
Animal Models in Diabetic Research—History, Presence, and Future Perspectives

Abstract
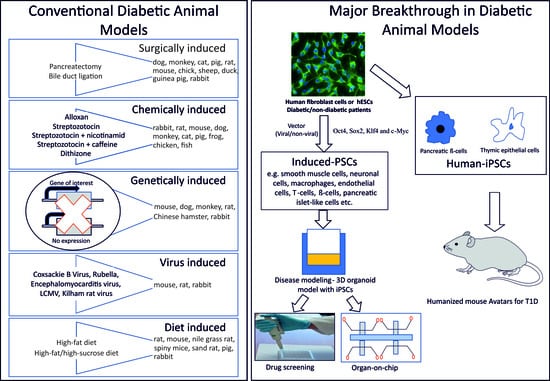
:
1. Introduction
2. History of Animal Models of Diabetes
3. Surgical Models
4. Chemical Models
5. Virus-Induced Models
6. Genetic Models
7. Diet-Induced Models
8. Gestational Diabetes Models
9. Experiments on Animals in the 21st Century
10. Organ-on-Chip Models in Diabetic Research
11. Insulin Resistant Adipose Model
12. Glomerulus-on-a-Chip
13. Pancreas-on-Chip
14. Diabetic Foot Ulcer on Chip
15. iPSCs and Organ-on-Chip
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pandey, S.; Dvorakova, M.C. Significance of animal models in the research of diabetes. Diabetol. Metab. Endokrinol. Vyz. 2019, 22, 65–71. [Google Scholar]
- Pandey, S.; Jirásko, M.; Lochman, J.; Chvátal, A.; Chottova Dvorakova, M.; Kučera, R. iPSCs in Neurodegenerative Disorders: A Unique Platform for Clinical Research and Personalized Medicine. J. Pers. Med. 2022, 12, 1485. [Google Scholar] [CrossRef] [PubMed]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 2. Classification and Diagnosis of Diabetes: Standards of Care in Diabetes-2023. Diabetes Care 2023, 46 (Suppl. 1), S19–S40. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, P.; Urmila, A.; Likhit, A.; Subhash, B.; Shailendra, G. Rodent models for diabetes. 3 Biotech 2023, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- de Leiva-Hidalgo, A.; de Leiva-Pérez, A.; Bruguès-Bruguès, E. From pancreatic extracts to artificial pancreas: History, science and controversies about the discovery of the pancreatic antidiabetic hormone. Av. Diabetol. 2011, 27, 15–26. [Google Scholar] [CrossRef]
- Luft, R. Oskar Minkowski: Discovery of the pancreatic origin of diabetes, 1889. Diabetologia 1989, 32, 399–401. [Google Scholar] [CrossRef]
- DeFronzo, R.A. The triumvirate: Beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988, 37, 667–687. [Google Scholar] [CrossRef]
- Murai, Y.; Ohta, T.; Tadaki, H.; Miyajima, K.; Shinohara, M.; Fatchiyah, F.; Yamada, T. Assessment of Pharmacological Responses to an Anti-diabetic Drug in a New Obese Type 2 Diabetic Rat Model. Med. Arh. 2017, 71, 380–384. [Google Scholar] [CrossRef]
- Rostène, W.; De Meyts, P. Insulin: A 100-Year-Old Discovery with a Fascinating History. Endocr. Rev. 2021, 42, 503–527. [Google Scholar] [CrossRef]
- Allen, F.M. Studies Concerning Glycosuria and Diabetes; Harvard University Press: Cambridge, UK, 1913. [Google Scholar]
- Mirsky, A.; Gitelson, S. The diabetic response of geese to pancreatectomy. Endocrinology 1958, 63, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.; Katz, L.N.; Bolene, C. The Effect of Pancreatectomy on Lipemia, Tissue Lipidosis and Atherogenesis in Chicks. Circulation 1951, 4, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Collins-Williams, J.; Renold, A.E.; Marble, A. Attempts to produce diabetes in guinea pigs by alloxan and pancreatectomy with observations on the effect of a diet deficient in cystine and methionine. Endocrinology 1950, 46, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pauls, F.; Bancroft, R.W. Production of diabetes in the mouse by partial pancreatectomy. Am. J. Physiol. 1950, 160, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Long, C.N.; Lukens, F.D. The effects of adrenalectomy and hypophysectomy upon experimental diabetes in the cat. J. Exp. Med. 1936, 63, 465–490. [Google Scholar] [CrossRef] [PubMed]
- Foglia, V.A. Caracteristicas del la diabetes en la rata. Rev. Soc. Argent. Biol. 1944, 20, 21–37. [Google Scholar]
- Imamura, T.; Koffler, M.; Helderman, J.H.; Prince, D.; Thirlby, R.; Inman, L.; Unger, R.H. Severe diabetes induced in subtotally depancreatized dogs by sustained hyperglycemia. Diabetes 1988, 37, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Kobayashi, N.; Okitsu, T.; Yong, C.; Fukazawa, T.; Ikeda, H.; Kosaka, Y.; Narushima, M.; Arata, T.; Tanaka, N. Development of a porcine model of type 1 diabetes by total pancreatectomy and establishment of a glucose tolerance evaluation method. Artif. Organs 2004, 28, 1035–1042. [Google Scholar] [CrossRef]
- Gao, X.; He, J.; Zhu, A.; Xie, K.; Yan, K.; Jiang, X.; Xu, Y.; Li, Q.; Xu, A.; Ye, D.; et al. Modelling gestational diabetes mellitus: Large animals hold great promise. Rev. Endocr. Metab. Disord. 2021, 22, 407–420. [Google Scholar] [CrossRef]
- Walpole, A.L.; Innes, J.R. Experimental diabetes: The effect of ligation of the pancreatic duct upon the action of alloxan in rabbits. Br. J. Pharmacol. Chemother. 1946, 1, 174–185. [Google Scholar] [CrossRef]
- Catala, J.; Bonnafous, R.; Hollande, E. Disturbances in the regulation of glycaemia in rabbits following pancreatic duct ligation. Biochemical and immunocytochemical studies. Diabetes Metab. 1986, 12, 203–211. [Google Scholar]
- Martin, J.M.; Lacy, P.E. The prediabetic period in partially pancreatectomized rats. Diabetes 1963, 12, 238–242. [Google Scholar] [CrossRef]
- Dhuria, R.S.; Singh, G.; Kaur, A.; Kaur, R.; Kaur, T. Current status and patent prospective of animal models in diabetic research. Adv. Biomed. Res. 2015, 4, 117. [Google Scholar]
- Jacobs, H.R. Hypoglycemic Action of Alloxan. Proc. Soc. Exp. Biol. Med. 1937, 37, 407–409. [Google Scholar] [CrossRef]
- Dunn, J.S.; Mcletchie, N.G.B. Experimental alloxan diabetes in the rat. Lancet 1943, 242, 384–387. [Google Scholar] [CrossRef]
- Goldner, M.G.; Gomori, G. Studies on the mechanism of alloxan diabetes. Endocrinology 1944, 35, 241–248. [Google Scholar] [CrossRef]
