
Interaction of Glutathione with MMACHC Arginine-Rich Pocket Variants Associated with Cobalamin C Disease: Insights from Molecular Modeling

Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Structure Preparation and Mutant Modeling
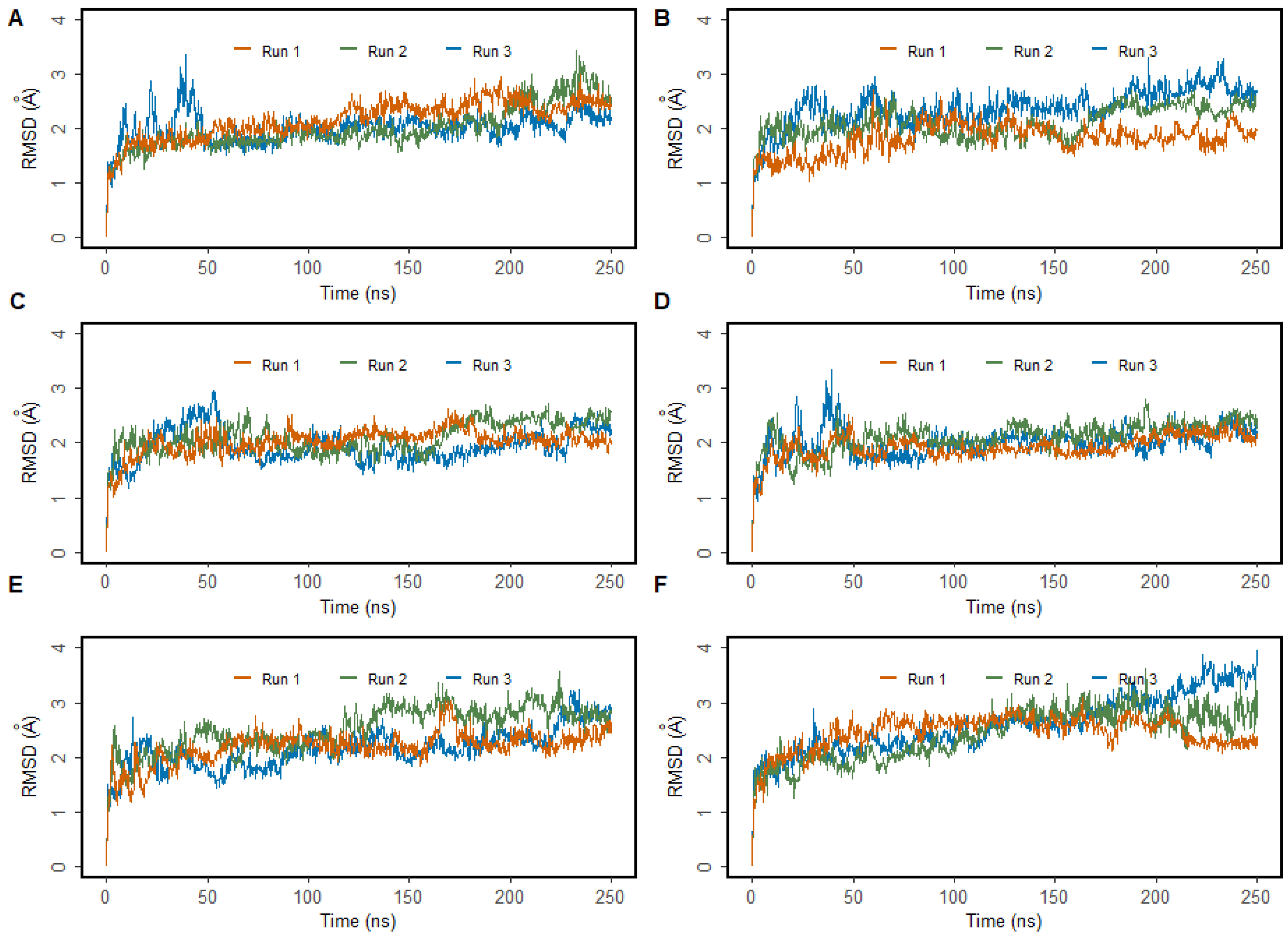
2.2. Molecular Dynamics Simulations
3. Results and Discussions
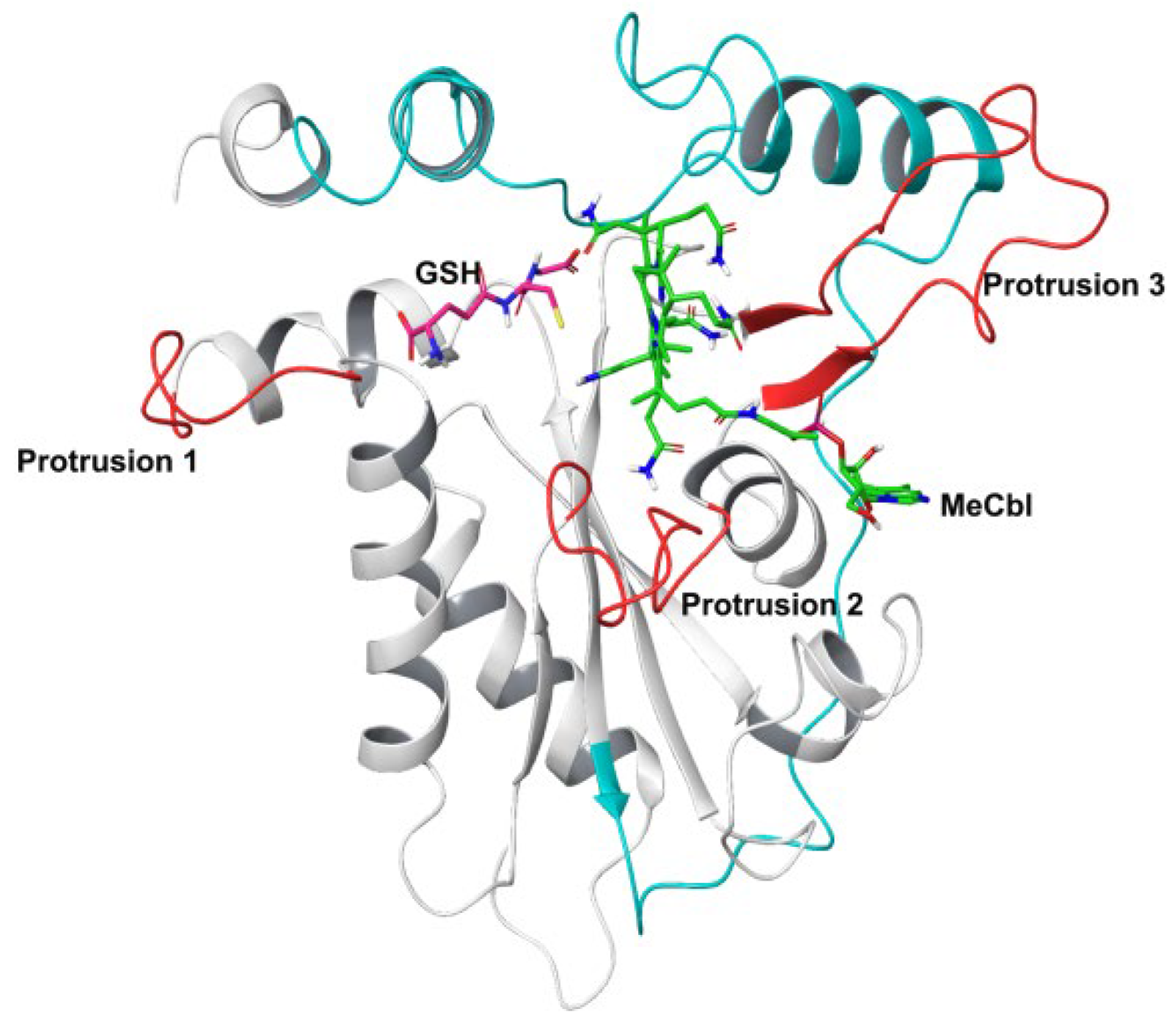
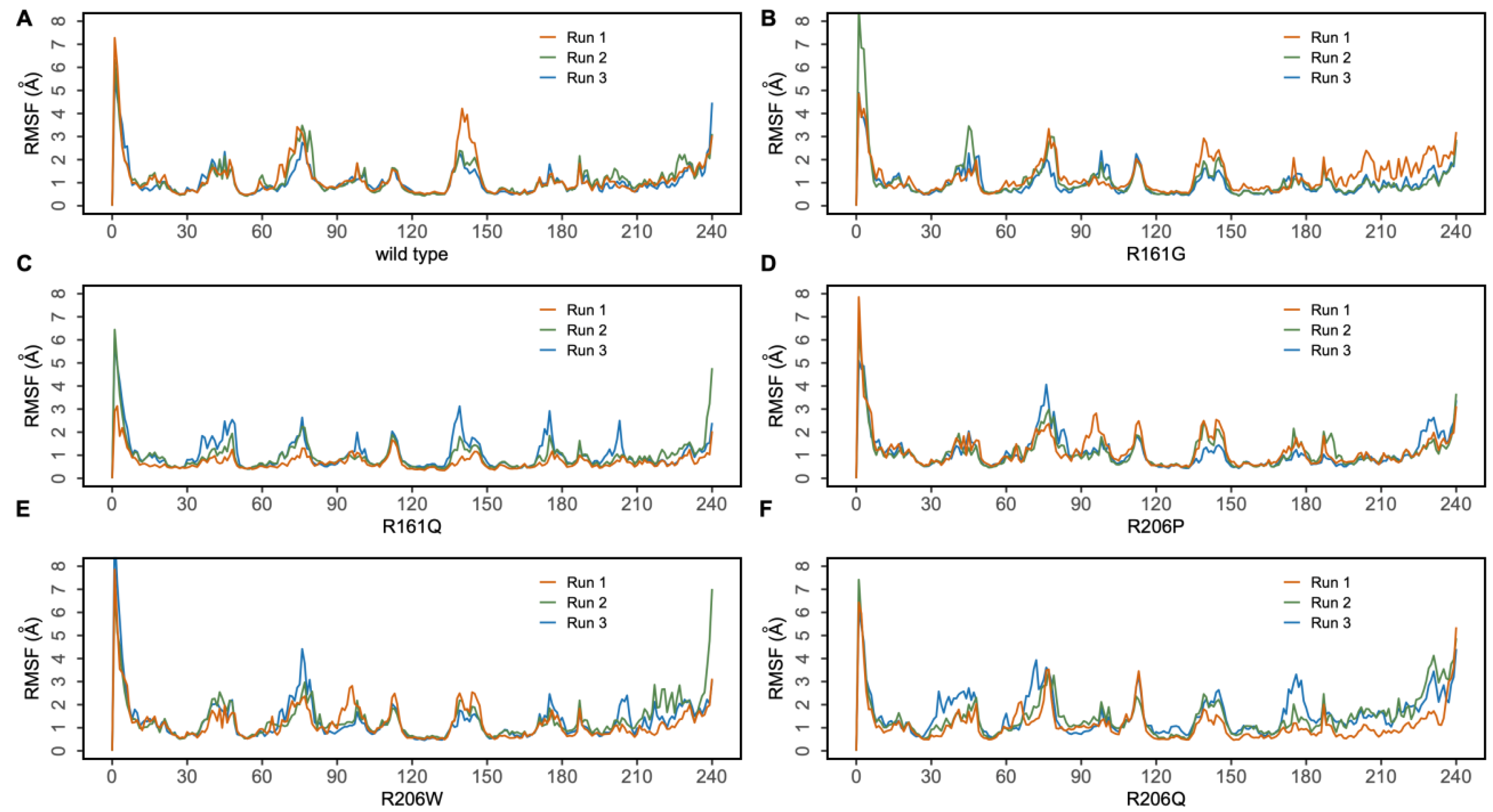
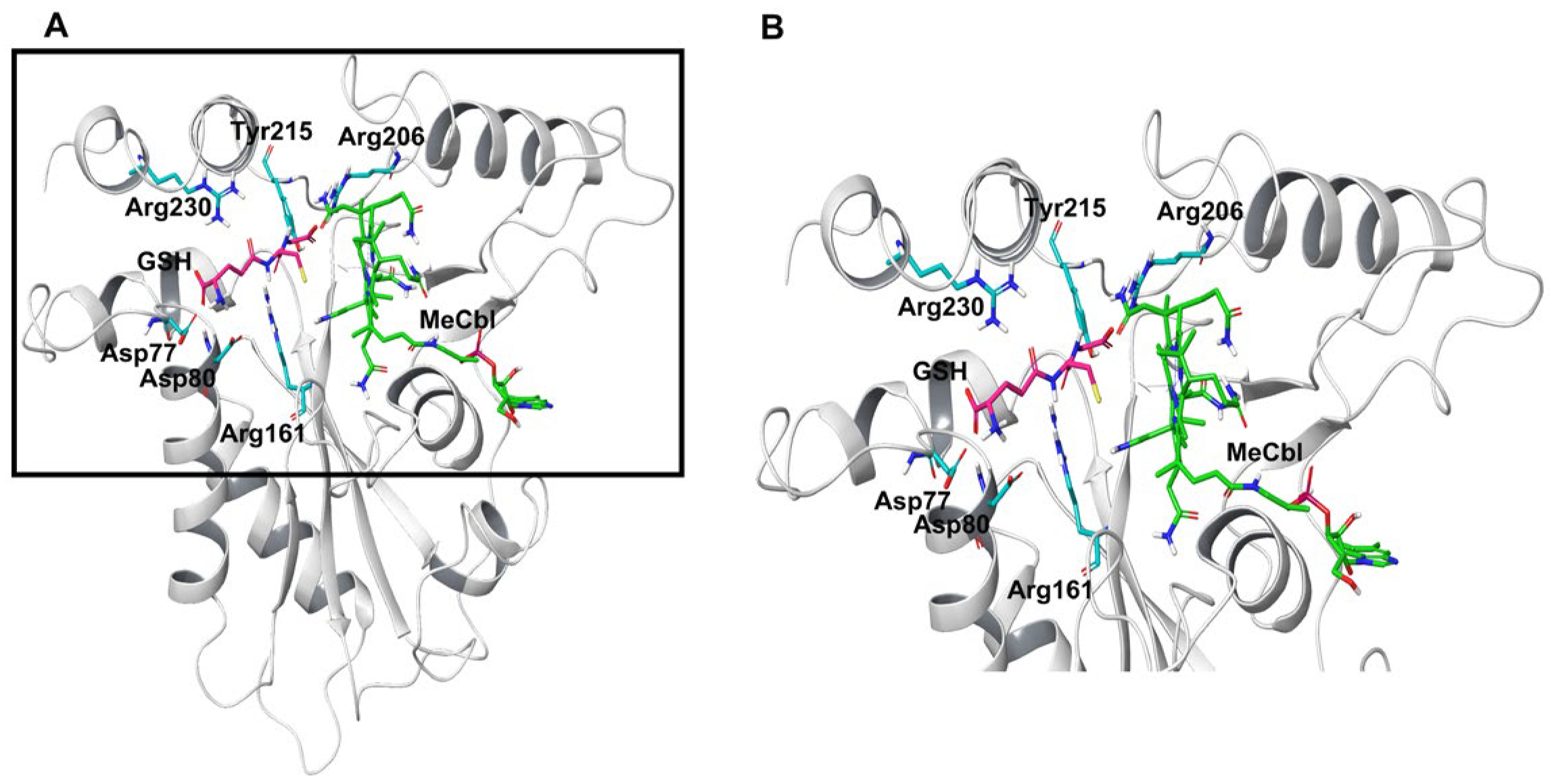
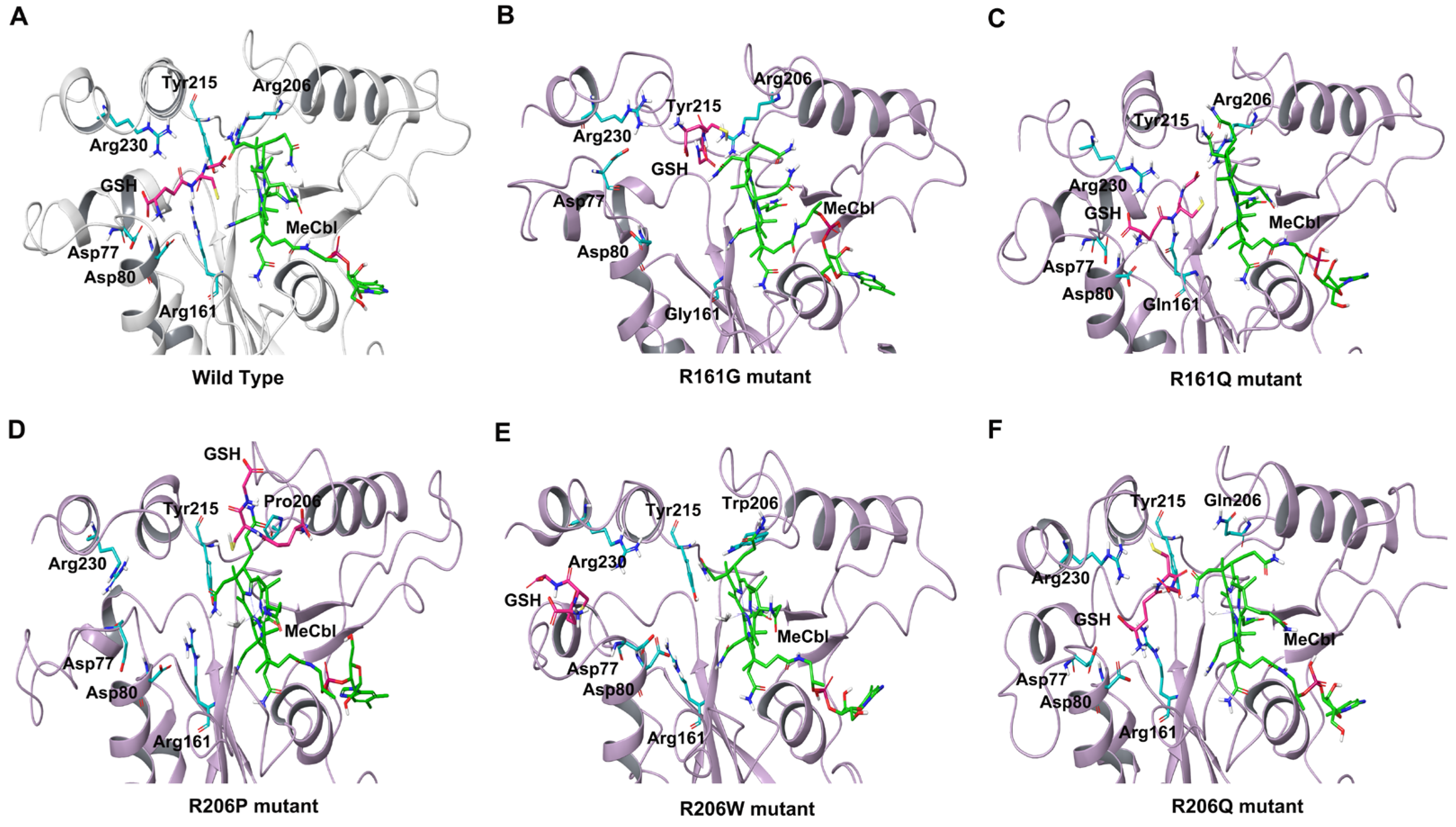
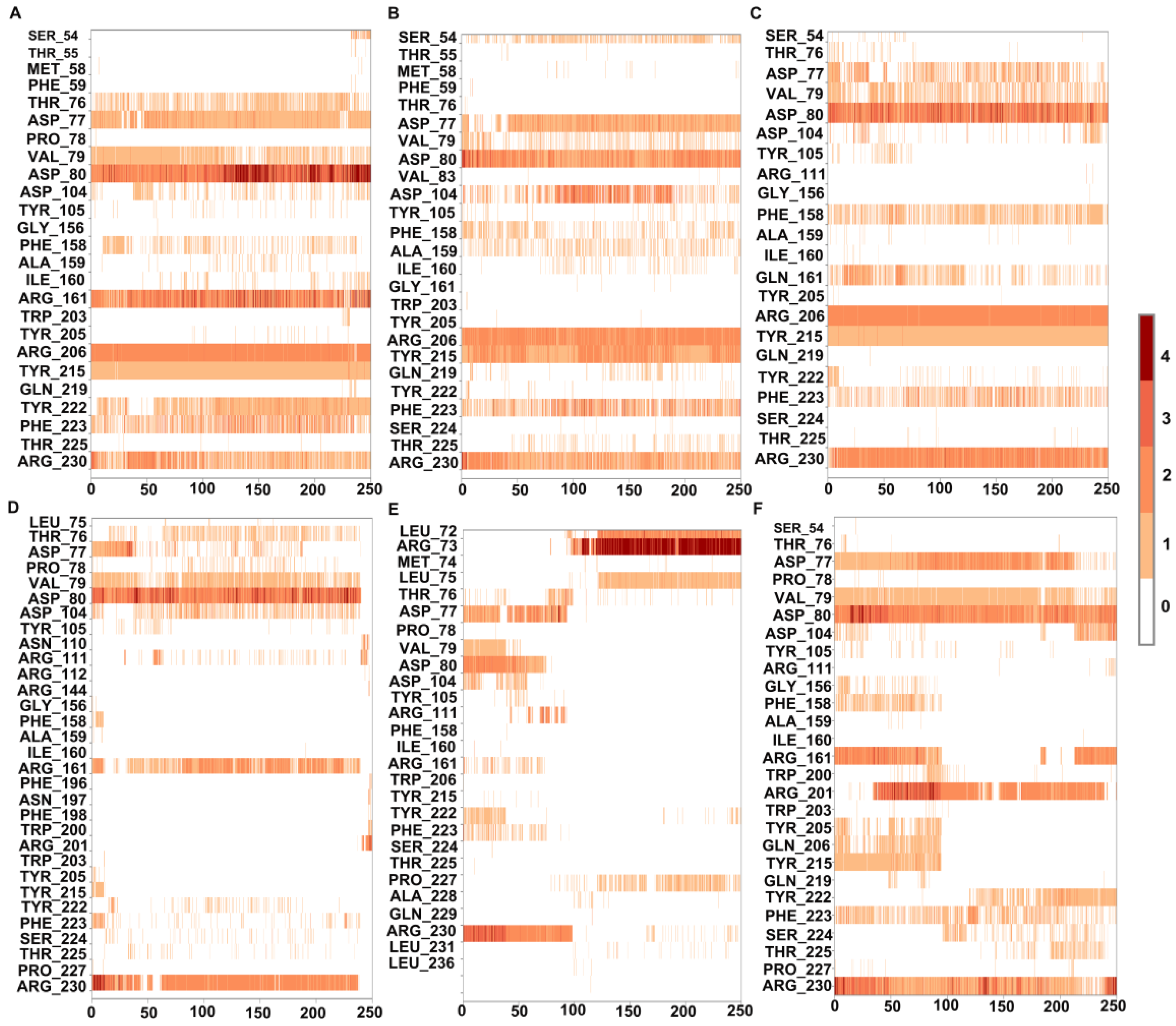
3.1. MD Simulations of Wild-Type MMACHC with MeCbl and GSH
3.2. Structural Insights into Pathogenic Mutations in MMACHC with MeCbl and GSH
3.2.1. R161G and R161Q
3.2.2. R206W, R206P, and R206Q
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, Folate, and the Methionine Remethylation Cycle—Biochemistry, Pathways, and Regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, L.; Jacobsen, D.W. Intracellular Processing of Vitamin B12 by MMACHC (CblC). In Vitamins and Hormones; Elsevier: Amsterdam, The Netherlands, 2022; Volume 119, pp. 275–298. ISBN 978-0-323-99223-7. [Google Scholar]
