
BRD3 Regulates the Inflammatory and Stress Response in Rheumatoid Arthritis Synovial Fibroblasts

 , , and
, , and 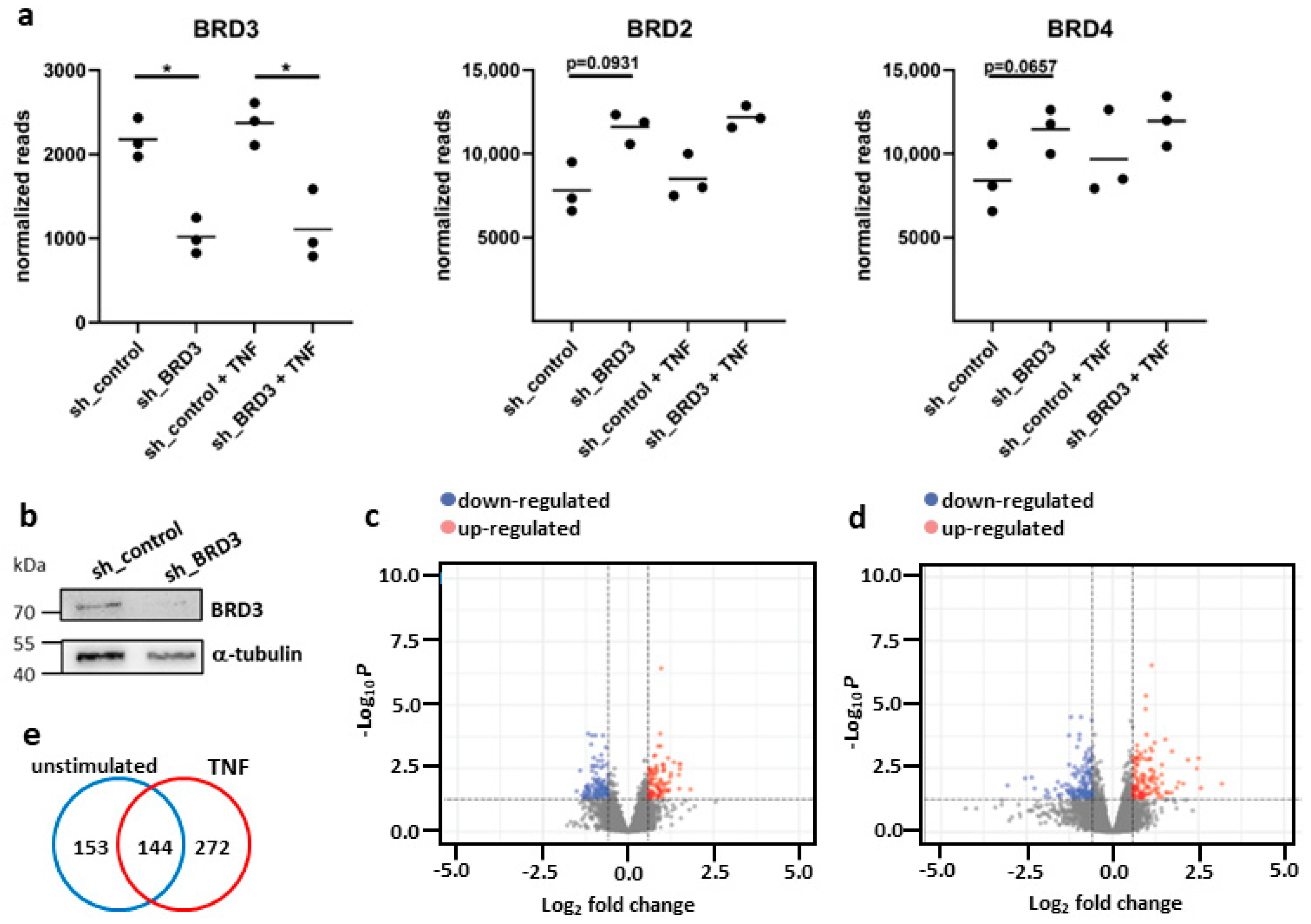{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples and Cell Preparation
2.2. Silencing of BRD3
2.3. RNA Sequencing of FLS
2.4. Analysis of RNAseq Results
2.5. Analysis of BRD2, BRD3 and BRD4 Expression in Subsets of FLS
2.6. Treatment of FLS
2.7. Western Blotting
2.8. Enzyme-Linked Immunosorbent Assay and L-Lactate Assay
2.9. Autophagy Live Cell Imaging
2.10. Statistical Analysis
3. Results
3.1. Silencing of BRD3 in FLS
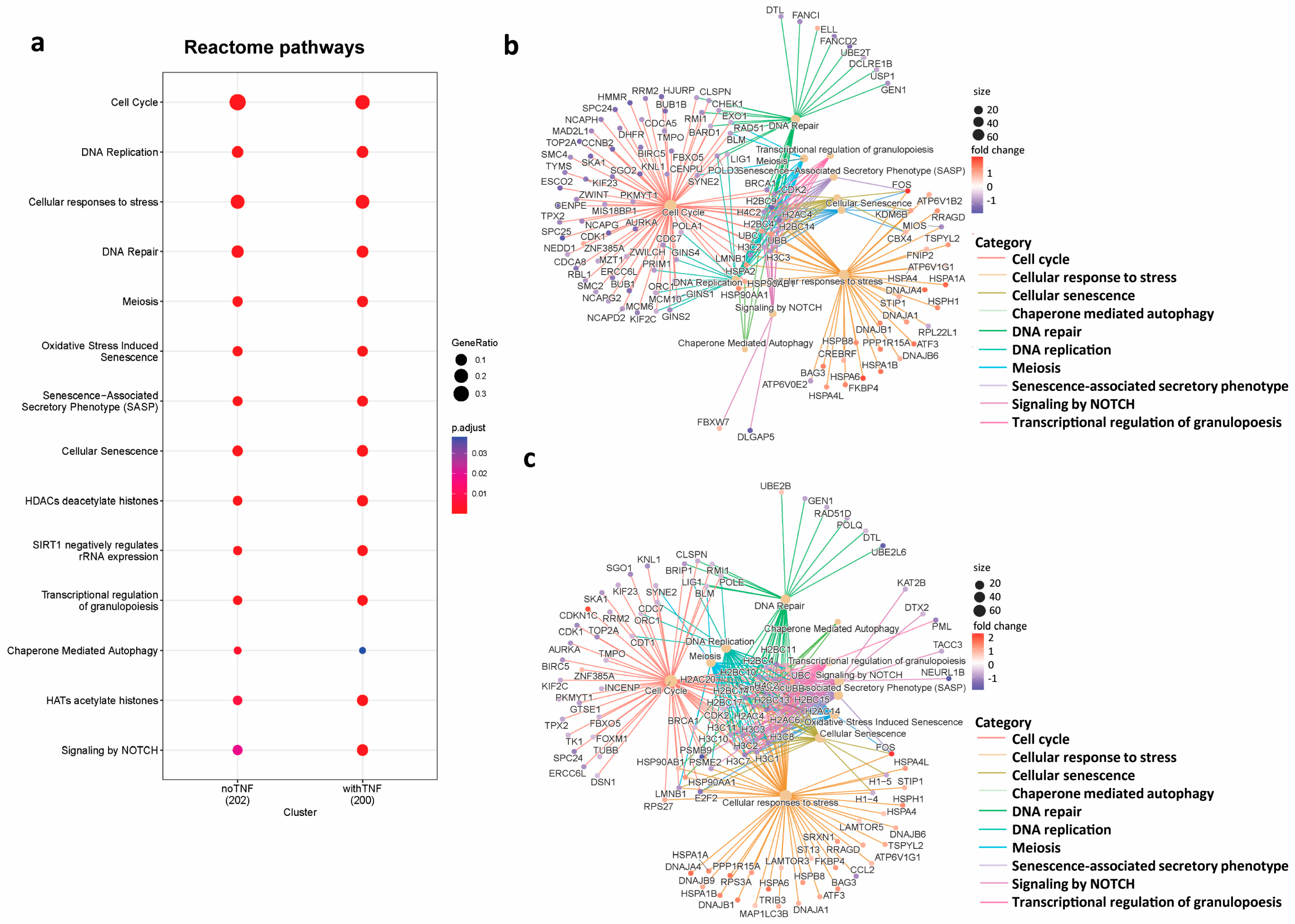
3.2. Identification of Pathways Regulated by BRD3
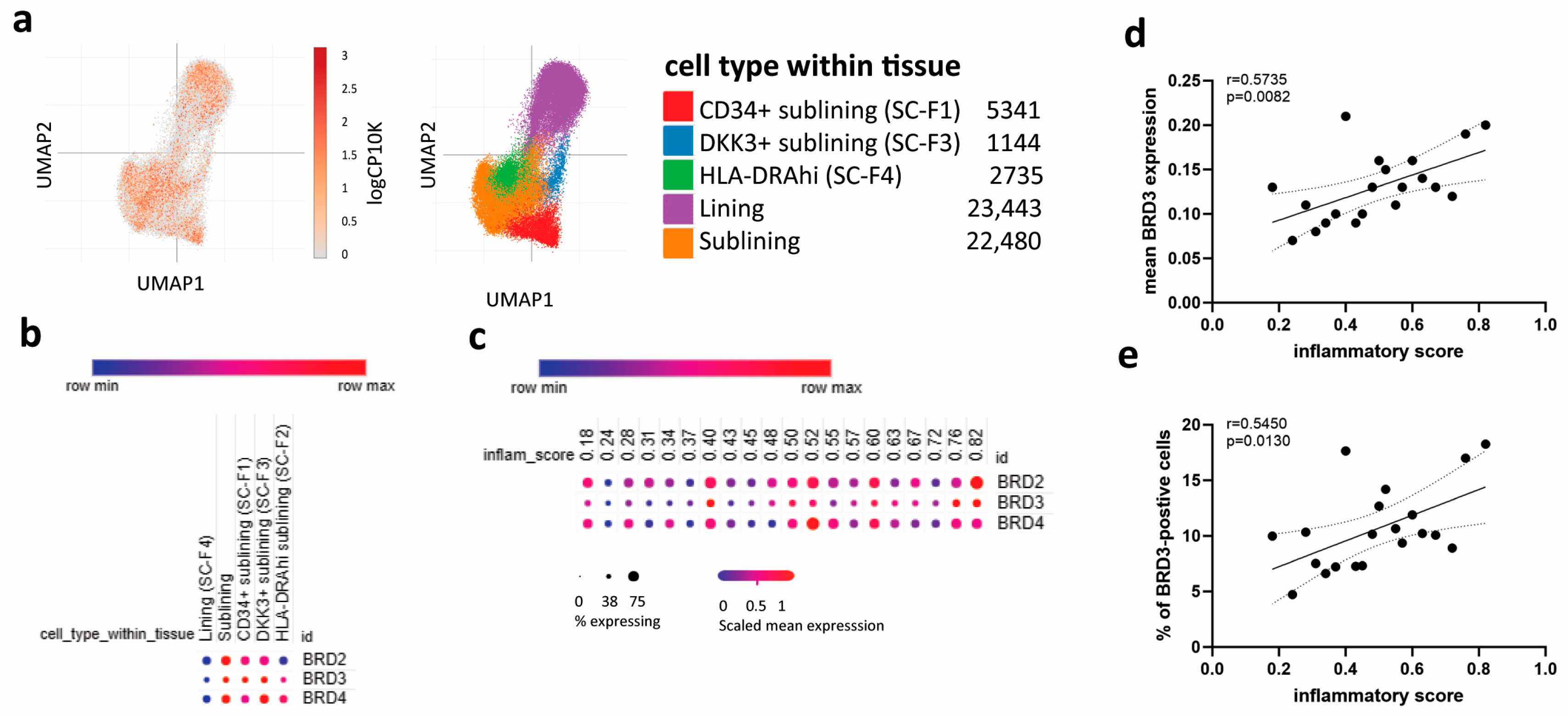
3.3. BRD3 Expression Is Increased in Sublining FLS
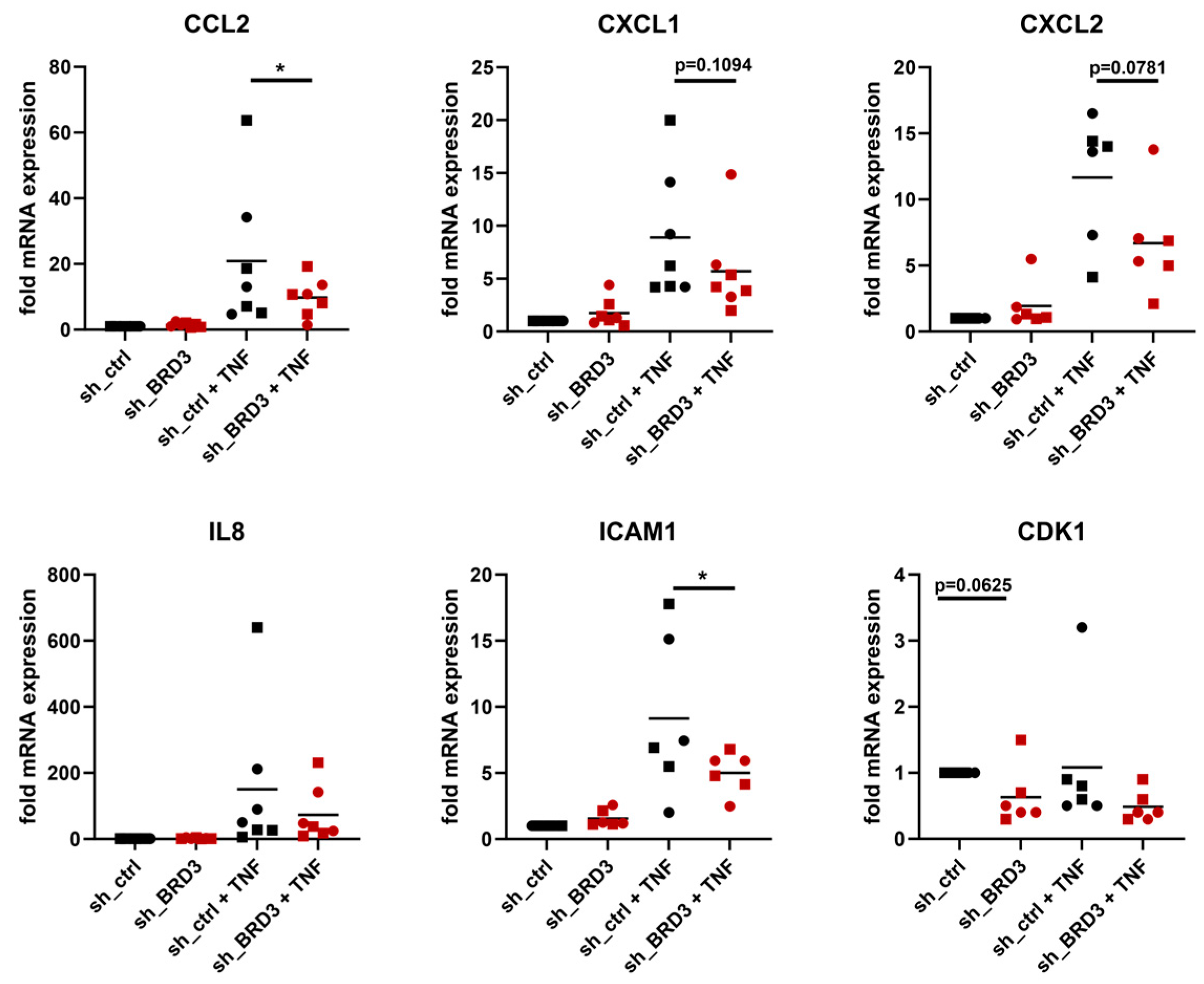
3.4. BRD3 Regulates Inflammatory Pathways in FLS
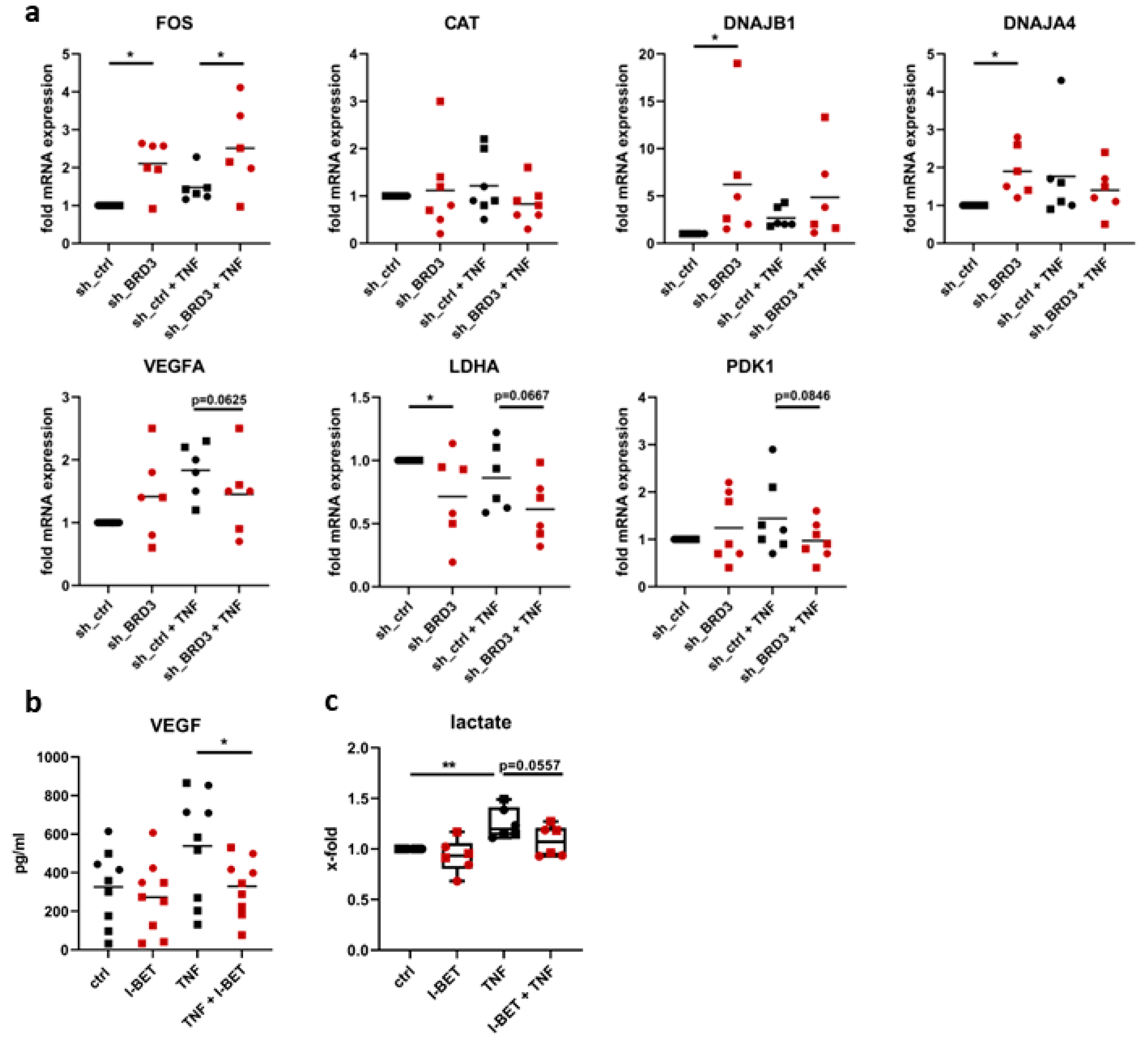
3.5. BRD3 Regulates Stress-Related Genes in FLS
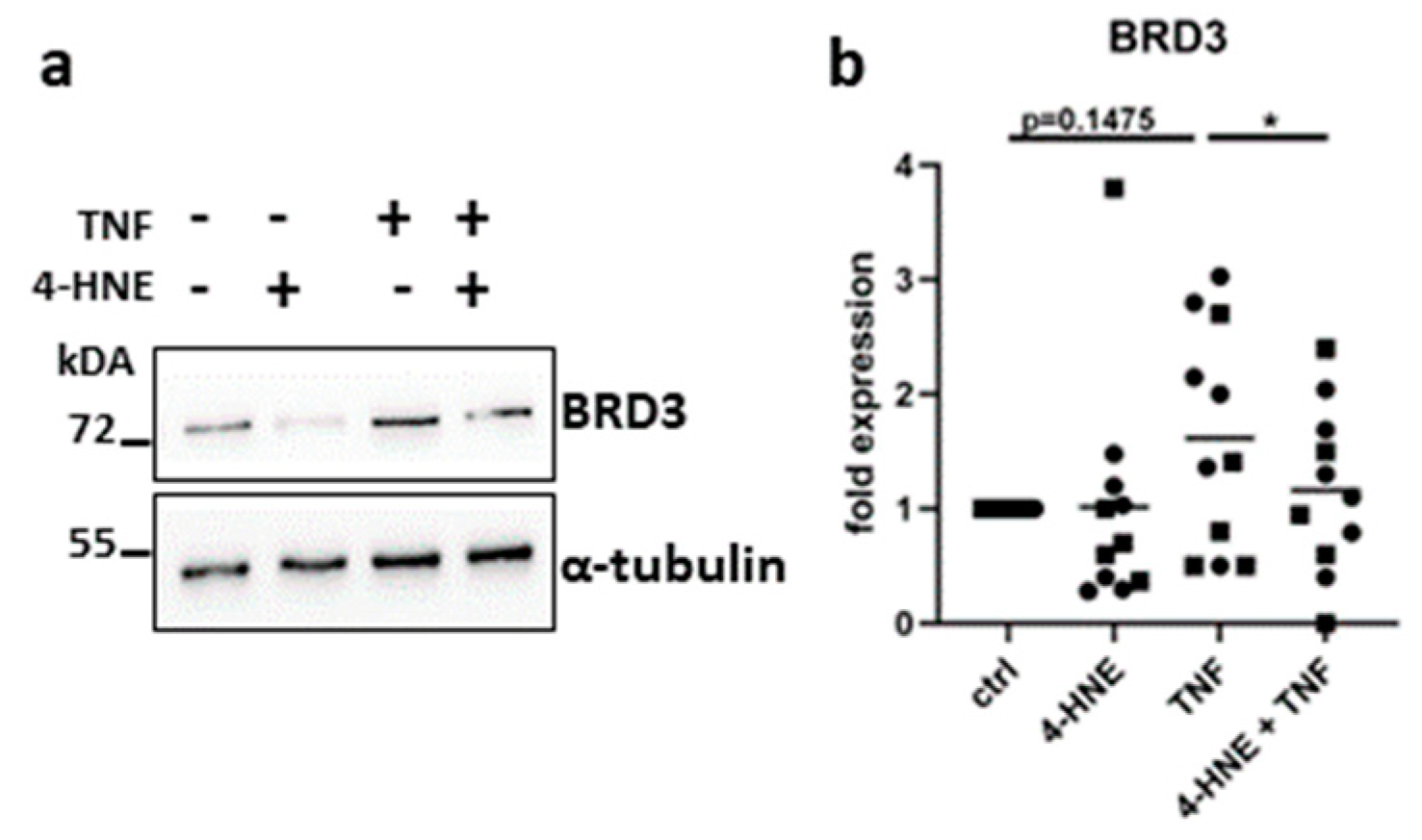
3.6. Oxidative Stress Suppresses the Expression of BRD3
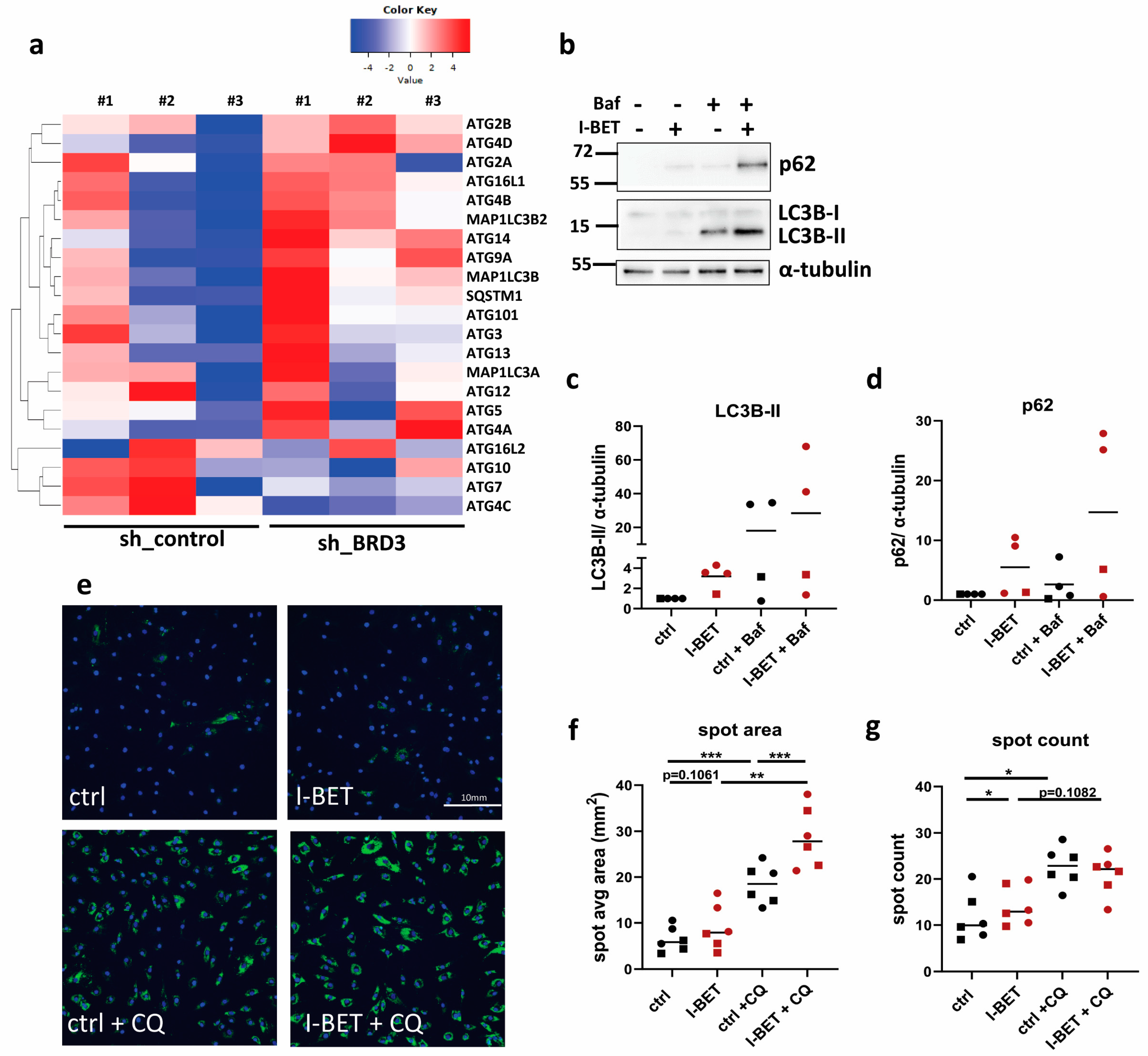
3.7. BET Proteins Regulate Autophagy in FLS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Ospelt, C. Synovial fibroblasts in 2017. RMD Open 2017, 3, e000471. [Google Scholar] [CrossRef]
- Zou, Z.; Li, H.; Yu, K.; Ma, K.; Wang, Q.; Tang, J.; Liu, G.; Lim, K.; Hooper, G.; Woodfield, T.; et al. The potential role of synovial cells in the progression and treatment of osteoarthritis. Exploration 2023, 3, 20220132. [Google Scholar] [CrossRef]