- Dunn, J.M.; Kirkpatrick, J.; Mcletchie, N.; Telfer, S. Necrosis of the islets of Langerhans produced experimentally. J. Pathol. 1943, 55, 245–257. [Google Scholar] [CrossRef]
- Rakieten, N.; Rakieten, M.L.; Nadkarni, M.R. Studies on the diabetogenic action of streptozotocin (NSC-37917). Cancer Chemother. Rep. 1963, 29, 91–98. [Google Scholar]
- Pandey, S.; Chottova Dvorakova, M. Future perspective of diabetic animal models. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 25–38. [Google Scholar] [CrossRef]
- Szkudelski, T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 2001, 50, 537–546. [Google Scholar]
- Gorus, F.K.; Malaisse, W.J.; Pipeleers, D.G. Selective uptake of alloxan by pancreatic B-cells. Biochem. J. 1982, 208, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Elsner, M.; Tiedge, M.; Guldbakke, B.; Munday, R.; Lenzen, S. Importance of the GLUT2 glucose transporter for pancreatic beta cell toxicity of alloxan. Diabetologia 2002, 45, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Jörns, A.; Munday, R.; Tiedge, M.; Lenzen, S. Comparative toxicity of alloxan, N-alkylalloxans and ninhydrin to isolated pancreatic islets in vitro. J. Endocrinol. 1997, 155, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 2008, 51, 216–226. [Google Scholar] [CrossRef]
- Gorray, K.C.; Baskin, D.; Brodsky, J.; Fujimoto, W.Y. Responses of pancreatic b cells to alloxan and streptozotocin in the guinea pig. Pancreas 1986, 1, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Ramfjord, S. Clinical and histologic effects of alloxan in rhesus monkeys. Am. J. Clin. Pathol. 1952, 22, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Dufrane, D.; van Steenberghe, M.; Guiot, Y.; Goebbels, R.M.; Saliez, A.; Gianello, P. Streptozotocin-Induced Diabetes in Large Animals (Pigs/Primates): Role of GLUT2 Transporter and β-cell Plasticity. Transplantation 2006, 81, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Romsos, D.R.; Leveille, G.A.; Allee, G.L. Alloxan diabetes in the pig (Sus domesticus). Response to glucose, tolbutamide and insulin administration. Comp. Biochem. Physiol. A Comp. Physiol. 1971, 40, 557–568. [Google Scholar] [CrossRef]
- Logothetopoulos, J.; Brosky, G. Mitotic activity of islet cells in alloxan and streptozotocin diabetic mice studies by radioautography. Diabetes 1968, 17, 306. [Google Scholar]
- Lazarus, S.S.; Shapiro, S.H. Comparison of morphologic changes in nuclei of rabbit pancreatic islet B-cells induced by streptozotocin, alloxan, and in vitro necrosis. Lab. Investig. 1973, 29, 90–98. [Google Scholar]
- Portha, B.; Picon, L.; Rosselin, G. Chemical diabetes in the adult rat as the spontaneous evolution of neonatal diabetes. Diabetologia 1979, 17, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Danby, R.; Bluff, L.; Deheny, T.P.; Gibson, W.R. Effects of alloxan and streptozotocin at high doses on blood glucose levels, glucose tolerance, and responsiveness to sulphonylureas in chickens. Gen. Comp. Endocrinol. 1982, 47, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Castiñeiras, M.J.; Boronat, A.; Itarte, E.; Guinovart, J.J.; Rosell-Pérez, M. Inducción de diabetes experimental en la rana [Induction of experimental diabetes in frog (author’s transl)]. Rev. Esp. Fisiol. 1978, 34, 385–388. [Google Scholar] [PubMed]
- Kumar, S.; Khanna, S.S. Influence of alloxan administration on the blood glucose, islets of langerhans and some other tissues of the frog, Rana tigrina. Anat. Anz. 1978, 143, 242–249. [Google Scholar] [PubMed]
- Kumar, S.; Khanna, S.S. Blood glucose and pancreatic islets in frogs after streptozotocin treatment. Anat. Anz. 1981, 150, 335–342. [Google Scholar] [PubMed]
- Kerns, K.C.; Farrar, E.S. Streptozotocin treated bullfrogs fail to develop insulin deficiency. Comp. Biochem. Physiol. A Comp. Physiol. 1986, 85, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Gill, T.S.; Khanna, S.S. Effect of streptozotocin on the blood glucose level and histology of the principal islets of Channa punctatus (Bloch). Z. Mikrosk. Anat. Forsch. 1975, 89, 319–326. [Google Scholar] [PubMed]
- Gill, T.S.; Khanna, S.S. Effect of alloxan administration upon Channa punctatus (Bloch). Z. Mikrosk. Anat. Forsch. 1974, 88, 673–680. [Google Scholar]
- Wright, J.R., Jr.; Abraham, C.; Dickson, B.C.; Yang, H.; Morrison, C.M. Streptozotocin dose-response curve in tilapia, a glucose-responsive teleost fish. Gen. Comp. Endocrinol. 1999, 114, 431–440. [Google Scholar] [CrossRef]
- Xu, B.Y.; Morrison, C.M.; Yang, H.; Wright, J.R., Jr. Tilapia islet grafts are highly alloxan-resistant. Gen. Comp. Endocrinol. 2004, 137, 132–140. [Google Scholar] [CrossRef]
- Sharchil, C.; Vijay, A.; Ramachandran, V.; Bhagavatheeswaran, S.; Devarajan, R.; Koul, B.; Yadav, D.; Balakrishnan, A. Zebrafish: A Model to Study and Understand the Diabetic Nephropathy and Other Microvascular Complications of Type 2 Diabetes Mellitus. Vet. Sci. 2022, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.S.; Sarras, M.P., Jr.; Intine, R.V. Limb regeneration is impaired in an adult zebrafish model of diabetes mellitus. Wound Repair Regen. 2010, 18, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Black, H.E.; Rosenblum, I.Y.; Capen, C.C. Chemically induced (streptozotocin-alloxan) diabetes mellitus in the dog. Biochemical and ultrastructural studies. Am. J. Pathol. 1980, 98, 295–310. [Google Scholar] [PubMed]
