

Small Molecule Targeting Immune Cells: A Novel Approach for Cancer Treatment
 , ,
, ,
Abstract
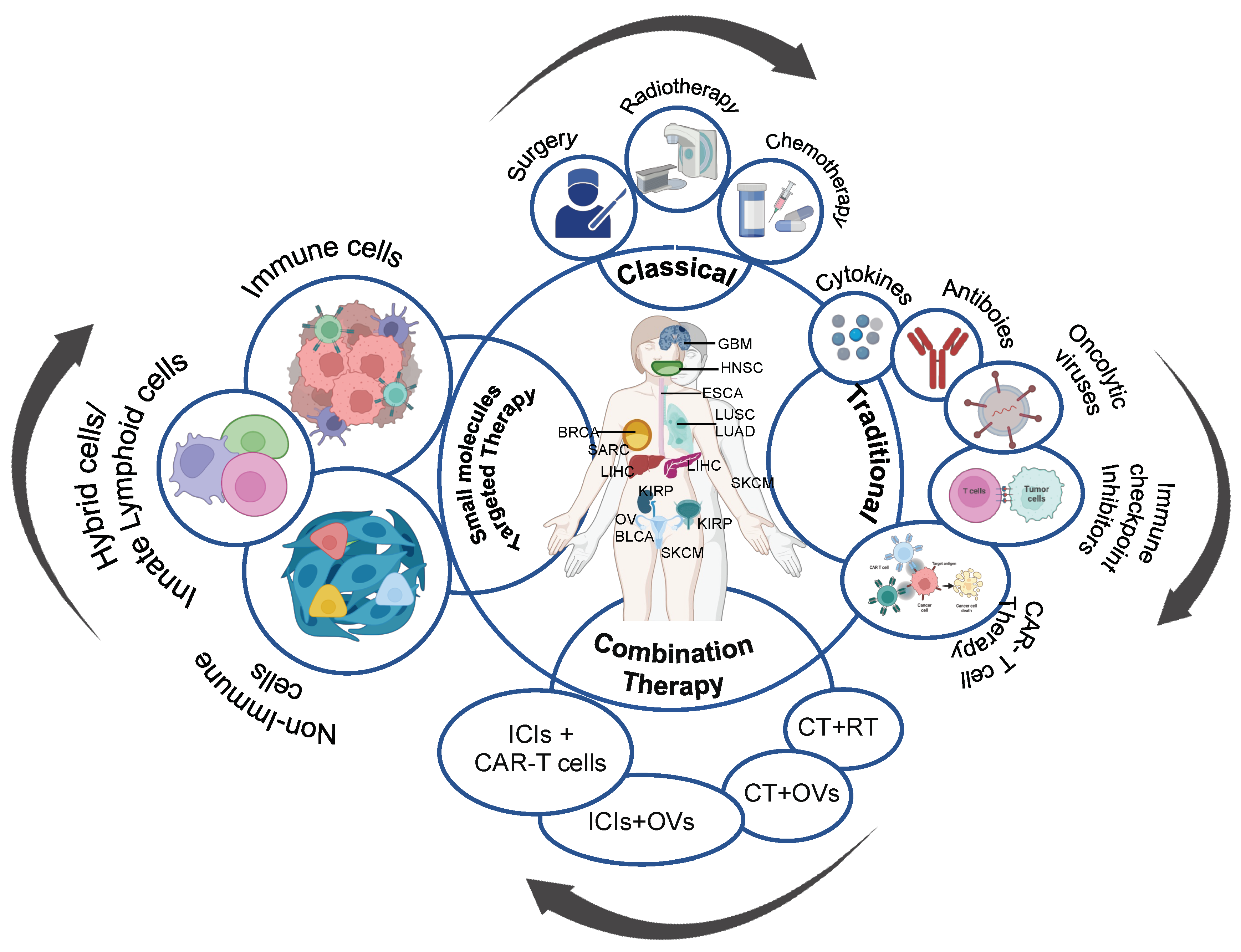
:1. Introduction
2. Targeted Small Molecule Therapy
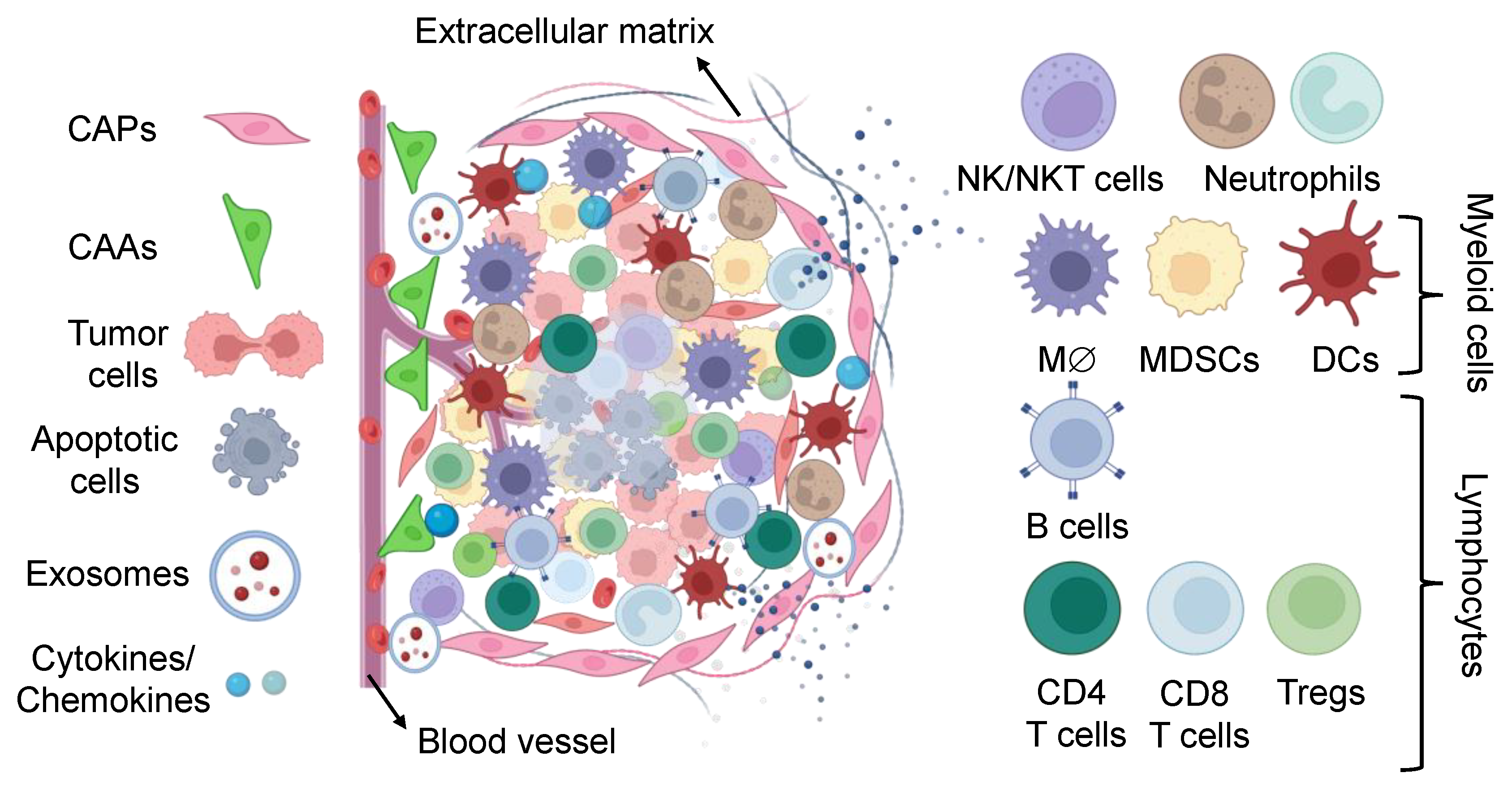
3. Targeting Tumor Microenvironment: A Possible Route for Tumor Containment
3.1. Targeting Non-Immune Cells for Cancer Therapy
3.1.1. Cancer-Associated Fibroblasts
3.1.2. Cancer-Associated Adipocytes
3.1.3. Tumor Endothelial Cells
3.1.4. Extracellular Vesicles
3.2. Targeting Immune Cells for Cancer Therapy
3.2.1. Tumor-Infiltrating Natural Killer Cells
3.2.2. Invariant Natural Killer T Cells
3.2.3. Natural Killer Cells
3.2.4. Neutrophils
3.2.5. Tumor-Associated Neutrophils
3.2.6. Tumor-Associated Macrophages
3.2.7. Myeloid-Derived Suppressor Cells
3.2.8. Dendritic Cells
3.2.9. B Cells
3.2.10. Helper CD4+ T Cell
3.2.11. Cytotoxic CD8+ T Cell
3.2.12. Tissue-Resident Memory T Cells
3.2.13. Regulatory T Cells
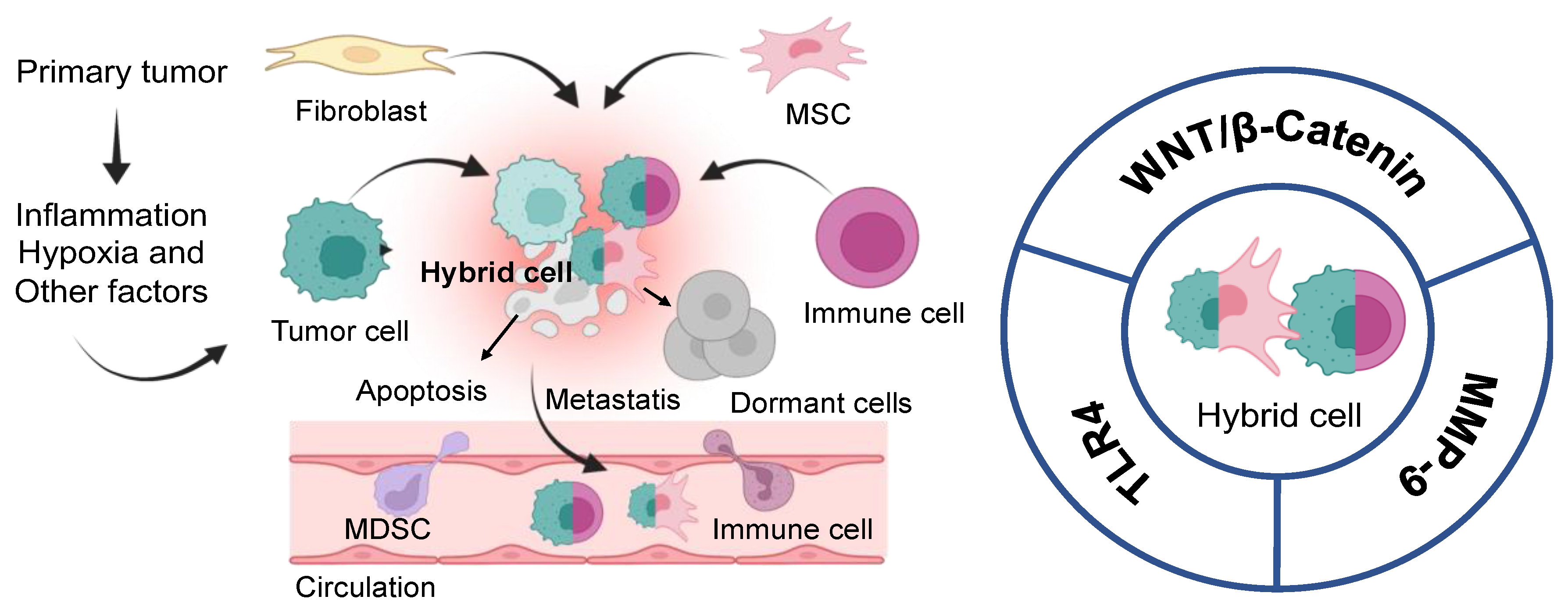
4. Hybrid Cells or New Immune Cells “Soldier”
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Björnmalm, M.; Thurecht, K.J.; Michael, M.; Scott, A.M.; Caruso, F. Bridging Bio-Nano Science and Cancer Nanomedicine. ACS Nano 2017, 11, 9594–9613. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Gun, S.Y.; Lee, S.W.L.; Sieow, J.L.; Wong, S.C. Targeting Immune Cells for Cancer Therapy. Redox Biol. 2019, 25, 101174. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Dobosz, P.; Stępień, M.; Golke, A.; Dzieciątkowski, T. Challenges of the Immunotherapy: Perspectives and Limitations of the Immune Checkpoint Inhibitor Treatment. Int. J. Mol. Sci. 2022, 23, 2847. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Kaufman, H.L.; Wigginton, J.M.; Wolchok, J.D. Combination Immunotherapy: A Road Map. J. Immunother. Cancer 2017, 5, 16. [Google Scholar] [CrossRef]
- Liu, G.-H.; Chen, T.; Zhang, X.; Ma, X.-L.; Shi, H.-S. Small Molecule Inhibitors Targeting the Cancers. MedComm (2020) 2022, 3, e181. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small Molecules, Big Impact: 20 Years of Targeted Therapy in Oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Osipov, A.; Saung, M.T.; Zheng, L.; Murphy, A.G. Small Molecule Immunomodulation: The Tumor Microenvironment and Overcoming Immune Escape. J. Immunother. Cancer 2019, 7, 224. [Google Scholar] [CrossRef]
- Van der Zanden, S.Y.; Luimstra, J.J.; Neefjes, J.; Borst, J.; Ovaa, H. Opportunities for Small Molecules in Cancer Immunotherapy. Trends Immunol. 2020, 41, 493–511. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Wolf, D.; Tilg, H.; Rumpold, H.; Gastl, G.; Wolf, A.M. The Kinase Inhibitor Imatinib—An Immunosuppressive Drug? Curr. Cancer Drug Targets 2007, 7, 251–258. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I Interferons in Anticancer Immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Bagheri, S.; Rahban, M.; Bostanian, F.; Esmaeilzadeh, F.; Bagherabadi, A.; Zolghadri, S.; Stanek, A. Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment. Pharmaceutics 2022, 14, 515. [Google Scholar] [CrossRef]
- Wang, N.; Ma, T.; Yu, B. Targeting Epigenetic Regulators to Overcome Drug Resistance in Cancers. Signal Transduct. Target. Ther. 2023, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Genova, C.; Dellepiane, C.; Carrega, P.; Sommariva, S.; Ferlazzo, G.; Pronzato, P.; Gangemi, R.; Filaci, G.; Coco, S.; Croce, M. Therapeutic Implications of Tumor Microenvironment in Lung Cancer: Focus on Immune Checkpoint Blockade. Front. Immunol. 2021, 12, 799455. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Netea-Maier, R.T.; Smit, J.W.A.; Netea, M.G. Metabolic Changes in Tumor Cells and Tumor-Associated Macrophages: A Mutual Relationship. Cancer Lett. 2018, 413, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-Associated Fibroblasts Enhance Tumor-Associated Macrophages Enrichment and Suppress NK Cells Function in Colorectal Cancer. Cell Death Dis. 2019, 10, 273. [Google Scholar] [CrossRef]
- Min, A.; Zhu, C.; Wang, J.; Peng, S.; Shuai, C.; Gao, S.; Tang, Z.; Su, T. Focal Adhesion Kinase Knockdown in Carcinoma-Associated Fibroblasts Inhibits Oral Squamous Cell Carcinoma Metastasis via Downregulating MCP-1/CCL2 Expression. J. Biochem. Mol. Toxicol. 2015, 29, 70–76. [Google Scholar] [CrossRef]
- Wright, K.; Ly, T.; Kriet, M.; Czirok, A.; Thomas, S.M. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers 2023, 15, 1899. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2020, 31, 148–161.e5. [Google Scholar] [CrossRef]
- Fain, J.N. Release of Interleukins and Other Inflammatory Cytokines by Human Adipose Tissue Is Enhanced in Obesity and Primarily Due to the Nonfat Cells. Vitam. Horm. 2006, 74, 443–477. [Google Scholar] [CrossRef]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity Promotes Breast Cancer by CCL2-Mediated Macrophage Recruitment and Angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor Endothelial Cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed]