- Yamasaki, S.; Yagishita, N.; Tsuchimochi, K.; Nishioka, K.; Nakajima, T. Rheumatoid arthritis as a hyper-endoplasmic-reticulum-associated degradation disease. Arthritis Res. Ther. 2005, 7, 181–186. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, H.; Wu, Y.; He, Z.; Qin, Y.; Shen, Q. The Autophagy Level Is Increased in the Synovial Tissues of Patients with Active Rheumatoid Arthritis and Is Correlated with Disease Severity. Mediat. Inflamm. 2017, 2017, 7623145. [Google Scholar] [CrossRef]
- Shin, Y.J.; Han, S.H.; Kim, D.S.; Lee, G.H.; Yoo, W.H.; Kang, Y.M.; Choi, J.Y.; Lee, Y.C.; Park, S.J.; Jeong, S.K.; et al. Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress. Arthritis Res. Ther. 2010, 12, R19. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Ospelt, C.; Gay, R.E.; Gay, S.; Klein, K. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 2014, 66, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Falconer, J.; Murphy, A.N.; Young, S.P.; Clark, A.R.; Tiziani, S.; Guma, M.; Buckley, C.D. Review: Synovial Cell Metabolism and Chronic Inflammation in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 984–999. [Google Scholar] [CrossRef]
- Friscic, J.; Bottcher, M.; Reinwald, C.; Bruns, H.; Wirth, B.; Popp, S.J.; Walker, K.I.; Ackermann, J.A.; Chen, X.; Turner, J.; et al. The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial fibroblasts. Immunity 2021, 54, 1002–1021.e10. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Klein, K. Bromodomain protein inhibition: A novel therapeutic strategy in rheumatic diseases. RMD Open 2018, 4, e000744. [Google Scholar] [CrossRef]