- Han, Q.; Sun, J.; Xie, W.; Bai, Y.; Wang, S.; Huang, J.; Zhou, S.; Li, Q.; Zhang, H.; Tang, Z. Repeated Low-Dose Streptozotocin and Alloxan Induced Long-Term and Stable Type 1 Diabetes Model in Beagle Dogs. Biomed. Res. Int. 2022, 2022, 5422287. [Google Scholar] [CrossRef] [PubMed]
- Tschoepe, D.; Job, F.P.; Huebinger, A.; Freytag, G.; Torsello, G.; Peter, B.; Gries, F.A. Combined subtotal pancreatectomy with selective streptozotocin infusion—A model for the induction of insulin deficiency in dogs. Res. Exp. Med. 1989, 189, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zeng, L.; He, S.; Chen, Y.; Tian, B.; Mai, G.; Yang, G.; Wei, L.; Zhang, Y.; Li, H.; et al. Comparison of single high-dose streptozotocin with partial pancreatectomy combined with low-dose streptozotocin for diabetes induction in rhesus monkeys. Exp. Biol. Med. 2010, 235, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Reiser, H.J.; Whitworth UGJr Hatchell, D.L.; Sutherland, F.S.; Nanda, S.; McAdoo, T.; Hardin, J.R. Experimental diabetes in cats induced by partial pancreatectomy alone or combined with local injection of alloxan. Lab. Anim. Sci. 1987, 37, 449–452. [Google Scholar] [PubMed]
- Phares, C.K. Streptozotocin-induced diabetes in Syrian hamsters: New model of diabetes mellitus. Experientia 1980, 36, 681–682. [Google Scholar] [CrossRef]
- Nakamura, T.; Terajima, T.; Ogata, T.; Ueno, K.; Hashimoto, N.; Ono, K.; Yano, S. Establishment and pathophysiological characterization of type 2 diabetic mouse model produced by streptozotocin and nicotinamide. Biol. Pharm. Bull. 2006, 29, 1167–1174. [Google Scholar] [CrossRef]
- Naidoo, P.; Islam, M.S. Development of an alternative non-obese non-genetic rat model of type 2 diabetes using caffeine and streptozotocin. Pharmacol. Rep. 2014, 66, 585–593. [Google Scholar] [CrossRef]
- Zhang, F.; Ye, C.; Li, G.; Ding, W.; Zhou, W.; Zhu, H.; Chen, G.; Luo, T.; Guang, M.; Liu, Y.; et al. The rat model of type 2 diabetic mellitus and its glycometabolism characters. Exp. Anim. 2003, 52, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.D.; Islam, M.S. Fructose-fed streptozotocin-injected rat: An alternative model for type 2 diabetes. Pharmacol. Rep. 2012, 64, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wang, N.; Li, G.; Guo, W.; Yang, C.; Liu, D. Establishment and Assessment of Mice Models of Type 2 Diabetes Mellitus. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2017, 39, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, S.J.; Mroz, Z.; Dekker, R.; Corbijn, H.; Ackermans, M.; Sauerwein, H. Association of insulin resistance with hyperglycemia in streptozotocin-diabetic pigs: Effects of metformin at isoenergetic feeding in a type 2-like diabetic pig model. Metabolism 2006, 55, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Capiotti, K.M.; Antonioli RJr Kist, L.W.; Bogo, M.R.; Bonan, C.D.; Da Silva, R.S. Persistent impaired glucose metabolism in a zebrafish hyperglycemia model. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2014, 171, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Engerman, R.L.; Kern, T.S. Experimental galactosemia produces diabetic-like retinopathy. Diabetes 1984, 33, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S.; Engerman, R.L. Comparison of retinal lesions in alloxan-diabetic rats and galactose-fed rats. Curr. Eye Res. 1994, 13, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Sidenius, P.; Jakobsen, J. Axonal transport in rats after galactose feeding. Diabetologia 1980, 19, 229–233. [Google Scholar] [CrossRef]
- Kadota, I. Studies on experimental diabetes mellitus, as produced by organic reagents; oxine diabetes and dithizone diabetes. J. Lab. Clin. Med. 1950, 35, 568–591. [Google Scholar]
- Epand, R.M.; Stafford, A.R.; Tyers, M.; Nieboer, E. Mechanism of action of diabetogenic zinc-chelating agents. Model system studies. Mol. Pharmacol. 1985, 27, 366–374. [Google Scholar]
- Kharat, A.; Sanap, A.; Kheur, S.; Shekatkar, M.; Bhonde, R. Insulin-producing cell clusters derived from human gingival mesenchymal stem cells as a model for diabetes research. Mol. Biol. Rep. 2022, 49, 11973–11982. [Google Scholar] [CrossRef]
- Haber, R.S.; Weinstein, S.P. Role of Glucose Transporters in Glucocorticoid-Induced Insulin Resistance: GLUT4 Isoform in Rat Skeletal Muscle is Not Decreased by Dexamethasone. Diabetes 1992, 41, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.R.; Turner, S.L.; Jefferson, W.H.; Bailey, C.J. Prevention of dexamethasone-induced insulin resistance by metformin. Biochem. Pharmacol. 1998, 56, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Furman, B.L. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. 2021, 1, e78. [Google Scholar] [CrossRef]
- Dekel, Y.; Glucksam, Y.; Elron-Gross, I.; Margalit, R. Insights into modeling streptozotocin-induced diabetes in ICR mice. Lab. Anim. 2009, 38, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Lukić, M.L.; Stosić-Grujicić, S.; Shahin, A. Effector mechanisms in low-dose streptozotocin-induced diabetes. Dev. Immunol. 1998, 6, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Haliga, R.; Mocanu, V.; Oboroceanu, T.; Stitt, P.A.; Luca, V.C. The effects of dietary flaxseed supplementation on lipid metabolism in streptozotocin-induced diabetic hamsters. Rev. Med. Chir. Soc. Med. Nat. Iasi 2007, 111, 472–476. [Google Scholar] [PubMed]