- Koutmos, M.; Gherasim, C.; Smith, J.L.; Banerjee, R. Structural Basis of Multifunctionality in a Vitamin B12-Processing Enzyme. J. Biol. Chem. 2011, 286, 29780–29787. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhou, H.; Li, M.; Wang, W.; Xu, M.; Zhu, Z.; Zhang, C.; Wang, Q.; Yu, F.; He, J. Structural Study of the Complex of cblC Methylmalonic Aciduria and Homocystinuria-Related Protein MMACHC with Cyanocobalamin. Crystals 2022, 12, 468. [Google Scholar] [CrossRef]
- Froese, D.S.; Gravel, R.A. Genetic Disorders of Vitamin B 12 Metabolism: Eight Complementation Groups—Eight Genes. Expert Rev. Mol. Med. 2010, 12, e37. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Ellis, J.P.; Tirone, J.C.; Pawelek, P.D.; Doré, C.; Atkinson, J.L.; Watkins, D.; Morel, C.F.; Fujiwara, T.M.; Moras, E.; Hosack, A.R.; et al. Identification of the Gene Responsible for Methylmalonic Aciduria and Homocystinuria, cblC Type. Nat. Genet. 2006, 38, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Morel, C.F.; Lerner-Ellis, J.P.; Rosenblatt, D.S. Combined Methylmalonic Aciduria and Homocystinuria (cblC): Phenotype–Genotype Correlations and Ethnic-Specific Observations. Mol. Genet. Metab. 2006, 88, 315–321. [Google Scholar] [CrossRef]
- Huemer, M.; Baumgartner, M.R. The Clinical Presentation of Cobalamin-related Disorders: From Acquired Deficiencies to Inborn Errors of Absorption and Intracellular Pathways. J. Inherit. Metab. Dis. 2019, 42, 686–705. [Google Scholar] [CrossRef]
- Rosenblatt, D.S.; Aspler, A.L.; Shevell, M.I.; Pletcher, B.A.; Fenton, W.A.; Seashore, M.R. Clinical Heterogeneity and Prognosis in Combined Methylmalonic Aciduria and Homocystinuria (cblC). J. Inherit. Metab. Dis. 1997, 20, 528–538. [Google Scholar] [CrossRef]
- Kalantari, S.; Brezzi, B.; Bracciamà, V.; Barreca, A.; Nozza, P.; Vaisitti, T.; Amoroso, A.; Deaglio, S.; Manganaro, M.; Porta, F.; et al. Adult-Onset CblC Deficiency: A Challenging Diagnosis Involving Different Adult Clinical Specialists. Orphanet J. Rare Dis. 2022, 17, 33. [Google Scholar] [CrossRef]
