 236, 3406–3419. [Google Scholar] [CrossRef]
- Wang, J.; Li, R.; Peng, Z.; Hu, B.; Rao, X.; Li, J. HMGB1 participates in LPS-induced acute lung injury by activating the AIM2 inflammasome in macrophages and inducing polarization of M1 macrophages via TLR2, TLR4, and RAGE/NF-κB signaling pathways. Int. J. Mol. Med. 2020, 45, 61–80. [Google Scholar] [CrossRef]
- Volchuk, A.; Ye, A.; Chi, L.; Steinberg, B.E.; Goldenberg, N.M. Indirect regulation of HMGB1 release by gasdermin D. Nat. Commun. 2020, 11, 4561. [Google Scholar] [CrossRef]
- Li, W.; Deng, M.; Loughran, P.A.; Yang, M.; Lin, M.; Yang, C.; Gao, W.; Jin, S.; Li, S.; Cai, J.; et al. LPS Induces Active HMGB1 Release from Hepatocytes into Exosomes Through the Coordinated Activities of TLR4 and Caspase-11/GSDMD Signaling. Front. Immunol. 2020, 11, 229. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, Q.; Cheng, Y.; Chen, X.; Wang, G.; Shi, M.; Zhang, T.; Cao, Y.; Pan, H.; Zhang, L.; et al. Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1(+) regulatory B cell expansion. J. Immunother. Cancer 2018, 6, 145. [Google Scholar] [CrossRef]
- Pucci, M.; Reclusa Asiain, P.; Durendez Saez, E.; Jantus-Lewintre, E.; Malarani, M.; Khan, S.; Fontana, S.; Naing, A.; Passiglia, F.; Raez, L.E.; et al. Extracellular Vesicles As miRNA Nano-Shuttles: Dual Role in Tumor Progression. Target. Oncol. 2018, 13, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhang, B.; Ocansey, D.K.W.; Xu, W.; Qian, H. Extracellular vesicles: A bright star of nanomedicine. Biomaterials 2021, 269, 120467. [Google Scholar] [CrossRef] [PubMed]
- Galvano, A.; Taverna, S.; Badalamenti, G.; Incorvaia, L.; Castiglia, M.; Barraco, N.; Passiglia, F.; Fulfaro, F.; Beretta, G.; Duro, G.; et al. Detection of RAS mutations in circulating tumor DNA: A new weapon in an old war against colorectal cancer. A systematic review of literature and meta-analysis. Ther. Adv. Med. Oncol. 2019, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Romano, R.; Bucci, C.; Marzetti, E. Extracellular Vesicles and Damage-Associated Molecular Patterns: A Pandora’s Box in Health and Disease. Front. Immunol. 2020, 11, 601740. [Google Scholar] [CrossRef]
- Fontana, S.; Saieva, L.; Taverna, S.; Alessandro, R. Contribution of proteomics to understanding the role of tumor-derived exosomes in cancer progression: State of the art and new perspectives. Proteomics 2013, 13, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol. Cancer. 2018, 17, 146. [Google Scholar] [CrossRef]
- Chen, M.; Liu, Y.; Varley, P.; Chang, Y.; He, X.-X.; Huang, H.; Tang, D.; Lotze, M.T.; Lin, J.; Tsung, A. High-Mobility Group Box 1 Promotes Hepatocellular Carcinoma Progression through miR-21-Mediated Matrix Metalloproteinase Activity. Cancer Res. 2015, 75, 1645–1656. [Google Scholar] [CrossRef]
- Yan, J.; Ying, S.; Cai, X. MicroRNA-Mediated Regulation of HMGB1 in Human Hepatocellular Carcinoma. Biomed. Res. Int. 2018, 2018, 2754941. [Google Scholar] [CrossRef]
- Li, J.; Kong, X.; Jiang, S.; Liao, W.; Zhang, Z.; Song, J.; Liang, Y.; Zhang, W. miR-627/HMGB1/NF-κB regulatory loop modulates TGF-β1-induced pulmonary fibrosis. J. Cell. Biochem. 2019, 120, 2983–2993. [Google Scholar] [CrossRef]
- Xu, B.; Gan, C.-X.; Chen, S.-S.; Li, J.-Q.; Liu, M.-Z.; Guo, G.-H. BMSC-derived exosomes alleviate smoke inhalation lung injury through blockade of the HMGB1/NF-κB pathway. Life Sci. 2020, 257, 118042. [Google Scholar] [CrossRef]
- Taverna, S.; Tonacci, A.; Ferraro, M.; Cammarata, G.; Cuttitta, G.; Bucchieri, S.; Pace, E.; Gangemi, S. High Mobility Group Box 1, Biological Functions and Relevance in Oxidative Stress Related Chronic Diseases. Cells 2022, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Agresti, A. HMG proteins: Dynamic players in gene regulation and differentiation. Curr. Opin. Genet. Dev. 2005, 15, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Hock, R.; Furusawa, T.; Ueda, T.; Bustin, M. HMG chromosomal proteins in development and disease. Trends Cell. Biol. 2007, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Parnes, J.R.; Molfino, N.A.; Colice, G.; Martin, U.; Corren, J.; Menzies-Gow, A. Targeting TSLP in Asthma. J. Asthma Allergy 2022, 15, 749–765. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furci, F.; Murdaca, G.; Pelaia, C.; Imbalzano, E.; Pelaia, G.; Caminati, M.; Allegra, A.; Senna, G.; Gangemi, S. TSLP and HMGB1: Inflammatory Targets and Potential Biomarkers for Precision Medicine in Asthma and COPD. Biomedicines 2023, 11, 437. https://doi.org/10.3390/biomedicines11020437
Furci F, Murdaca G, Pelaia C, Imbalzano E, Pelaia G, Caminati M, Allegra A, Senna G, Gangemi S. TSLP and HMGB1: Inflammatory Targets and Potential Biomarkers for Precision Medicine in Asthma and COPD. Biomedicines. 2023; 11(2):437. https://doi.org/10.3390/biomedicines11020437
Chicago/Turabian StyleFurci, Fabiana, Giuseppe Murdaca, Corrado Pelaia, Egidio Imbalzano, Girolamo Pelaia, Marco Caminati, Alessandro Allegra, Gianenrico Senna, and Sebastiano Gangemi. 2023. "TSLP and HMGB1: Inflammatory Targets and Potential Biomarkers for Precision Medicine in Asthma and COPD" Biomedicines 11, no. 2: 437. https://doi.org/10.3390/biomedicines11020437