- Georganaki, M.; Ramachandran, M.; Tuit, S.; Núñez, N.G.; Karampatzakis, A.; Fotaki, G.; van Hooren, L.; Huang, H.; Lugano, R.; Ulas, T.; et al. Tumor Endothelial Cell Up-Regulation of IDO1 Is an Immunosuppressive Feed-Back Mechanism That Reduces the Response to CD40-Stimulating Immunotherapy. Oncoimmunology 2020, 9, 1730538. [Google Scholar] [CrossRef]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.-M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-like Character in PDGF-Induced Glioma Cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.M.; Mehdipour, M.; Monfaredan, A.; Jahanban-Esfahlan, R. Hesa-A Down-Regulates Erb/B2 Oncogene Expression and Improves Outcome of Oral Carcinoma in a Rat Model. Asian Pac. J. Cancer Prev. 2015, 16, 6947–6951. [Google Scholar] [CrossRef] [PubMed]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural Killer Cell Cytotoxicity Is Suppressed by Exposure to the Human NKG2D Ligand MICA*008 That Is Shed by Tumor Cells in Exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, H.; Zhangyuan, G.; Huang, R.; Zhang, W.; He, Q.; Jin, K.; Zhuo, H.; Zhang, Z.; Wang, J.; et al. 14-3-3ζ Delivered by Hepatocellular Carcinoma-Derived Exosomes Impaired Anti-Tumor Function of Tumor-Infiltrating T Lymphocytes. Cell Death Dis. 2018, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Battke, C.; Ruiss, R.; Welsch, U.; Wimberger, P.; Lang, S.; Jochum, S.; Zeidler, R. Tumour Exosomes Inhibit Binding of Tumour-Reactive Antibodies to Tumour Cells and Reduce ADCC. Cancer Immunol. Immunother. 2011, 60, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-Derived Exosomes Induce N2 Polarization of Neutrophils to Promote Gastric Cancer Cell Migration. Mol. Cancer 2018, 17, 146. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.-P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D. Membrane-Associated Hsp72 from Tumor-Derived Exosomes Mediates STAT3-Dependent Immunosuppressive Function of Mouse and Human Myeloid-Derived Suppressor Cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-Derived Microvesicles Induce, Expand and up-Regulate Biological Activities of Human Regulatory T Cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, X.; Zhang, B.; Shi, H.; Yuan, X.; Sun, Y.; Pan, Z.; Qian, H.; Xu, W. Exosomes Derived from Gastric Cancer Cells Activate NF-ΚB Pathway in Macrophages to Promote Cancer Progression. Tumor Biol. 2016, 37, 12169–12180. [Google Scholar] [CrossRef]
- Wu, C.-H.; Li, J.; Li, L.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C.; Jong, A.Y. Extracellular Vesicles Derived from Natural Killer Cells Use Multiple Cytotoxic Proteins and Killing Mechanisms to Target Cancer Cells. J. Extracell. Vesicles 2019, 8, 1588538. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR Exosomes Derived from Effector CAR-T Cells Have Potent Antitumour Effects and Low Toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef]
- Wu, S.-W.; Li, L.; Wang, Y.; Xiao, Z. CTL-Derived Exosomes Enhance the Activation of CTLs Stimulated by Low-Affinity Peptides. Front. Immunol. 2019, 10, 1274. [Google Scholar] [CrossRef]
- Bruno, A.; Ferlazzo, G.; Albini, A.; Noonan, D.M. A Think Tank of TINK/TANKs: Tumor-Infiltrating/Tumor-Associated Natural Killer Cells in Tumor Progression and Angiogenesis. J. Natl. Cancer Inst. 2014, 106, dju200. [Google Scholar] [CrossRef]
- Park, Y.-J.; Song, B.; Kim, Y.-S.; Kim, E.-K.; Lee, J.-M.; Lee, G.-E.; Kim, J.-O.; Kim, Y.-J.; Chang, W.-S.; Kang, C.-Y. Tumor Microenvironmental Conversion of Natural Killer Cells into Myeloid-Derived Suppressor Cells. Cancer Res. 2013, 73, 5669–5681. [Google Scholar] [CrossRef]
- Artis, D.; Spits, H. The Biology of Innate Lymphoid Cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Bernink, J.H.; Lanier, L. NK Cells and Type 1 Innate Lymphoid Cells: Partners in Host Defense. Nat. Immunol. 2016, 17, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Irshad, S.; Flores-Borja, F.; Lawler, K.; Monypenny, J.; Evans, R.; Male, V.; Gordon, P.; Cheung, A.; Gazinska, P.; Noor, F.; et al. RORγt+ Innate Lymphoid Cells Promote Lymph Node Metastasis of Breast Cancers. Cancer Res. 2017, 77, 1083–1096. [Google Scholar] [CrossRef]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-Based Mechanisms of Cytotoxic Chemotherapy: Implications for the Design of Novel and Rationale-Based Combined Treatments against Cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef]
- Childs, R.W.; Carlsten, M. Therapeutic Approaches to Enhance Natural Killer Cell Cytotoxicity against Cancer: The Force Awakens. Nat. Rev. Drug Discov. 2015, 14, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Lagrue, K.; Carisey, A.; Morgan, D.J.; Chopra, R.; Davis, D.M. Lenalidomide Augments Actin Remodeling and Lowers NK-Cell Activation Thresholds. Blood 2015, 126, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fayad, L.; Wagner-Bartak, N.; Zhang, L.; Hagemeister, F.; Neelapu, S.S.; Samaniego, F.; McLaughlin, P.; Fanale, M.; Younes, A.; et al. Lenalidomide in Combination with Rituximab for Patients with Relapsed or Refractory Mantle-Cell Lymphoma: A Phase 1/2 Clinical Trial. Lancet Oncol. 2012, 13, 716–723. [Google Scholar] [CrossRef]
- Badoux, X.C.; Keating, M.J.; Wen, S.; Wierda, W.G.; O’Brien, S.M.; Faderl, S.; Sargent, R.; Burger, J.A.; Ferrajoli, A. Phase II Study of Lenalidomide and Rituximab as Salvage Therapy for Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2013, 31, 584–591. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Sagiv-Barfi, I.; Rafiq, S.; Herman, S.E.M.; Butchar, J.P.; Cheney, C.; Zhang, X.; Buggy, J.J.; Muthusamy, N.; Levy, R.; et al. Ibrutinib Antagonizes Rituximab-Dependent NK Cell-Mediated Cytotoxicity. Blood 2014, 123, 1957–1960. [Google Scholar] [CrossRef]