- Xiao, Y.; Liang, L.; Huang, M.; Qiu, Q.; Zeng, S.; Shi, M.; Zou, Y.; Ye, Y.; Yang, X.; Xu, H. Bromodomain and extra-terminal domain bromodomain inhibition prevents synovial inflammation via blocking IkappaB kinase-dependent NF-kappaB activation in rheumatoid fibroblast-like synoviocytes. Rheumatology 2016, 55, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Krishna, V.; Yin, X.; Song, Q.; Walsh, A.; Pocalyko, D.; Bachman, K.; Anderson, I.; Madakamutil, L.; Nagpal, S. Integration of the Transcriptome and Genome-Wide Landscape of BRD2 and BRD4 Binding Motifs Identifies Key Superenhancer Genes and Reveals the Mechanism of Bet Inhibitor Action in Rheumatoid Arthritis Synovial Fibroblasts. J. Immunol. 2021, 206, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Hussong, M.; Borno, S.T.; Kerick, M.; Wunderlich, A.; Franz, A.; Sultmann, H.; Timmermann, B.; Lehrach, H.; Hirsch-Kauffmann, M.; Schweiger, M.R. The bromodomain protein BRD4 regulates the KEAP1/NRF2-dependent oxidative stress response. Cell Death Dis. 2014, 5, e1195. [Google Scholar] [CrossRef]
- Hussong, M.; Kaehler, C.; Kerick, M.; Grimm, C.; Franz, A.; Timmermann, B.; Welzel, F.; Isensee, J.; Hucho, T.; Krobitsch, S.; et al. The bromodomain protein BRD4 regulates splicing during heat shock. Nucleic Acids Res. 2017, 45, 382–394. [Google Scholar] [CrossRef]
- Col, E.; Hoghoughi, N.; Dufour, S.; Penin, J.; Koskas, S.; Faure, V.; Ouzounova, M.; Hernandez-Vargash, H.; Reynoird, N.; Daujat, S.; et al. Bromodomain factors of BET family are new essential actors of pericentric heterochromatin transcriptional activation in response to heat shock. Sci. Rep. 2017, 7, 5418. [Google Scholar] [CrossRef]