- Carrillo-Carrasco, N.; Chandler, R.J.; Venditti, C.P. Combined Methylmalonic Acidemia and Homocystinuria, cblC Type. I. Clinical Presentations, Diagnosis and Management. J. Inherit. Metab. Dis. 2012, 35, 91–102. [Google Scholar] [CrossRef]
- Wang, S.; Yan, C.; Wen, B.; Zhao, Y. Clinical Feature and Outcome of Late-Onset Cobalamin C Disease Patients with Neuropsychiatric Presentations: A Chinese Case Series. Neuropsychiatr. Dis. Treat. 2019, 15, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gherasim, C.; Banerjee, R. Decyanation of Vitamin B 12 by a Trafficking Chaperone. Proc. Natl. Acad. Sci. USA 2008, 105, 14551–14554. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gherasim, C.; Lesniak, N.A.; Banerjee, R. Glutathione-Dependent One-Electron Transfer Reactions Catalyzed by a B12 Trafficking Protein. J. Biol. Chem. 2014, 289, 16487–16497. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hannibal, L.; Gherasim, C.; Jacobsen, D.W.; Banerjee, R. A Human Vitamin B12 Trafficking Protein Uses Glutathione Transferase Activity for Processing Alkylcobalamins. J. Biol. Chem. 2009, 284, 33418–33424. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Kopec, J.; Fitzpatrick, F.; Schuller, M.; McCorvie, T.J.; Chalk, R.; Plessl, T.; Fettelschoss, V.; Fowler, B.; Baumgartner, M.R.; et al. Structural Insights into the MMACHC-MMADHC Protein Complex Involved in Vitamin B12 Trafficking. J. Biol. Chem. 2015, 290, 29167–29177. [Google Scholar] [CrossRef]
- Beck, B.B.; Van Spronsen, F.; Diepstra, A.; Berger, R.M.F.; Kömhoff, M. Renal Thrombotic Microangiopathy in Patients with cblC Defect: Review of an under-Recognized Entity. Pediatr. Nephrol. 2017, 32, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.; Aiello, C.; Cerone, R.; Martins, E.; Caruso, U.; Moroni, I.; Rizzo, C.; Diogo, L.; Leão, E.; Kok, F. Spectrum of MMACHC Mutations in Italian and Portuguese Patients with Combined Methylmalonic Aciduria and Homocystinuria, cblC Type. Mol. Genet. Metab. 2008, 93, 475–480. [Google Scholar] [CrossRef]