- Xu, Z.; Zhu, X.; Su, L.; Zou, C.; Chen, X.; Hou, Y.; Gong, C.; Ng, W.; Ni, Z.; Wang, L.; et al. A High-Throughput Assay for Screening Natural Products That Boost NK Cell-Mediated Killing of Cancer Cells. Pharm. Biol. 2020, 58, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.P.; Emens, L.A. The Multikinase Inhibitor Sorafenib Reverses the Suppression of IL-12 and Enhancement of IL-10 by PGE2 in Murine Macrophages. Int. Immunopharmacol. 2010, 10, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Zollo, M.; Di Dato, V.; Spano, D.; De Martino, D.; Liguori, L.; Marino, N.; Vastolo, V.; Navas, L.; Garrone, B.; Mangano, G.; et al. Targeting Monocyte Chemotactic Protein-1 Synthesis with Bindarit Induces Tumor Regression in Prostate and Breast Cancer Animal Models. Clin. Exp. Metastasis 2012, 29, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Liguori, M.; Buracchi, C.; Pasqualini, F.; Bergomas, F.; Pesce, S.; Sironi, M.; Grizzi, F.; Mantovani, A.; Belgiovine, C.; Allavena, P. Functional TRAIL Receptors in Monocytes and Tumor-Associated Macrophages: A Possible Targeting Pathway in the Tumor Microenvironment. Oncotarget 2016, 7, 41662–41676. [Google Scholar] [CrossRef]
- Shen, L.; Sundstedt, A.; Ciesielski, M.; Miles, K.M.; Celander, M.; Adelaiye, R.; Orillion, A.; Ciamporcero, E.; Ramakrishnan, S.; Ellis, L.; et al. Tasquinimod Modulates Suppressive Myeloid Cells and Enhances Cancer Immunotherapies in Murine Models. Cancer Immunol. Res. 2015, 3, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Li, H.-J.; Chen, K.-G.; Wang, Y.-C.; Yang, X.-Z.; Lian, Z.-X.; Du, J.-Z.; Wang, J. Spatial Targeting of Tumor-Associated Macrophages and Tumor Cells with a PH-Sensitive Cluster Nanocarrier for Cancer Chemoimmunotherapy. Nano Lett. 2017, 17, 3822–3829. [Google Scholar] [CrossRef]
- Binnemars-Postma, K.; Bansal, R.; Storm, G.; Prakash, J. Targeting the Stat6 Pathway in Tumor-Associated Macrophages Reduces Tumor Growth and Metastatic Niche Formation in Breast Cancer. FASEB J. 2018, 32, 969–978. [Google Scholar] [CrossRef]
- Zhong, H.; Gutkin, D.W.; Han, B.; Ma, Y.; Keskinov, A.A.; Shurin, M.R.; Shurin, G.V. Origin and Pharmacological Modulation of Tumor-Associated Regulatory Dendritic Cells. Int. J. Cancer 2014, 134, 2633–2645. [Google Scholar] [CrossRef]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 Inhibition Augments Endogenous Antitumor Immunity by Reducing Myeloid-Derived Suppressor Cell Function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib Inhibition of Stat3 Induces Renal Cell Carcinoma Tumor Cell Apoptosis and Reduces Immunosuppressive Cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Niccoli, P.; Castellano, D.; Valle, J.W.; Hammel, P.; Raoul, J.-L.; Vinik, A.; Van Cutsem, E.; Bang, Y.-J.; Lee, S.-H.; et al. Sunitinib in Pancreatic Neuroendocrine Tumors: Updated Progression-Free Survival and Final Overall Survival from a Phase III Randomized Study. Ann. Oncol. 2017, 28, 339–343. [Google Scholar] [CrossRef]
- Xu, R.; Zhang, Y.; Ye, X.; Xue, S.; Shi, J.; Pan, J.; Chen, Q. Inhibition Effects and Induction of Apoptosis of Flavonoids on the Prostate Cancer Cell Line PC-3 in Vitro. Food Chem. 2013, 138, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Cai, P.; Li, Q.; Wang, W.; Sun, Y.; Xu, Q.; Gu, Y. Axitinib Augments Antitumor Activity in Renal Cell Carcinoma via STAT3-Dependent Reversal of Myeloid-Derived Suppressor Cell Accumulation. Biomed. Pharmacother. 2014, 68, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Abu-Eid, R.; Samara, R.N.; Ozbun, L.; Abdalla, M.Y.; Berzofsky, J.A.; Friedman, K.M.; Mkrtichyan, M.; Khleif, S.N. Selective Inhibition of Regulatory T Cells by Targeting the PI3K-Akt Pathway. Cancer Immunol. Res. 2014, 2, 1080–1089. [Google Scholar] [CrossRef]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.-Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef]
- Taylor, A.; Rothstein, D.; Rudd, C.E. Small-Molecule Inhibition of PD-1 Transcription Is an Effective Alternative to Antibody Blockade in Cancer Therapy. Cancer Res. 2018, 78, 706–717. [Google Scholar] [CrossRef]
- Suvarna, K.; Honda, K.; Kondoh, Y.; Osada, H.; Watanabe, N. Identification of a Small-Molecule Ligand of β-Arrestin1 as an Inhibitor of Stromal Fibroblast Cell Migration Accelerated by Cancer Cells. Cancer Med. 2018, 7, 883–893. [Google Scholar] [CrossRef]
- Grither, W.R.; Longmore, G.D. Inhibition of Tumor-Microenvironment Interaction and Tumor Invasion by Small-Molecule Allosteric Inhibitor of DDR2 Extracellular Domain. Proc. Natl. Acad. Sci. USA 2018, 115, E7786–E7794. [Google Scholar] [CrossRef]
- Kim, D.J.; Dunleavey, J.M.; Xiao, L.; Ollila, D.W.; Troester, M.A.; Otey, C.A.; Li, W.; Barker, T.H.; Dudley, A.C. Suppression of TGFβ-Mediated Conversion of Endothelial Cells and Fibroblasts into Cancer Associated (Myo)Fibroblasts via HDAC Inhibition. Br. J. Cancer 2018, 118, 1359–1368. [Google Scholar] [CrossRef]
- Mertens, J.C.; Fingas, C.D.; Christensen, J.D.; Smoot, R.L.; Bronk, S.F.; Werneburg, N.W.; Gustafson, M.P.; Dietz, A.B.; Roberts, L.R.; Sirica, A.E.; et al. Therapeutic Effects of Deleting Cancer-Associated Fibroblasts in Cholangiocarcinoma. Cancer Res. 2013, 73, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In Vivo Antitumor Activity of SU11248, a Novel Tyrosine Kinase Inhibitor Targeting Vascular Endothelial Growth Factor and Platelet-Derived Growth Factor Receptors: Determination of a Pharmacokinetic/Pharmacodynamic Relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R.; et al. Pazopanib versus Sunitinib in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Kujawski, M.; Wu, J.; Lu, J.; Herrmann, A.; Loera, S.; Yen, Y.; Lee, F.; Yu, H.; Wen, W.; et al. Antitumor Activity of Targeting Src Kinases in Endothelial and Myeloid Cell Compartments of the Tumor Microenvironment. Clin. Cancer Res. 2010, 16, 924. [Google Scholar] [CrossRef]
- Satchi-Fainaro, R.; Mamluk, R.; Wang, L.; Short, S.M.; Nagy, J.A.; Feng, D.; Dvorak, A.M.; Dvorak, H.F.; Puder, M.; Mukhopadhyay, D.; et al. Inhibition of Vessel Permeability by TNP-470 and Its Polymer Conjugate, Caplostatin. Cancer Cell 2005, 7, 251–261. [Google Scholar] [CrossRef]