- Sakamaki, J.I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517–532.e9. [Google Scholar] [CrossRef]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S.; et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Frank-Bertoncelj, M.; Trenkmann, M.; Klein, K.; Karouzakis, E.; Rehrauer, H.; Bratus, A.; Kolling, C.; Armaka, M.; Filer, A.; Michel, B.A.; et al. Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions. Nat. Commun. 2017, 8, 14852. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Korsunsky, I.; Wei, K.; Pohin, M.; Kim, E.Y.; Barone, F.; Major, T.; Taylor, E.; Ravindran, R.; Kemble, S.; Watts, G.F.M.; et al. Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases. Med 2022, 3, 481–518. [Google Scholar] [CrossRef]
- Loh, C.; Park, S.H.; Lee, A.; Yuan, R.; Ivashkiv, L.B.; Kalliolias, G.D. TNF-induced inflammatory genes escape repression in fibroblast-like synoviocytes: Transcriptomic and epigenomic analysis. Ann. Rheum. Dis. 2019, 78, 1205–1214. [Google Scholar] [CrossRef]
- Krošel, M.; Larissa, M.; Miranda, H.; Jasna, F.; Matija, T.; Oliver, D.; Markus, H.; Hoffmann, C.O.; Kerstin, K. Bromodomain Protein Inhibitors Reorganize the Chromatin of Synovial Fibroblasts. Cells 2023, 12, 1149. [Google Scholar] [CrossRef]
- Balogh, E.; Veale, D.J.; McGarry, T.; Orr, C.; Szekanecz, Z.; Ng, C.T.; Fearon, U.; Biniecka, M. Oxidative stress impairs energy metabolism in primary cells and synovial tissue of patients with rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 95. [Google Scholar] [CrossRef]