- Sako, T.; Mori, A.; Lee, P.; Goto, H.; Fukuta, H.; Oda, H.; Saeki, K.; Miki, Y.; Makino, Y.; Ishioka, K.; et al. Supplementing transglucosidase with a high-fiber diet for prevention of postprandial hyperglycemia in streptozotocin-induced diabetic dogs. Vet. Res. Commun. 2010, 34, 161–172. [Google Scholar] [CrossRef]
- Grüssner, R.; Nakhleh, R.; Grüssner, A.; Tomadze, G.; Diem, P.; Sutherland, D. Streptozotocin-induced diabetes mellitus in pigs. Horm. Metab. Res. 1993, 25, 199–203. [Google Scholar] [CrossRef]
- Rahman, S.; Jan, G.; Jan, F.G.; Rahim, H.U. Phytochemical Analysis and hypoglycemic potential of Filago hurdwarica (Wall. ex DC.) Wagenitz in alloxan induced diabetic mice. Braz. J. Biol. 2022, 84, e261518. [Google Scholar] [CrossRef]
- Galagudza, M.M.; Nekrasova, M.K.; Syrenskii, A.V.; Nifontov, E.M. Resistance of the myocardium to ischemia and the efficacy of ischemic preconditioning in experimental diabetes mellitus. Neurosci. Behav. Physiol. 2007, 37, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Federiuk, I.F.; Casey, H.M.; Quinn, M.J.; Wood, M.D.; Ward, W.K. Induction of type-1 diabetes mellitus in laboratory rats by use of alloxan: Route of administration, pitfalls, and insulin treatment. Comp. Med. 2004, 54, 252–257. [Google Scholar] [PubMed]
- Vieira, G.T.; de Oliveira, T.T.; Carneiro, M.A.A.; Cangussu, S.D.; Humberto, G.A.P.; Taylor, J.G.; Humberto, J.L. Antidiabetic effect of Equisetum giganteum L. extract on alloxan-diabetic rabbit. J. Ethnopharmacol. 2020, 260, 112898. [Google Scholar] [CrossRef] [PubMed]
- Badin, J.K.; Kole, A.; Stivers, B.; Progar, V.; Pareddy, A.; Alloosh, M.; Sturek, M. Alloxan-induced diabetes exacerbates coronary atherosclerosis and calcification in Ossabaw miniature swine with metabolic syndrome. J. Transl. Med. 2018, 16, 58. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Viswanad, B.; Asrat, L.; Kaul, C.L.; Ramarao, P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: A model for type 2 diabetes and pharmacological screening. Pharmacol. Res. 2005, 52, 313–320. [Google Scholar] [CrossRef] [PubMed]
- From, G.L.; Craighead, J.E.; McLane, M.F.; Steinke, J. Virus-induced diabetes in mice. Metabolism 1968, 17, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Onodera, T.; Notkins, A.L. Virus-induced diabetes mellitus. XV. Beta cell damage and insulin-dependent hyperglycemia in mice infected with coxsackie virus B4. J. Exp. Med. 1978, 148, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Jenson, A.B.; Yoon, J.W.; Notkins, A.L. Virus-induced diabetes mellitus: Reovirus infection of pancreatic beta cells in mice. Science 1978, 201, 529–531. [Google Scholar] [CrossRef]
- Menser, M.A.; Forrest, J.M.; Bransby, R.D. Rubella infection and diabetes mellitus. Lancet 1978, 1, 57–60. [Google Scholar] [CrossRef]
- Filippi, C.M.; von Herrath, M.G. Viral trigger for type 1 diabetes: Pros and cons. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef]
- Mine, K.; Takahashi, H.; Nagafuchi, S. Model Animal Mimicking Human Virus-induced Diabetes. eBioMedicine 2018, 32, 8. [Google Scholar] [CrossRef] [PubMed]
- Meier, H.; Yerganian, G.A. Spontaneous hereditary diabetes mellitus in Chinese hamster (Cricetulus griseus). 1. Pathological findings. Proc. Soc. Exp. Biol. Med. 1959, 100, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Kunimoto, K.; Muraoka, Y.; Mizushima, Y.; Katagiri, K.; Tochino, Y. Breeding of a non-obese, diabetic strain of mice. Exp. Anim. 1980, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, H.T.; Petersen, E.S.; Steiner, P.E.; Tupikova, N. Spontaneous diabetes mellitus in the dog: An account of eight cases. Diabetes 1953, 2, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Lutty, G.A.; McLeod, D.S.; Otsuji, T.; Flower, R.W.; Sandagar, G.; Alexander, T.; Steidl, S.M.; Hansen, B.C. Ocular structure and function in an aged monkey with spontaneous diabetes mellitus. Exp. Eye Res. 2005, 80, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, N.; Kulkarni, R.N. Molecular approaches to study control of glucose homeostasis. ILAR J. 2006, 47, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Kawano, K.; Hirashima, T.; Mori, S.; Saitoh, Y.; Kurosumi, M.; Natori, T. New inbred strain of Long-Evans Tokushima lean rats with IDDM without lymphopenia. Diabetes 1991, 40, 1375–1381. [Google Scholar] [CrossRef]
- Yokoi, N.; Kanazawa, M.; Kitada, K.; Tanaka, A.; Kanazawa, Y.; Suda, S.; Ito, H.; Serikawa, T.; Komeda, K. A non-MHC locus essential for autoimmune type I diabetes in the Komeda Diabetes-Prone rat. J. Clin. Investig. 1997, 100, 2015–2021. [Google Scholar] [CrossRef]
- Lenzen, S.; Tiedge, M.; Elsner, M.; Lortz, S.; Weiss, H.; Jörns, A.; Klöppel, G.; Wedekind, D.; Prokop, C.M.; Hedrich, H.J. The LEW.1AR1/Ztm-iddm rat: A new model of spontaneous insulin-dependent diabetes mellitus. Diabetologia 2001, 44, 1189–1196. [Google Scholar] [CrossRef]
- Kramer, J.W.; Nottingham, S.; Robinette, J.; Lenz, G.; Sylvester, S.; Dessouky, M.I. Inherited, early onset, insulin-requiring diabetes mellitus of Keeshond dogs. Diabetes 1980, 29, 558–565. [Google Scholar] [CrossRef]