- Soares, J.; Espadinha, M.; Raimundo, L.; Ramos, H.; Gomes, A.S.; Gomes, S.; Loureiro, J.B.; Inga, A.; Reis, F.; Gomes, C.; et al. DIMP53-1: A Novel Small-Molecule Dual Inhibitor of P53-MDM2/X Interactions with Multifunctional P53-Dependent Anticancer Properties. Mol. Oncol. 2017, 11, 612–627. [Google Scholar] [CrossRef]
- Podar, K.; Tonon, G.; Sattler, M.; Tai, Y.-T.; LeGouill, S.; Yasui, H.; Ishitsuka, K.; Kumar, S.; Kumar, R.; Pandite, L.N.; et al. The Small-Molecule VEGF Receptor Inhibitor Pazopanib (GW786034B) Targets Both Tumor and Endothelial Cells in Multiple Myeloma. Proc. Natl. Acad. Sci. USA 2006, 103, 19478–19483. [Google Scholar] [CrossRef]
- Vu, H.N.; Miller, W.J.; O’Connor, S.A.; He, M.; Schafer, P.H.; Payvandi, F.; Muller, G.W.; Stirling, D.I.; Libutti, S.K. CC-5079: A Small Molecule with MKP1, Antiangiogenic, and Antitumor Activity. J. Surg. Res. 2010, 164, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an Orally Bioavailable Selective Inhibitor of Protein Kinase CK2, Inhibits Prosurvival and Angiogenic Signaling and Exhibits Antitumor Efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef]
- Bid, H.K.; Oswald, D.; Li, C.; London, C.A.; Lin, J.; Houghton, P.J. Anti-Angiogenic Activity of a Small Molecule STAT3 Inhibitor LLL12. PLoS ONE 2012, 7, e35513. [Google Scholar] [CrossRef]
- Jain, A.; Lai, J.C.K.; Bhushan, A. Biochanin A Inhibits Endothelial Cell Functions and Proangiogenic Pathways: Implications in Glioma Therapy. Anticancer. Drugs 2015, 26, 323–330. [Google Scholar] [CrossRef]
- Sweeny, L.; Liu, Z.; Lancaster, W.; Hart, J.; Hartman, Y.E.; Rosenthal, E.L. Inhibition of Fibroblasts Reduced Head and Neck Cancer Growth by Targeting Fibroblast Growth Factor Receptor. Laryngoscope 2012, 122, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn-Cymerman, D.; Rizzuto, G.A.; Merghoub, T.; Cohen, A.D.; Avogadri, F.; Lesokhin, A.M.; Weinberg, A.D.; Wolchok, J.D.; Houghton, A.N. OX40 Engagement and Chemotherapy Combination Provides Potent Antitumor Immunity with Concomitant Regulatory T Cell Apoptosis. J. Exp. Med. 2009, 206, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, B.D.; Spalding, A.C.; Campos, M.S.; Ashimori, N.; Dong, Z.; Wang, S.; Lawrence, T.S.; Nör, J.E. Metronomic Small Molecule Inhibitor of Bcl-2 (TW-37) Is Anti-Angiogenic and Potentiates the Anti-Tumor Effect of Ionizing Radiation. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 879–887. [Google Scholar] [CrossRef]
- Kraus, D.; Palasuberniam, P.; Chen, B. Targeting Phosphatidylinositol 3-Kinase Signaling Pathway for Therapeutic Enhancement of Vascular-Targeted Photodynamic Therapy. Mol. Cancer Ther. 2017, 16, 2422–2431. [Google Scholar] [CrossRef]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.-B.; Bousquet-Dubouch, M.-P.; et al. Pharmacological Targeting of the Protein Synthesis MTOR/4E-BP1 Pathway in Cancer-Associated Fibroblasts Abrogates Pancreatic Tumour Chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, M.; Yin, T.; Shi, H.; Wen, Y.; Zhang, B.; Chen, M.; Xu, G.; Ren, K.; Wei, Y. Targeting of Cancer-Associated Fibroblasts Enhances the Efficacy of Cancer Chemotherapy by Regulating the Tumor Microenvironment. Mol. Med. Rep. 2016, 13, 2476–2484. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhou, X.; Liu, X.; Jia, H.-H.; Zhao, X.-H.; Wang, Q.-X.; Han, L.; Song, X.; Zhu, Z.-Y.; Sun, T.; et al. Reprogramming Carcinoma Associated Fibroblasts by AC1MMYR2 Impedes Tumor Metastasis and Improves Chemotherapy Efficacy. Cancer Lett. 2016, 374, 96–106. [Google Scholar] [CrossRef]
- Godfrey, D.I.; MacDonald, H.R.; Kronenberg, M.; Smyth, M.J.; Van Kaer, L. NKT Cells: What’s in a Name? Nat. Rev. Immunol. 2004, 4, 231–237. [Google Scholar] [CrossRef]
- Carreño, L.J.; Saavedra-Ávila, N.A.; Porcelli, S.A. Synthetic Glycolipid Activators of Natural Killer T Cells as Immunotherapeutic Agents. Clin. Transl. Immunol. 2016, 5, e69. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Le Nours, J.; Andrews, D.M.; Uldrich, A.P.; Rossjohn, J. Unconventional T Cell Targets for Cancer Immunotherapy. Immunity 2018, 48, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, T.; Kamata, T.; Tanaka, K.; Ihara, F.; Takami, M.; Suzuki, H.; Nakajima, T.; Ikeuchi, T.; Kawasaki, Y.; Hanaoka, H.; et al. Phase II Study of α-Galactosylceramide-Pulsed Antigen-Presenting Cells in Patients with Advanced or Recurrent Non-Small Cell Lung Cancer. J. Immunother. Cancer 2020, 8, e000316. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, M.S.; Karadimitris, A. Role and Regulation of CD1d in Normal and Pathological B Cells. J. Immunol. 2014, 193, 4761–4768. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” Killer Cells in the Mouse. I. Cytotoxic Cells with Specificity for Mouse Moloney Leukemia Cells. Specificity and Distribution According to Genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Ivison, G.T.; Vendrame, E.; Martínez-Colón, G.J.; Ranganath, T.; Vergara, R.; Zhao, N.Q.; Martin, M.P.; Bendall, S.C.; Carrington, M.; Cyktor, J.C.; et al. Natural Killer Cell Receptors and Ligands Are Associated With Markers of HIV-1 Persistence in Chronically Infected ART Suppressed Patients. Front. Cell. Infect. Microbiol. 2022, 12, 757846. [Google Scholar] [CrossRef]
- Moretta, A. Natural Killer Cells and Dendritic Cells: Rendezvous in Abused Tissues. Nat. Rev. Immunol. 2002, 2, 957–964. [Google Scholar] [CrossRef]
- Martinet, L.; Smyth, M.J. Balancing Natural Killer Cell Activation through Paired Receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef]