- Ren, W.; Wang, C.; Wang, Q.; Zhao, D.; Zhao, K.; Sun, D.; Liu, X.; Han, C.; Hou, J.; Li, X.; et al. Bromodomain protein Brd3 promotes Ifnb1 transcription via enhancing IRF3/p300 complex formation and recruitment to Ifnb1 promoter in macrophages. Sci. Rep. 2017, 7, 39986. [Google Scholar] [CrossRef]
- Dai, J.; Zhou, S.; Ge, Q.; Qin, J.; Li, J.; Ju, H.; Cao, Y.; Zheng, M.; Li, C.; Gao, X.; et al. Recruitment of Brd3 and Brd4 to acetylated chromatin is essential for proinflammatory cytokine-induced matrix-degrading enzyme expression. J. Orthop. Surg. Res. 2019, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Daneshvar, K.; Ardehali, M.B.; Klein, I.A.; Hsieh, F.K.; Kratkiewicz, A.J.; Mahpour, A.; Cancelliere, S.O.L.; Zhou, C.; Cook, B.M.; Li, W.; et al. lncRNA DIGIT and BRD3 protein form phase-separated condensates to regulate endoderm differentiation. Nat. Cell Biol. 2020, 22, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Etxaniz, U.; Dall’Agnese, A.; Wu, S.Y.; Chiang, C.M.; Brennan, P.E.; Wood, M.J.A.; Puri, P.L. BRD3 and BRD4 BET Bromodomain Proteins Differentially Regulate Skeletal Myogenesis. Sci. Rep. 2017, 7, 6153. [Google Scholar] [CrossRef] [PubMed]
- Lamonica, J.M.; Deng, W.; Kadauke, S.; Campbell, A.E.; Gamsjaeger, R.; Wang, H.; Cheng, Y.; Billin, A.N.; Hardison, R.C.; Mackay, J.P.; et al. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. USA 2011, 108, E159–E168. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, Q.; Zhang, X.; Day, D.S.; Abraham, B.J.; Lun, K.; Chen, L.; Huang, J.; Ji, X. BRD2 interconnects with BRD3 to facilitate Pol II transcription initiation and elongation to prime promoters for cell differentiation. Cell Mol. Life Sci. 2022, 79, 338. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Kabala, P.A.; Grabiec, A.M.; Gay, R.E.; Kolling, C.; Lin, L.L.; Gay, S.; Tak, P.P.; Prinjha, R.K.; Ospelt, C.; et al. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann. Rheum. Dis. 2016, 75, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Friscic, J.; Reinwald, C.; Bottcher, M.; Houtman, M.; Euler, M.; Chen, X.; Walker, K.I.; Kirchner, P.; Zhu, H.; Wirth, B.; et al. Reset of inflammatory priming of joint tissue and reduction of the severity of arthritis flares by bromodomain inhibition. Arthritis Rheumatol. 2023, 75, 517–532. [Google Scholar] [CrossRef]
- Biniecka, M.; Canavan, M.; McGarry, T.; Gao, W.; McCormick, J.; Cregan, S.; Gallagher, L.; Smith, T.; Phelan, J.J.; Ryan, J.; et al. Dysregulated bioenergetics: A key regulator of joint inflammation. Ann. Rheum. Dis. 2016, 75, 2192–2200. [Google Scholar] [CrossRef]
- Akgol, G.; Ulusoy, H.; Telo, S.; Gulkesen, A.; Yildirim, T.; Poyraz, A.K.; Kaya, A. Is 4-Hydroxynonenal a Predictive Parameter for the Development of Joint Erosion in Patients with Rheumatoid Arthritis? Arch. Rheumatol. 