- Conaway, H.H.; Brown, C.J.; Sanders, L.L.; Cernosek, S.F.; Farris, H.E.; Roth, S.I. Spontaneous diabetes mellitus in the New Zealand white rabbit: History, classification, and genetic analysis. J. Hered. 1980, 71, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Cefalu, W.T. Animal models of type 2 diabetes: Clinical presentation and pathophysiological relevance to the human condition. ILAR J. 2006, 47, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Suzuki, K.; Ono, T.; Sasaki, M.; Toyota, T. Development of diabetes in the non-obese NIDDM rat (GK rat). Adv. Exp. Med. Biol. 1988, 246, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Shafrir, E.; Gutman, A. Psammomys obesus of the Jerusalem colony: A model for nutritionally induced, non-insulin-dependent diabetes. J. Basic. Clin. Physiol. Pharmacol. 1993, 4, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Kawano, K.; Hirashima, T.; Mori, S.; Saitoh, Y.; Kurosumi, M.; Natori, T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes 1992, 41, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Weksler-Zangen, S.; Yagil, C.; Zangen, D.H.; Ornoy, A.; Jacob, H.J.; Yagil, Y. The newly inbred cohen diabetic rat: A nonobese normolipidemic genetic model of diet-induced type 2 diabetes expressing sex differences. Diabetes 2001, 50, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Masuyama, T.; Shoda, T.; Takahashi, T.; Katsuda, Y.; Komeda, K.; Kuroki, M.; Kakehashi, A.; Kanazawa, Y. A new spontaneously diabetic non-obese Torii rat strain with severe ocular complications. Int. J. Exp. Diabetes Res. 2000, 1, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.G.; Jackson, C.V.; Zimmerman, K.; de Winter, W.; Huebert, N.; Hansen, M.K. Characterization of the ZDSD Rat: A Translational Model for the Study of Metabolic Syndrome and Type 2 Diabetes. J. Diabetes Res. 2015, 2015, 487816. [Google Scholar] [CrossRef]
- Wang, A.N.; Carlos, J.; Fraser, G.M.; McGuire, J.J. Zucker Diabetic-Sprague Dawley (ZDSD) rat: Type 2 diabetes translational research model. Exp. Physiol. 2022, 107, 265–282. [Google Scholar] [CrossRef]
- Cummings, B.P.; Digitale, E.K.; Stanhope, K.L.; Graham, J.L.; Baskin, D.G.; Reed, B.J.; Sweet, I.R.; Griffen, S.C.; Havel, P.J. Development and characterization of a novel rat model of type 2 diabetes mellitus: The UC Davis type 2 diabetes mellitus UCD-T2DM rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1782–R1793. [Google Scholar] [CrossRef]
- Nakamura, M.; Yamada, K. Studies on a diabetic (KK) strain of the mouse. Diabetologia 1967, 3, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuka, H.; Shino, A.; Suzuoki, Z. General survey of diabetic features of yellow KK mice. Endocrinol. Jpn. 1970, 17, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, W.; Iizuka, S.; Tabuchi, M.; Funo, S.; Yanagisawa, T.; Kimura, M.; Sato, T.; Endo, T.; Kawamura, H. A new mouse model of spontaneous diabetes derived from ddY strain. Exp. Anim. 1999, 48, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Bielschowsky, F.; Bielschowsky, M. The New Zealand strain of obese mice; their response to stilboestrol and to insulin. Aust. J. Exp. Biol. Med. Sci. 1956, 34, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.W.; Panepinto, L.M.; Spangler, R.; Westmoreland, N. Yucatan miniature swine as a model for the study of human diabetes mellitus. Diabetes 1982, 31 (Suppl. 1), 30–36. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.A.; Merricks, E.P.; Nichols, T.C. Swine models of type 2 diabetes mellitus: Insulin resistance, glucose tolerance, and cardiovascular complications. ILAR J. 2006, 47, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, M.; Kayo, T.; Ikeda, T.; Koizumi, A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 1997, 46, 887–894. [Google Scholar] [CrossRef]
- Matveyenko, A.V.; Butler, P.C. Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J. 2006, 47, 225–233. [Google Scholar] [CrossRef]
- Shibata, M.; Yasuda, B. Spontaneously occurring diabetes in NSY mice. Jikken Dobutsu 1979, 28, 584–590. [Google Scholar]
- Kaneko, K.; Chikamoto, A.; Hsu, J.C.; Tochinai, R.; Sekizawa, S.I.; Yamamoto, M.; Kuwahara, M. Effects of environmental enrichment on autonomic nervous activity in NSY mice. Exp. Anim. 2020, 69, 161–167. [Google Scholar] [CrossRef]
- Babaya, N.; Ueda, H.; Noso, S.; Hiromine, Y.; Itoi-Babaya, M.; Kobayashi, M.; Fujisawa, T.; Ikegami, H. Genetic dissection of susceptibility genes for diabetes and related phenotypes on mouse chromosome 14 by means of congenic strains. BMC Genet. 2014, 15, 93. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Miyasaka, Y.; Yoshida, K.; Kobayashi, M.; Horio, F.; Yokoi, N.; Mizuno, M.; Ikegami, H. A novel model mouse for type 2 diabetes mellitus with early onset and persistent hyperglycemia. Exp. Anim. 2022, 71, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Houssay, B.A.; Martínez, C. Experimental Diabetes and Diet. Science 1947, 105, 548–549. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Nielsen, K.; Haines, H.B.; Hackel, D.B. Diabetes mellitus in the sand rat induced by standard laboratory diets. Science 1964, 143, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Kalman, R.; Ziv, E.; Shafrir, E.; Bar-On, H.; Perez, R. Psammomys obesus and the albino rat—Two different models of nutritional insulin resistance, representing two different types of human populations. Lab. Anim. 2001, 35, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Alexander, J.T.; Zheng, P.; Yu, H.J.; Dourmashkin, J.; Leibowitz, S.F. Behavioral and endocrine traits of obesity-prone and obesity-resistant rats on macronutrient diets. Am. J. Physiol. 1998, 274, E1057–E1066. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Goriya, Y.; Sekimoto, H. Osteopenic changes in high sugar diet-induced diabetic rabbits (HSDD-R). Diabetes Res. 1991, 18, 115–122. [Google Scholar] [PubMed]