- Romero, D. Immunotherapy: PD-1 Says Goodbye, TIM-3 Says Hello. Nat. Rev. Clin. Oncol. 2016, 13, 202–203. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Grote, D.M.; Ziesmer, S.C.; Niki, T.; Hirashima, M.; Novak, A.J.; Witzig, T.E.; Ansell, S.M. IL-12 Upregulates TIM-3 Expression and Induces T Cell Exhaustion in Patients with Follicular B Cell Non-Hodgkin Lymphoma. J. Clin. Investig. 2012, 122, 1271–1282. [Google Scholar] [CrossRef]
- Barrow, A.D.; Colonna, M. Exploiting NK Cell Surveillance Pathways for Cancer Therapy. Cancers 2019, 11, 55. [Google Scholar] [CrossRef]
- Bi, J.; Zheng, X.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. TIGIT Safeguards Liver Regeneration through Regulating Natural Killer Cell-Hepatocyte Crosstalk. Hepatology 2014, 60, 1389–1398. [Google Scholar] [CrossRef]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.-G.; Teh, C.; Firth, M.; et al. CIS Is a Potent Checkpoint in NK Cell-Mediated Tumor Immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef]
- Kargl, J.; Busch, S.E.; Yang, G.H.Y.; Kim, K.-H.; Hanke, M.L.; Metz, H.E.; Hubbard, J.J.; Lee, S.M.; Madtes, D.K.; McIntosh, M.W.; et al. Neutrophils Dominate the Immune Cell Composition in Non-Small Cell Lung Cancer. Nat. Commun. 2017, 8, 14381. [Google Scholar] [CrossRef]
- Templeton, A.J.; McNamara, M.G.; Šeruga, B.; Vera-Badillo, F.E.; Aneja, P.; Ocaña, A.; Leibowitz-Amit, R.; Sonpavde, G.; Knox, J.J.; Tran, B.; et al. Prognostic Role of Neutrophil-to-Lymphocyte Ratio in Solid Tumors: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2014, 106, dju124. [Google Scholar] [CrossRef]
- Powell, D.; Lou, M.; Barros Becker, F.; Huttenlocher, A. Cxcr1 Mediates Recruitment of Neutrophils and Supports Proliferation of Tumor-Initiating Astrocytes in Vivo. Sci. Rep. 2018, 8, 13285. [Google Scholar] [CrossRef] [PubMed]
- Timaxian, C.; Vogel, C.F.A.; Orcel, C.; Vetter, D.; Durochat, C.; Chinal, C.; NGuyen, P.; Aknin, M.-L.; Mercier-Nomé, F.; Davy, M.; et al. Pivotal Role for Cxcr2 in Regulating Tumor-Associated Neutrophil in Breast Cancer. Cancers 2021, 13, 2584. [Google Scholar] [CrossRef]
- Purohit, A.; Saxena, S.; Varney, M.; Prajapati, D.R.; Kozel, J.A.; Lazenby, A.; Singh, R.K. Host Cxcr2-Dependent Regulation of Pancreatic Cancer Growth, Angiogenesis, and Metastasis. Am. J. Pathol. 2021, 191, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, D.R.; Molczyk, C.; Purohit, A.; Saxena, S.; Sturgeon, R.; Dave, B.J.; Kumar, S.; Batra, S.K.; Singh, R.K. Small Molecule Antagonist of CXCR2 and CXCR1 Inhibits Tumor Growth, Angiogenesis, and Metastasis in Pancreatic Cancer. Cancer Lett. 2023, 563, 216185. [Google Scholar] [CrossRef] [PubMed]
- Grecian, R.; Whyte, M.K.B.; Walmsley, S.R. The Role of Neutrophils in Cancer. Br. Med. Bull. 2018, 128, 5–14. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils Escort Circulating Tumour Cells to Enable Cell Cycle Progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-Associated Neutrophils in Patients with Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Dallavalasa, S.; Beeraka, N.M.; Basavaraju, C.G.; Tulimilli, S.V.; Sadhu, S.P.; Rajesh, K.; Aliev, G.; Madhunapantula, S.V. The Role of Tumor Associated Macrophages (TAMs) in Cancer Progression, Chemoresistance, Angiogenesis and Metastasis—Current Status. Curr. Med. Chem. 2021, 28, 8203–8236. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting Macrophages: Therapeutic Approaches in Cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.-Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages Impede CD8 T Cells from Reaching Tumor Cells and Limit the Efficacy of Anti-PD-1 Treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.-J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef]
- Brown, J.M.; Recht, L.; Strober, S. The Promise of Targeting Macrophages in Cancer Therapy. Clin. Cancer Res. 2017, 23, 3241–3250. [Google Scholar] [CrossRef]
- Cabrales, P. RRx-001 Acts as a Dual Small Molecule Checkpoint Inhibitor by Downregulating CD47 on Cancer Cells and SIRP-α on Monocytes/Macrophages. Transl. Oncol. 2019, 12, 626–632. [Google Scholar] [CrossRef]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming Resistance to Checkpoint Blockade Therapy by Targeting PI3Kγ in Myeloid Cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting Both Tumour-Associated CXCR2+ Neutrophils and CCR2+ Macrophages Disrupts Myeloid Recruitment and Improves Chemotherapeutic Responses in Pancreatic Ductal Adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages Mediate Gemcitabine Resistance of Pancreatic Adenocarcinoma by Upregulating Cytidine Deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of Myeloid-Derived Suppressor Cells in Cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef]
- Yang, Z.; Guo, J.; Weng, L.; Tang, W.; Jin, S.; Ma, W. Myeloid-Derived Suppressor Cells—New and Exciting Players in Lung Cancer. J. Hematol. Oncol. 2020, 13, 10. [Google Scholar] [CrossRef]
- Deng, Y.; Cheng, J.; Fu, B.; Liu, W.; Chen, G.; Zhang, Q.; Yang, Y. Hepatic Carcinoma-Associated Fibroblasts Enhance Immune Suppression by Facilitating the Generation of Myeloid-Derived Suppressor Cells. Oncogene 2017, 36, 1090–1101. [Google Scholar] [CrossRef]
- Li, A.; Chen, P.; Leng, Y.; Kang, J. Histone Deacetylase 6 Regulates the Immunosuppressive Properties of Cancer-Associated Fibroblasts in Breast Cancer through the STAT3–COX2-Dependent Pathway. Oncogene 2018, 37, 5952–5966. [Google Scholar] [CrossRef]