2016, 31, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Korb, A.; Pavenstadt, H.; Pap, T. Cell death in rheumatoid arthritis. Apoptosis 2009, 14, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, E.; Kato, M.; Kudo, Y.; Lee, W.; Hisada, R.; Fujieda, Y.; Oku, K.; Bohgaki, T.; Amengual, O.; Yasuda, S.; et al. Autophagy promotes citrullination of VIM (vimentin) and its interaction with major histocompatibility complex class II in synovial fibroblasts. Autophagy 2020, 16, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Mercado, N.; Clarke, C.; Bhavsar, P.K.; Adcock, I.M.; Barnes, P.J.; Chung, K.F. Bromodomain and extraterminal proteins suppress NF-E2-related factor 2-mediated antioxidant gene expression. J. Immunol. 2014, 192, 4913–4920. [Google Scholar] [CrossRef]
- An, Q.D.; Li, Y.Y.; Zhang, H.X.; Lu, J.; Yu, X.D.; Lin, Q.; Zhang, Y.G. Inhibition of bromodomain-containing protein 4 ameliorates oxidative stress-mediated apoptosis and cartilage matrix degeneration through activation of NF-E2-related factor 2-heme oxygenase-1 signaling in rat chondrocytes. J. Cell. Biochem. 2018, 119, 7719–7728. [Google Scholar] [CrossRef]
- Ospelt, C.; Camici, G.G.; Engler, A.; Kolling, C.; Vogetseder, A.; Gay, R.E.; Michel, B.A.; Gay, S. Smoking induces transcription of the heat shock protein system in the joints. Ann. Rheum. Dis. 2014, 73, 1423–1426. [Google Scholar] [CrossRef]
- Kallberg, H.; Ding, B.; Padyukov, L.; Bengtsson, C.; Ronnelid, J.; Klareskog, L.; Alfredsson, L.; Group, E.S. Smoking is a major preventable risk factor for rheumatoid arthritis: Estimations of risks after various exposures to cigarette smoke. Ann. Rheum. Dis. 2011, 70, 508–511. [Google Scholar] [CrossRef]