- Yin, W.; Yuan, Z.; Tsutsumi, K.; Xie, Y.; Zhang, Q.; Wang, Z.; Fu, G.; Long, G.; Yang, Y. A lipoprotein lipase-promoting agent, NO-1886, improves glucose and lipid metabolism in high fat, high sucrose-fed New Zealand white rabbits. Int. J. Exp. Diabesity Res. 2003, 4, 27–34. [Google Scholar] [CrossRef]
- Xi, S.; Yin, W.; Wang, Z.; Kusunoki, M.; Lian, X.; Koike, T.; Fan, J.; Zhang, Q. A minipig model of high-fat/high-sucrose diet-induced diabetes and atherosclerosis. Int. J. Exp. Pathol. 2004, 85, 223–231. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Yin, W.; Li, Q.; Cai, M.; Zhang, C.; Xiao, J.; Hou, H.; Li, H.; Zu, X. Severe insulin resistance and moderate glomerulosclerosis in a minipig model induced by high-fat/high-sucrose/ high-cholesterol diet. Exp. Anim. 2007, 56, 11–20. [Google Scholar] [CrossRef]
- Chen, H.; Liu, Y.Q.; Li, C.H.; Guo, X.M.; Huang, L.J. The susceptibility of three strains of Chinese minipigs to diet-induced type 2 diabetes mellitus. Lab. Anim. 2009, 38, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, S.J.; Schuurman, T. Considerations on pig models for appetite, metabolic syndrome and obese type 2 diabetes: From food intake to metabolic disease. Eur. J. Pharmacol. 2015, 759, 231–239. [Google Scholar] [CrossRef]
- Hummel, K.P.; Coleman, D.L.; Lane, P.W. The influence of genetic background on expression of mutations at the diabetes locus in the mouse. I. C57BL-KsJ and C57BL-6J strains. Biochem. Genet. 1972, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Shafrir, E.; Ziv, E.; Kalman, R. Nutritionally induced diabetes in desert rodents as models of type 2 diabetes: Acomys cahirinus (spiny mice) and Psammomys obesus (desert gerbil). ILAR J. 2006, 47, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Melhorn, M.I.; Zandi, S.; Frimmel, S.; Tayyari, F.; Hisatomi, T.; Almulki, L.; Pronczuk, A.; Hayes, K.C.; Hafezi-Moghadam, A. An animal model of spontaneous metabolic syndrome: Nile grass rat. FASEB J. 2010, 24, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhu, X.; Odiba, A.S.; Lin, Z.; Wen, J.; Gong, D.; Liang, J.; Wu, S.; Lan, G. Comparatively analyzing the liver-specific transcriptomic profiles in Kunming mice afflicted with streptozotocin- and natural food-induced type 2 diabetes mellitus. Mol. Biol. Rep. 2022, 49, 1369–1377. [Google Scholar] [CrossRef]
- Carlson, A.J.; Drennan, F.M. The control of pancreatic diabetes in pregnancy by the passage of the internal secretion of the pancreas of the fetus to the blood of the mother. Am. J. Physiol. 1911, 28, 391–395. [Google Scholar] [CrossRef]
- Markowitz, J.; Soskin, S. Pancreatic diabetes and pregnancy. Am. J. Physiol. 1927, 79, 553–558. [Google Scholar] [CrossRef]
- He, Y.; Wu, N.; Yu, W.; Li, L.; OuYang, H.; Liu, X.; Qian, M.; Al-Mureish, A. Research Progress on the Experimental Animal Model of Gestational Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 4235–4247. [Google Scholar] [CrossRef]
- Chandrasekera, P.C.; Pippin, J.J. Of rodents and men: Species-specific glucose regulation and type 2 diabetes research. ALTEX 2014, 31, 157–176. [Google Scholar] [CrossRef] [PubMed]
- Brissova, M.; Fowler, M.J.; Nicholson, W.E.; Chu, A.; Hirshberg, B.; Harlan, D.M.; Powers, A.C. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem. 2005, 53, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.J.; Kim, A.; Miller, K.; Hara, M. Pancreatic islet plasticity: Interspecies comparison of islet architecture and composition. Islets 2010, 2, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Joksimovic, S.L.; Jevtovic-Todorovic, V.; Todorovic, S.M. The Mechanisms of Plasticity of Nociceptive Ion Channels in Painful Diabetic Neuropathy. Front. Pain Res. 2022, 3, 869735. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Sin, A.; Chin, K.C.; Jamil, M.F.; Kostov, Y.; Rao, G.; Shuler, M.L. The design and fabrication of three-chamber microscale cell culture analog devices with integrated dissolved oxygen sensors. Biotechnol. Prog. 2004, 20, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Li, H.; Collins, J.J.; Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA 2016, 113, E7–E15. [Google Scholar] [CrossRef]
- Nakao, Y.; Kimura, H.; Sakai, Y.; Fujii, T. Bile canaliculi formation by aligning rat primary hepatocytes in a microfluidic device. Biomicrofluidics 2011, 5, 22212. [Google Scholar] [CrossRef]
- Gröger, M.; Rennert, K.; Giszas, B.; Weiß, E.; Dinger, J.; Funke, H.; Kiehntopf, M.; Peters, F.T.; Lupp, A.; Bauer, M.; et al. Corrigendum: Monocyte-induced recovery of inflammation-associated hepatocellular dysfunction in a biochip-based human liver model. Sci. Rep. 2018, 8, 46988. [Google Scholar] [CrossRef]
- Vedula, E.M.; Alonso, J.L.; Arnaout, M.A.; Charest, J.L. A microfluidic renal proximal tubule with active reabsorptive function. PLoS ONE 2017, 12, e0184330. [Google Scholar] [CrossRef]