- Thevenot, P.T.; Sierra, R.A.; Raber, P.L.; Al-Khami, A.A.; Trillo-Tinoco, J.; Zarreii, P.; Ochoa, A.C.; Cui, Y.; Del Valle, L.; Rodriguez, P.C. The Stress-Response Sensor Chop Regulates the Function and Accumulation of Myeloid-Derived Suppressor Cells in Tumors. Immunity 2014, 41, 389–401. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 Is a Novel Direct Target of HIF-1α, and Its Blockade under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Younis, R.H.; Han, K.L.; Webb, T.J. Human Head and Neck Squamous Cell Carcinoma–Associated Semaphorin 4D Induces Expansion of Myeloid-Derived Suppressor Cells. J. Immunol. 2016, 196, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Vadrevu, S.K.; Sharma, S.K.; Chintala, N.K.; Ghouse, S.; Cho, J.-H.; Fairlie, D.P.; Paterson, Y.; Astrinidis, A.; Karbowniczek, M. The Ribosomal Protein S19 Suppresses Antitumor Immune Responses via the Complement C5a Receptor 1. J. Immunol. 2017, 198, 2989–2999. [Google Scholar] [CrossRef] [PubMed]
- Drabczyk-Pluta, M.; Werner, T.; Hoffmann, D.; Leng, Q.; Chen, L.; Dittmer, U.; Zelinskyy, G. Granulocytic Myeloid-Derived Suppressor Cells Suppress Virus-Specific CD8+ T Cell Responses during Acute Friend Retrovirus Infection. Retrovirology 2017, 14, 42. [Google Scholar] [CrossRef]
- Zhang, Z.-N.; Yi, N.; Zhang, T.-W.; Zhang, L.-L.; Wu, X.; Liu, M.; Fu, Y.-J.; He, S.-J.; Jiang, Y.-J.; Ding, H.-B.; et al. Myeloid-Derived Suppressor Cells Associated With Disease Progression in Primary HIV Infection: PD-L1 Blockade Attenuates Inhibition. J. Acquir. Immune Defic. Syndr. 2017, 76, 200–208. [Google Scholar] [CrossRef] [PubMed]
- De Coaña, Y.P.; Poschke, I.; Gentilcore, G.; Mao, Y.; Nyström, M.; Hansson, J.; Masucci, G.V.; Kiessling, R. Ipilimumab Treatment Results in an Early Decrease in the Frequency of Circulating Granulocytic Myeloid-Derived Suppressor Cells as Well as Their Arginase1 ProductionIpilimumab Decreases GrMDSC Frequency and ARG1 Production. Cancer Immunol. Res. 2013, 1, 158–162. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Lesokhin, A.; Merghoub, T.; Wolchok, J.D. Targeting Myeloid-Derived Suppressor Cells with Colony Stimulating Factor-1 Receptor Blockade Can Reverse Immune Resistance to Immunotherapy in Indoleamine 2,3-Dioxygenase-Expressing Tumors. EBioMedicine 2016, 6, 50–58. [Google Scholar] [CrossRef]
- Guislain, A.; Gadiot, J.; Kaiser, A.; Jordanova, E.S.; Broeks, A.; Sanders, J.; van Boven, H.; de Gruijl, T.D.; Haanen, J.B.A.G.; Bex, A.; et al. Sunitinib Pretreatment Improves Tumor-Infiltrating Lymphocyte Expansion by Reduction in Intratumoral Content of Myeloid-Derived Suppressor Cells in Human Renal Cell Carcinoma. Cancer Immunol. Immunother. 2015, 64, 1241–1250. [Google Scholar] [CrossRef]
- Heine, A.; Schilling, J.; Grünwald, B.; Krüger, A.; Gevensleben, H.; Held, S.A.E.; Garbi, N.; Kurts, C.; Brossart, P.; Knolle, P.; et al. The Induction of Human Myeloid Derived Suppressor Cells through Hepatic Stellate Cells Is Dose-Dependently Inhibited by the Tyrosine Kinase Inhibitors Nilotinib, Dasatinib and Sorafenib, but Not Sunitinib. Cancer Immunol. Immunother. 2016, 65, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Du Four, S.; Maenhout, S.K.; De Pierre, K.; Renmans, D.; Niclou, S.P.; Thielemans, K.; Neyns, B.; Aerts, J.L. Axitinib Increases the Infiltration of Immune Cells and Reduces the Suppressive Capacity of Monocytic MDSCs in an Intracranial Mouse Melanoma Model. Oncoimmunology 2015, 4, e998107. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ye, T.H.; Han, Y.P.; Song, H.; Zhang, Y.K.; Xia, Y.; Wang, N.Y.; Xiong, Y.; Song, X.J.; Zhu, Y.X.; et al. Reductions in Myeloid-Derived Suppressor Cells and Lung Metastases Using AZD4547 Treatment of a Metastatic Murine Breast Tumor Model. Cell Physiol. Biochem. 2014, 33, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Saegusa, J.; Matsuki, F.; Akashi, K.; Kageyama, G.; Morinobu, A. Tofacitinib Facilitates the Expansion of Myeloid-Derived Suppressor Cells and Ameliorates Arthritis in SKG Mice. Arthritis Rheumatol. 2015, 67, 893–902. [Google Scholar] [CrossRef]
- Okuma, A.; Hanyu, A.; Watanabe, S.; Hara, E. P16Ink4a and P21Cip1/Waf1 Promote Tumour Growth by Enhancing Myeloid-Derived Suppressor Cells Chemotaxis. Nat. Commun. 2017, 8, 2050. [Google Scholar] [CrossRef]
- Bu, L.-L.; Yu, G.-T.; Deng, W.-W.; Mao, L.; Liu, J.-F.; Ma, S.-R.; Fan, T.-F.; Hall, B.; Kulkarni, A.B.; Zhang, W.-F.; et al. Targeting STAT3 Signaling Reduces Immunosuppressive Myeloid Cells in Head and Neck Squamous Cell Carcinoma. Oncoimmunology 2016, 5, e1130206. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of Metastatic Mouse Cancers Resistant to Immune Checkpoint Blockade by Suppression of Myeloid-Derived Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic Cells and the Control of Immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice. I. Morphology, Quantitation, Tissue Distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Xu, D.-P.; Shi, W.-W.; Zhang, T.-T.; Lv, H.-Y.; Li, J.-B.; Lin, A.; Yan, W.-H. Elevation of HLA-G-Expressing DC-10 Cells in Patients with Gastric Cancer. Hum. Immunol. 2016, 77, 800–804. [Google Scholar] [CrossRef]
- Mestrallet, G.; Sone, K.; Bhardwaj, N. Strategies to Overcome DC Dysregulation in the Tumor Microenvironment. Front. Immunol. 2022, 13, 980709. [Google Scholar] [CrossRef]
- Sánchez-León, M.L.; Jiménez-Cortegana, C.; Cabrera, G.; Vermeulen, E.M.; de la Cruz-Merino, L.; Sánchez-Margalet, V. The Effects of Dendritic Cell-Based Vaccines in the Tumor Microenvironment: Impact on Myeloid-Derived Suppressor Cells. Front. Immunol. 2022, 13, 1050484. [Google Scholar] [CrossRef]
- Huai, G.; Markmann, J.F.; Deng, S.; Rickert, C.G. TGF-β-secreting Regulatory B Cells: Unsung Players in Immune Regulation. Clin. Transl. Immunol. 2021, 10, e1270. [Google Scholar] [CrossRef]
- Michaud, D.; Steward, C.R.; Mirlekar, B.; Pylayeva-Gupta, Y. Regulatory B Cells in Cancer. Immunol. Rev. 2021, 299, 74–92. [Google Scholar] [CrossRef]