- Carnevali, S.; Petruzzelli, S.; Longoni, B.; Vanacore, R.; Barale, R.; Cipollini, M.; Scatena, F.; Paggiaro, P.; Celi, A.; Giuntini, C. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L955–L963. [Google Scholar] [CrossRef]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Krosel, M.; Gabathuler, M.; Moser, L.; Maciukiewicz, M.; Zullig, T.; Seifritz, T.; Tomsic, M.; Distler, O.; Ospelt, C.; Klein, K. The histone acetyl transferases CBP and p300 regulate stress response pathways in synovial fibroblasts at transcriptional and functional levels. Sci. Rep. 2023, 13, 17112. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seifritz, T.; Brunner, M.; Camarillo Retamosa, E.; Maciukiewicz, M.; Krošel, M.; Moser, L.; Züllig, T.; Tomšič, M.; Distler, O.; Ospelt, C.; et al. BRD3 Regulates the Inflammatory and Stress Response in Rheumatoid Arthritis Synovial Fibroblasts. Biomedicines 2023, 11, 3188. https://doi.org/10.3390/biomedicines11123188
Seifritz T, Brunner M, Camarillo Retamosa E, Maciukiewicz M, Krošel M, Moser L, Züllig T, Tomšič M, Distler O, Ospelt C, et al. BRD3 Regulates the Inflammatory and Stress Response in Rheumatoid Arthritis Synovial Fibroblasts. Biomedicines. 2023; 11(12):3188. https://doi.org/10.3390/biomedicines11123188
Chicago/Turabian StyleSeifritz, Tanja, Matthias Brunner, Eva Camarillo Retamosa, Malgorzata Maciukiewicz, Monika Krošel, Larissa Moser, Thomas Züllig, Matija Tomšič, Oliver Distler, Caroline Ospelt, and et al. 2023. "BRD3 Regulates the Inflammatory and Stress Response in Rheumatoid Arthritis Synovial Fibroblasts" Biomedicines 11, no. 12: 3188. https://doi.org/10.3390/biomedicines11123188