- Ligresti, G.; Nagao, R.J.; Xue, J.; Choi, Y.J.; Xu, J.; Ren, S.; Aburatani, T.; Anderson, S.K.; MacDonald, J.W.; Bammler, T.K.; et al. A novel three-dimensional human peritubular microvascular system. J. Am. Soc. Nephrol. 2016, 27, 2370–2381. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.J.; Suh, K.Y. A multi-layer microfluidic device for efficient culture and analysis of renal tubular cells. Lab Chip 2010, 10, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.J.; Mehr, A.P.; Hamilton, G.A.; McPartlin, L.A.; Chung, S.; Suh, K.Y.; Ingber, D.E. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. 2013, 5, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Villenave, R.; Lucchesi, C.; Varone, A.; Hubeau, C.; Lee, H.H.; Alves, S.E.; Salmon, M.; Ferrante, T.C.; Weaver, J.C.; et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 2016, 13, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.W.; Olefsky, J.M. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 2005, 280, 35361–35371. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Hacohen, N.; Golub, T.R.; Van Parijs, L.; Lodish, H.F. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3t3-l1 adipocytes: Nuclear factor-kappab activation by tnf-alpha is obligatory. Diabetes 2002, 51, 1319–1336. [Google Scholar] [CrossRef] [PubMed]
- Rotter, V.; Nagaev, I.; Smith, U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J. Biol. Chem. 2003, 278, 45777–45784. [Google Scholar] [CrossRef] [PubMed]
- Godwin, L.A.; Brooks, J.C.; Hoepfner, L.D.; Wanders, D.; Judd, R.L.; Easley, C.J. A microfluidic interface for the culture and sampling of adiponectin from primary adipocytes. Analyst 2015, 140, 1019–1025. [Google Scholar] [CrossRef]
- Li, X.; Easley, C.J. Microfluidic systems for studying dynamic function of adipocytes and adipose tissue. Anal. Bioanal. Chem. 2018, 410, 791–800. [Google Scholar] [CrossRef]
- Liu, Y.; Kongsuphol, P.; Chiam, S.Y.; Zhang, Q.X.; Gourikutty, S.B.N.; Saha, S.; Biswas, S.K.; Ramadan, Q. Adipose-on-a-chip: A dynamic microphysiological in vitro model of the human adipose for immune-metabolic analysis in type II diabetes. Lab Chip 2019, 19, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Kongsuphol, P.; Gupta, S.; Liu, Y.; Bhuvanendran Nair Gourikutty, S.; Biswas, S.K.; Ramadan, Q. In vitro micro-physiological model of the inflamed human adipose tissue for immune-metabolic analysis in type II diabetes. Sci. Rep. 2019, 9, 4887. [Google Scholar] [CrossRef]
- Tanataweethum, N.; Zhong, F.; Trang, A.; Lee, C.; Cohen, R.N.; Bhushan, A. Towards an Insulin Resistant Adipose Model on a Chip. Cell. Mol. Bioeng. 2020, 14, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; LesherPerez, S.C.; Kim, B.C.; Yamanishi, C.; Labuz, J.M.; Leung, B.; Takayama, S. Pharmacokinetic profile that reduces nephrotoxicity of gentamicin in a perfused kidney-on-a-chip. Biofabrication 2016, 8, 015021. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.J.; Cho, H.S.; Kang, D.H.; Bae, W.G.; Kwon, T.H.; Suh, K.Y. Fluid-shear-stress-induced translocation of aquaporin-2 and reorganization of actin cytoskeleton in renal tubular epithelial cells. Integr. Biol. 2011, 3, 134–141. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, X.; Wen, X.; Wu, T.; Wang, W.; Yang, M.; Wang, J.; Fang, M.; Lin, B.; Lin, H. Development of a Functional Glomerulus at the Organ Level on a Chip to Mimic Hypertensive Nephropathy. Sci. Rep. 2016, 6, 31771. [Google Scholar] [CrossRef]
- Wang, L.; Tao, T.; Su, W.; Yu, H.; Yu, Y.; Qin, J. A disease model of diabetic nephropathy in a glomerulus-on-a-chip microdevice. Lab Chip 2017, 17, 1749–1760. [Google Scholar] [CrossRef]
- Petrosyan, A.; Cravedi, P.; Villani, V.; Angeletti, A.; Manrique, J.; Renieri, A.; De Filippo, R.E.; Perin, L.; Da Sacco, S. A glomerulus-on-a-chip to recapitulate the human glomerular filtration barrier. Nat. Commun. 2019, 10, 3656. [Google Scholar] [CrossRef]
- Xie, R.; Korolj, A.; Liu, C.; Song, X.; Lu, R.X.Z.; Zhang, B.; Ramachandran, A.; Liang, Q.; Radisic, M. h-FIBER: Microfluidic Topographical Hollow Fiber for Studies of Glomerular Filtration Barrier. ACS Cent. Sci. 2020, 6, 903–912. [Google Scholar] [CrossRef]
- Perin, L.; Da Sacco, S. Generation of a Glomerular Filtration Barrier on a Glomerulus-on-a-Chip Platform. Methods Mol. Biol. 2022, 2373, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Nourmohammadzadeh, M.; Elias, J.E.; Chan, M.; Chen, Z.; McGarrigle, J.J.; Oberholzer, J.; Wang, Y. A pumpless microfluidic device driven by surface tension for pancreatic islet analysis. Biomed. Microdevices 2016, 18, 80. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Wennberg Huldt, C.; Kanebratt, K.P.; Durieux, I.; Gunne, D.; Andersson, S.; Ewart, L.; Haynes, W.G.; Maschmeyer, I.; Winter, A.; et al. Functional coupling of human pancreatic islets and liver spheroids on-a-chip: Towards a novel human ex vivo type 2 diabetes model. Sci. Rep. 2017, 7, 14620. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moncayo, R.; Jimenez-Valdes, R.J.; Gonzalez-Suarez, A.M.; Garcia-Cordero, J.L. Integrated Microfluidic Device for Functional Secretory Immunophenotyping of Immune Cells. ACS Sens. 2020, 5, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Comas, J.; Ramón-Azcón, J. Islet-on-a-chip for the study of pancreatic β-cell function. In Vitro Models 2022, 1, 41–57. [Google Scholar] [CrossRef]
- Abadpour, S.; Aizenshtadt, A.; Olsen, P.A.; Shoji, K.; Wilson, S.R.; Krauss, S.; Scholz, H. Pancreas-on-a-Chip Technology for Transplantation Applications. Curr. Diabetes Rep. 2020, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Alberti, M.; Dancik, Y.; Wu, B.; Wu, R.; Feng, Z.; Ramasamy, S.; Bigliardi, P.L.; Qi, M.B.; Wang, Z. Full-thickness human skin-on-chip with enhanced epidermal morphogenesis and barrier function. Mater. Today 2018, 21, 326–340. [Google Scholar] [CrossRef]
- Lukács, B.; Bajza, Á.; Kocsis, D.; Csorba, A.; Antal, I.; Iván, K.; Laki, A.J.; Erdő, F. Skin-on-a-Chip Device for Ex Vivo Monitoring of Transdermal Delivery of Drugs-Design, Fabrication, and Testing. Pharmaceutics 2019, 11, 445. [Google Scholar] [CrossRef]