- Narita, Y.; Nagane, M.; Mishima, K.; Terui, Y.; Arakawa, Y.; Yonezawa, H.; Asai, K.; Fukuhara, N.; Sugiyama, K.; Shinojima, N.; et al. Phase I/II Study of Tirabrutinib, a Second-Generation Bruton’s Tyrosine Kinase Inhibitor, in Relapsed/Refractory Primary Central Nervous System Lymphoma. Neuro Oncol. 2020, 23, 122–133. [Google Scholar] [CrossRef]
- Dhillon, S. Orelabrutinib: First Approval. Drugs 2021, 81, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, R.; Naseem, M.; Lo, J.H.; Battaglin, F.; Soni, S.; Puccini, A.; Berger, M.D.; Zhang, W.; Baba, H.; Lenz, H.-J. B Cell and B Cell-Related Pathways for Novel Cancer Treatments. Cancer Treat. Rev. 2019, 73, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Ramamoorthi, G.; Albert, G.; Gallen, C.; Beyer, A.; Snyder, C.; Koski, G.; Disis, M.L.; Czerniecki, B.J.; Kodumudi, K. Differentiation and Regulation of TH Cells: A Balancing Act for Cancer Immunotherapy. Front. Immunol. 2021, 12, 669474. [Google Scholar] [CrossRef] [PubMed]
- Tauber, P.A.; Kratzer, B.; Schatzlmaier, P.; Smole, U.; Köhler, C.; Rausch, L.; Kranich, J.; Trapin, D.; Neunkirchner, A.; Zabel, M.; et al. The Small Molecule Inhibitor BX-795 Uncouples IL-2 Production from Inhibition of Th2 Inflammation and Induces CD4+ T Cells Resembling ITreg. Front. Immunol. 2023, 14, 1094694. [Google Scholar] [CrossRef]
- Lei, X.; Khatri, I.; de Wit, T.; de Rink, I.; Nieuwland, M.; Kerkhoven, R.; van Eenennaam, H.; Sun, C.; Garg, A.D.; Borst, J.; et al. CD4+ Helper T Cells Endow CDC1 with Cancer-Impeding Functions in the Human Tumor Micro-Environment. Nat. Commun. 2023, 14, 217. [Google Scholar] [CrossRef]
- Li, T.-T.; Jiang, J.-W.; Qie, C.-X.; Xuan, C.-X.; Hu, X.-L.; Liu, W.-M.; Chen, W.-T.; Liu, J. Identification of Active Small-Molecule Modulators Targeting the Novel Immune Checkpoint VISTA. BMC Immunol. 2021, 22, 55. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The Role of Interleukin-2 during Homeostasis and Activation of the Immune System. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, A.; Barbon, C.M.; Wang, Y.; Ye, M.; Prickett, L.; Chandra, D.; Shaw, J.; Deng, N.; Sachsenmeier, K.; Clarke, J.D.; et al. Small Molecule AZD4635 Inhibitor of A2AR Signaling Rescues Immune Cell Function Including CD103+ Dendritic Cells Enhancing Anti-Tumor Immunity. J. Immunother. Cancer 2020, 8, e000417. [Google Scholar] [CrossRef] [PubMed]
- Brightman, S.E.; Naradikian, M.S.; Miller, A.M.; Schoenberger, S.P. Harnessing Neoantigen Specific CD4 T Cells for Cancer Immunotherapy. J. Leukoc. Biol. 2020, 107, 625–633. [Google Scholar] [CrossRef]
- Song, Y.; Margolles-Clark, E.; Bayer, A.; Buchwald, P. Small-Molecule Modulators of the OX40-OX40 Ligand Co-Stimulatory Protein-Protein Interaction. Br. J. Pharmacol. 2014, 171, 4955–4969. [Google Scholar] [CrossRef] [PubMed]
- Schoenberger, S.P.; Toes, R.E.; van der Voort, E.I.; Offringa, R.; Melief, C.J. T-Cell Help for Cytotoxic T Lymphocytes Is Mediated by CD40-CD40L Interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the Role of CD4+ T Cells in Cancer Immunotherapy-New Insights into Old Paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Cenerenti, M.; Saillard, M.; Romero, P.; Jandus, C. The Era of Cytotoxic CD4 T Cells. Front. Immunol. 2022, 13, 867189. [Google Scholar] [CrossRef]
- Gabr, M.T.; Gambhir, S.S. Discovery and Optimization of Small-Molecule Ligands for V-Domain Ig Suppressor of T-Cell Activation (VISTA). J. Am. Chem. Soc. 2020, 142, 16194–16198. [Google Scholar] [CrossRef]
- Lizotte, P.H.; Hong, R.-L.; Luster, T.A.; Cavanaugh, M.E.; Taus, L.J.; Wang, S.; Dhaneshwar, A.; Mayman, N.; Yang, A.; Kulkarni, M.; et al. A High-Throughput Immune-Oncology Screen Identifies EGFR Inhibitors as Potent Enhancers of Antigen-Specific Cytotoxic T-Lymphocyte Tumor Cell Killing. Cancer Immunol. Res. 2018, 6, 1511–1523. [Google Scholar] [CrossRef]
- Kursunel, M.A.; Taskiran, E.Z.; Tavukcuoglu, E.; Yanik, H.; Demirag, F.; Karaosmanoglu, B.; Ozbay, F.G.; Uner, A.; Esendagli, D.; Kizilgoz, D.; et al. Small Cell Lung Cancer Stem Cells Display Mesenchymal Properties and Exploit Immune Checkpoint Pathways in Activated Cytotoxic T Lymphocytes. Cancer Immunol. Immunother. 2022, 71, 445–459. [Google Scholar] [CrossRef]
- Yonesaka, K.; Haratani, K.; Takamura, S.; Sakai, H.; Kato, R.; Takegawa, N.; Takahama, T.; Tanaka, K.; Hayashi, H.; Takeda, M.; et al. B7-H3 Negatively Modulates CTL-Mediated Cancer Immunity. Clin. Cancer Res. 2018, 24, 2653–2664. [Google Scholar] [CrossRef]
- Iglesias-Escudero, M.; Arias-González, N.; Martínez-Cáceres, E. Regulatory Cells and the Effect of Cancer Immunotherapy. Mol. Cancer 2023, 22, 26. [Google Scholar] [CrossRef]
- Lu, C.; Yang, D.; Klement, J.D.; Oh, I.K.; Savage, N.M.; Waller, J.L.; Colby, A.H.; Grinstaff, M.W.; Oberlies, N.H.; Pearce, C.J.; et al. SUV39H1 Represses the Expression of Cytotoxic T-Lymphocyte Effector Genes to Promote Colon Tumor Immune Evasion. Cancer Immunol. Res. 2019, 7, 414–427. [Google Scholar] [CrossRef]
- Liu, X.; Yin, M.; Dong, J.; Mao, G.; Min, W.; Kuang, Z.; Yang, P.; Liu, L.; Zhang, N.; Deng, H. Tubeimoside-1 Induces TFEB-Dependent Lysosomal Degradation of PD-L1 and Promotes Antitumor Immunity by Targeting MTOR. Acta Pharm. Sin. B 2021, 11, 3134–3149. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Castro, I.; Levay, A.; Malek, T.R.; Gilboa, E. Aptamer-Targeted Inhibition of MTOR in T Cells Enhances Antitumor Immunity. J. Clin. Investig. 2014, 124, 188–197. [Google Scholar] [CrossRef]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Zeng, Z.; Bagati, A.; Tay, R.E.; Sanz, L.A.; Hartono, S.R.; Ito, Y.; Abderazzaq, F.; Hatchi, E.; Jiang, P.; et al. CARM1 Inhibition Enables Immunotherapy of Resistant Tumors by Dual Action on Tumor Cells and T Cells. Cancer Discov. 2021, 11, 2050–2071. [Google Scholar] [CrossRef]