- Fischer, S.; Huemer, M.; Baumgartner, M.; Deodato, F.; Ballhausen, D.; Boneh, A.; Burlina, A.B.; Cerone, R.; Garcia, P.; Gökçay, G.; et al. Clinical Presentation and Outcome in a Series of 88 Patients with the cblC Defect. J. Inherit. Metab. Dis. 2014, 37, 831–840. [Google Scholar] [CrossRef]
- Passantino, R.; Mangione, M.R.; Ortore, M.G.; Costa, M.A.; Provenzano, A.; Amenitsch, H.; Sabbatella, R.; Alfano, C.; Martorana, V.; Vilasi, S. Investigation on a MMACHC Mutant from cblC Disease: The c.394C>T Variant. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2022, 1870, 140793. [Google Scholar] [CrossRef]
- Almannai, M.; Marom, R.; Divin, K.; Scaglia, F.; Sutton, V.R.; Craigen, W.J.; Lee, B.; Burrage, L.C.; Graham, B.H. Milder Clinical and Biochemical Phenotypes Associated with the c.482G > A (p.Arg161Gln) Pathogenic Variant in Cobalamin C Disease: Implications for Management and Screening. Mol. Genet. Metab. 2017, 122, 60–66. [Google Scholar] [CrossRef]
- Froese, D.S.; Krojer, T.; Wu, X.; Shrestha, R.; Kiyani, W.; von Delft, F.; Gravel, R.A.; Oppermann, U.; Yue, W.W. Structure of MMACHC Reveals an Arginine-Rich Pocket and a Domain-Swapped Dimer for Its B 12 Processing Function. Biochemistry 2012, 51, 5083–5090. [Google Scholar] [CrossRef] [PubMed]
- Ruetz, M.; Shanmuganathan, A.; Gherasim, C.; Karasik, A.; Salchner, R.; Kieninger, C.; Wurst, K.; Banerjee, R.; Koutmos, M.; Kräutler, B. Antivitamin B12 Inhibition of the Human B12-Processing Enzyme CblC: Crystal Structure of an Inactive Ternary Complex with Glutathione as the Cosubstrate. Angew. Chem. Int. Ed. 2017, 56, 7387–7392. [Google Scholar] [CrossRef] [PubMed]
- Alogheli, H.; Olanders, G.; Schaal, W.; Brandt, P.; Karlén, A. Docking of Macrocycles: Comparing Rigid and Flexible Docking in Glide. J. Chem. Inf. Model. 2017, 57, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-1: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA, 2021.