- Ejiugwo, M.; Rochev, Y.; Gethin, G.; O’Connor, G. Toward Developing Immunocompetent Diabetic Foot Ulcer-on-a-Chip Models for Drug Testing. Tissue Eng. Part C Methods 2021, 27, 77–88. [Google Scholar] [CrossRef]
- Fanizza, F.; Campanile, M.; Forloni, G.; Giordano, C.; Albani, D. Induced pluripotent stem cell-based organ-on-a-chip as personalized drug screening tools: A focus on neurodegenerative disorders. J. Tissue Eng. 2022, 13, 20417314221095339. [Google Scholar] [CrossRef]
- Tsamandouras, N.; Chen, W.L.K.; Edington, C.D.; Stokes, C.L.; Griffith, L.G.; Cirit, M. Integrated Gut and Liver Microphysiological Systems for Quantitative In Vitro Pharmacokinetic Studies. AAPS J. 2017, 19, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Shinha, K.; Nihei, W.; Ono, T.; Nakazato, R.; Kimura, H. A pharmacokinetic-pharmacodynamic model based on multi-organ-on-a-chip for drug-drug interaction studies. Biomicrofluidics 2020, 14, 044108. [Google Scholar] [CrossRef] [PubMed]
- Saiding, Q.; Ma, J.; Ke, C.; Cui, W. From “organs on a chip” to “patient on a chip”. Innovation 2022, 3, 100282. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Kam, C.; Shuler, M.L. A microfluidic device for a pharmacokinetic-pharmacodynamic (PK-PD) model on a chip. Lab Chip 2010, 10, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Musah, S.; Mammoto, A.; Ferrante, T.C.; Jeanty, S.S.F.; Hirano-Kobayashi, M.; Mammoto, T.; Roberts, K.; Chung, S.; Novak, R.; Ingram, M.; et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat. Biomed. Eng. 2017, 1, 0069. [Google Scholar] [CrossRef] [PubMed]
- Naumovska, E.; Aalderink, G.; Wong Valencia, C.; Kosim, K.; Nicolas, A.; Brown, S.; Vulto, P.; Erdmann, K.S.; Kurek, D. Direct On-Chip Differentiation of Intestinal Tubules from Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 4964. [Google Scholar] [CrossRef] [PubMed]
- Kujala, V.J.; Pasqualini, F.S.; Goss, J.A.; Nawroth, J.C.; Parker, K.K. Laminar ventricular myocardium on a microelectrode array-based chip. J. Mater. Chem. B 2016, 4, 3534–3543. [Google Scholar] [CrossRef]
- Marsano, A.; Conficconi, C.; Lemme, M.; Occhetta, P.; Gaudiello, E.; Votta, E.; Cerino, G.; Redaelli, A.; Rasponi, M. Beating heart on a chip: A novel microfluidic platform to generate functional 3D cardiac microtissues. Lab Chip 2016, 16, 599–610. [Google Scholar] [CrossRef]
- Qian, F.; Huang, C.; Lin, Y.D.; Ivanovskaya, A.N.; O’Hara, T.J.; Booth, R.H.; Creek, C.J.; Enright, H.A.; Soscia, D.A.; Belle, A.M.; et al. Simultaneous electrical recording of cardiac electrophysiology and contraction on chip. Lab Chip 2017, 17, 1732–1739. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Deng, P.; Chen, W.; Guo, Y.; Tao, T.; Qin, J. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab Chip 2018, 18, 3606–3616. [Google Scholar] [CrossRef]
- Sakolish, C.; Reese, C.E.; Luo, Y.S.; Valdiviezo, A.; Schurdak, M.E.; Gough, A.; Taylor, D.L.; Chiu, W.A.; Vernetti, L.A.; Rusyn, I. Analysis of reproducibility and robustness of a human microfluidic four-cell liver acinus microphysiology system (LAMPS). Toxicology 2021, 448, 152651. [Google Scholar] [CrossRef]
- Tissue Chips for Modeling Diabetes. 2018. Available online: https://ncats.nih.gov/tissuechip/projects/modeling/2018 (accessed on 18 September 2023).
- Cheatham, W.W. Peroxisome proliferator-activated receptor translational research and clinical experience. Am. J. Clin. Nutr. 2010, 91, 262S–266S. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Animal | Dose | Reference |
|---|---|---|---|
| STZ | Mouse | Multiple low-dose: 40 mg/kg i.p. for 5 consecutive days | [75] |
| Single high-dose: 100–200 mg/kg i.v. or i.p. | [76] | ||
| Rat | Multiple low-dose: 20 mg/kg i.p. for 5 consecutive days | [77] | |
| Single high-dose: 40–70 mg/kg i.v. or i.p. | [75] | ||
| Hamster | 50 mg/kg i.p. | [78] | |
| Dog | 25 mg/kg i.v. | [79] | |
| Pig | 150 mg/kg i.v. | [80] | |
| Primates | 50–150 mg/kg i.v. | [24] | |
| Alloxan | Mouse | 150 mg/kg i.p. | [81] |
| Rat | 125 mg/kg s.c. | [82] | |
| 200 mg/kg i.p. | [83] | ||
| Rabbit | 100 mg/kg i.p. | [84] | |
| Dog | 50–75 mg/kg i.v. | [24] | |
| Pig | 100–175 mg/kg i.v. | [85] |
| Animal | Chemical | Diet | Reference |
|---|---|---|---|
| Mouse | NA 240 mg/kg i.p. (15 min before STZ) STZ 100 mg/kg i.p. (twice on day 0 and 2) | HFD | [60] |
| Rat | NA 230 mg/kg i.p. (15 min before STZ) STZ 65 mg/kg i.v. | Normal | [24] |
| STZ 35 mg/kg i.p. (after 2 weeks of HFD) | HFD (58% calories as fat) | [86] | |
| STZ 40 mg/kg i.p. (after 2 weeks of FDS10, then normal water) | FDS10 | [63] | |
| Caffeine 20 mg/kg i.p. (15 min before STZ) STZ 65 mg/kg i.p. | Normal | [61] | |
| Pig | STZ 130 mg/kg as 30 min slow infusion | Low fat diet (5% calories as fat) | [65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, S.; Chmelir, T.; Chottova Dvorakova, M. Animal Models in Diabetic Research—History, Presence, and Future Perspectives. Biomedicines 2023, 11, 2852. https://doi.org/10.3390/biomedicines11102852
Pandey S, Chmelir T, Chottova Dvorakova M. Animal Models in Diabetic Research—History, Presence, and Future Perspectives. Biomedicines. 2023; 11(10):2852. https://doi.org/10.3390/biomedicines11102852
Chicago/Turabian StylePandey, Shashank, Tomas Chmelir, and Magdalena Chottova Dvorakova. 2023. "Animal Models in Diabetic Research—History, Presence, and Future Perspectives" Biomedicines 11, no. 10: 2852. https://doi.org/10.3390/biomedicines11102852