- Schrödinger Release 2020-4: Desmond Molecular Dynamics System, D.E. Shaw Research New York, NY, 2021; Maestro-Desmond Interoperability Tools, Schrödinger: New York, NY, USA, 2020.
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; IEEE: New York, NY, USA, 2006; p. 43. [Google Scholar]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover Chains: The Canonical Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible Multiple Time Scale Molecular Dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A Fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of Gyration as an Indicator of Protein Structure Compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed]
- Gherasim, C.; Ruetz, M.; Li, Z.; Hudolin, S.; Banerjee, R. Pathogenic Mutations Differentially Affect the Catalytic Activities of the Human B12-Processing Chaperone CblC and Increase Futile Redox Cycling. J. Biol. Chem. 2015, 290, 11393–11402. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Healy, S.; McDonald, M.; Kochan, G.; Oppermann, U.; Niesen, F.H.; Gravel, R.A. Thermolability of Mutant MMACHC Protein in the Vitamin B12-Responsive cblC Disorder. Mol. Genet. Metab. 2010, 100, 29–36. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Type | MeCbl (kcal/mol) | GSH (kcal/mol) |
|---|---|---|
| Run 1 | –140.03 ± 14.92 | –36.31 ± 21.43 |
| Run 2 | –146.06 ± 18.62 | –34.29 ± 18.12 |
| Run 3 | –140.95 ± 15.04 | –33.38 ± 19.65 |
| R161G | MeCbl (kcal/mol) | GSH (kcal/mol) |
| Run 1 | –140.78 ± 16.10 | –35.78 ± 20.97 |
| Run 2 | –138.14 ± 18.43 | –24.69 ± 17.05 |
| Run 3 | –131.61 ± 15.51 | –26.13 ± 18.40 |
| R161Q | MeCbl (kcal/mol) | GSH (kcal/mol) |
| Run 1 | –145.53 ± 14.02 | –36.09 ± 16.44 |
| Run 2 | –148.82 ± 13.18 | –26.54 ± 17.12 |
| Run 3 | –141.20 ± 14.54 | –33.82 ± 15.77 |
| R206P | MeCbl (kcal/mol) | GSH (kcal/mol) |
| Run 1 | –144.50 ± 13.60 | –22.62 ± 14.31 |
| Run 2 | –155.96 ± 17.16 | –25.66 ± 16.48 |
| Run 3 | –145.21 ± 14.16 | –20.68 ± 16.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antony, P.; Baby, B.; Ali, A.; Vijayan, R.; Al Jasmi, F. Interaction of Glutathione with MMACHC Arginine-Rich Pocket Variants Associated with Cobalamin C Disease: Insights from Molecular Modeling. Biomedicines 2023, 11, 3217. https://doi.org/10.3390/biomedicines11123217
Antony P, Baby B, Ali A, Vijayan R, Al Jasmi F. Interaction of Glutathione with MMACHC Arginine-Rich Pocket Variants Associated with Cobalamin C Disease: Insights from Molecular Modeling. Biomedicines. 2023; 11(12):3217. https://doi.org/10.3390/biomedicines11123217
Chicago/Turabian StyleAntony, Priya, Bincy Baby, Amanat Ali, Ranjit Vijayan, and Fatma Al Jasmi. 2023. "Interaction of Glutathione with MMACHC Arginine-Rich Pocket Variants Associated with Cobalamin C Disease: Insights from Molecular Modeling" Biomedicines 11, no. 12: 3217. https://doi.org/10.3390/biomedicines11123217