- Liu, Y.; Debo, B.; Li, M.; Shi, Z.; Sheng, W.; Shi, Y. LSD1 Inhibition Sustains T Cell Invigoration with a Durable Response to PD-1 Blockade. Nat. Commun. 2021, 12, 6831. [Google Scholar] [CrossRef] [PubMed]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef]
- Marro, B.S.; Zak, J.; Zavareh, R.B.; Teijaro, J.R.; Lairson, L.L.; Oldstone, M.B.A. Discovery of Small Molecules for the Reversal of T Cell Exhaustion. Cell Rep. 2019, 29, 3293–3302.e3. [Google Scholar] [CrossRef]
- You, D.; Hillerman, S.; Locke, G.; Chaudhry, C.; Stromko, C.; Murtaza, A.; Fan, Y.; Koenitzer, J.; Chen, Y.; Briceno, S.; et al. Enhanced Antitumor Immunity by a Novel Small Molecule HPK1 Inhibitor. J. Immunother. Cancer 2021, 9, e001402. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Bevan, M.J. Proinflammatory Microenvironments within the Intestine Regulate the Differentiation of Tissue-Resident CD8+ T Cells Responding to Infection. Nat. Immunol. 2015, 16, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Lokensgard, J.R. Brain-Resident T Cells Following Viral Infection. Viral Immunol. 2019, 32, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Yu, Y.; Zeng, H.; Liu, Z.; You, R.; Zhang, H.; Liu, C.; Su, X.; Yan, S.; Chang, Y.; et al. CD103+CD8+ Tissue-Resident Memory T Cell Infiltration Predicts Clinical Outcome and Adjuvant Therapeutic Benefit in Muscle-Invasive Bladder Cancer. Br. J. Cancer 2022, 126, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Okła, K.; Farber, D.L.; Zou, W. Tissue-Resident Memory T Cells in Tumor Immunity and Immunotherapy. J. Exp. Med. 2021, 218, e20201605. [Google Scholar] [CrossRef]
- Kozik, P.; Gros, M.; Itzhak, D.N.; Joannas, L.; Heurtebise-Chrétien, S.; Krawczyk, P.A.; Rodríguez-Silvestre, P.; Alloatti, A.; Magalhaes, J.G.; Del Nery, E.; et al. Small Molecule Enhancers of Endosome-to-Cytosol Import Augment Anti-Tumor Immunity. Cell Rep. 2020, 32, 107905. [Google Scholar] [CrossRef]
- Van der Gracht, E.T.I.; Behr, F.M.; Arens, R. Functional Heterogeneity and Therapeutic Targeting of Tissue-Resident Memory T Cells. Cells 2021, 10, 164. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T Cells in Tumor Microenvironment: New Mechanisms, Potential Therapeutic Strategies and Future Prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An Essential Role for the IL-2 Receptor in Treg Cell Function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef]
- Chen, Y.; Xin, Z.; Huang, L.; Zhao, L.; Wang, S.; Cheng, J.; Wu, P.; Chai, Y. CD8+ T Cells Form the Predominant Subset of NKG2A+ Cells in Human Lung Cancer. Front. Immunol. 2019, 10, 3002. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, S.; Brown, M.L. Harnessing Proteases for T Regulatory Cell Immunotherapy. Eur. J. Immunol. 2020, 50, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Tian, E.-M.; Yu, M.-C.; Feng, M.; Lu, L.-X.; Liu, C.-L.; Shen, L.-A.; Wang, Y.-H.; Xie, Q.; Zhu, D. RORγt Agonist Synergizes with CTLA-4 Antibody to Inhibit Tumor Growth through Inhibition of Treg Cells via TGF-β Signaling in Cancer. Pharmacol. Res. 2021, 172, 105793. [Google Scholar] [CrossRef]
- Zou, H.; Li, R.; Hu, H.; Hu, Y.; Chen, X. Modulation of Regulatory T Cell Activity by TNF Receptor Type II-Targeting Pharmacological Agents. Front. Immunol. 2018, 9, 594. [Google Scholar] [CrossRef]
- Premkumar, K.; Shankar, B.S. TGF-ΒR Inhibitor SB431542 Restores Immune Suppression Induced by Regulatory B-T Cell Axis and Decreases Tumour Burden in Murine Fibrosarcoma. Cancer Immunol. Immunother. 2021, 70, 153–168. [Google Scholar] [CrossRef]
- Kujawski, M.; Zhang, C.; Herrmann, A.; Reckamp, K.; Scuto, A.; Jensen, M.; Deng, J.; Forman, S.; Figlin, R.; Yu, H. Targeting STAT3 in Adoptively Transferred T Cells Promotes Their in Vivo Expansion and Antitumor Effects. Cancer Res. 2010, 70, 9599–9610. [Google Scholar] [CrossRef]
- Ranasinghe, R.; Eri, R. Modulation of the CCR6-CCL20 Axis: A Potential Therapeutic Target in Inflammation and Cancer. Medicina 2018, 54, 88. [Google Scholar] [CrossRef]
- Melms, J.C.; Vallabhaneni, S.; Mills, C.E.; Yapp, C.; Chen, J.-Y.; Morelli, E.; Waszyk, P.; Kumar, S.; Deming, D.; Moret, N.; et al. Inhibition of Haspin Kinase Promotes Cell-Intrinsic and Extrinsic Antitumor Activity. Cancer Res. 2020, 80, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Kwilas, A.R.; Ardiani, A.; Donahue, R.N.; Aftab, D.T.; Hodge, J.W. Dual Effects of a Targeted Small-Molecule Inhibitor (Cabozantinib) on Immune-Mediated Killing of Tumor Cells and Immune Tumor Microenvironment Permissiveness When Combined with a Cancer Vaccine. J. Transl. Med. 2014, 12, 294. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.M.; Abdel-Hafiz, H.A.; Jones, R.T.; Jean, A.; Hoff, K.J.; Duex, J.E.; Chauca-Diaz, A.; Costello, J.C.; Dancik, G.M.; Tamburini, B.A.J.; et al. Inhibition of the CCL2 Receptor, CCR2, Enhances Tumor Response to Immune Checkpoint Therapy. Commun. Biol. 2020, 3, 720. [Google Scholar] [CrossRef] [PubMed]
- Atif, M.; Alsrhani, A.; Naz, F.; Ullah, M.I.; Alameen, A.A.M.; Imran, M.; Ejaz, H. Adenosine A2A Receptor as a Potential Target for Improving Cancer Immunotherapy. Mol. Biol. Rep. 2022, 49, 10677–10687. [Google Scholar] [CrossRef]
- Yuan, L.; Ye, J.; Fan, D. The B7-H4 Gene Induces Immune Escape Partly via Upregulating the PD-1/Stat3 Pathway in Non-Small Cell Lung Cancer. Hum. Immunol. 2020, 81, 254–261. [Google Scholar] [CrossRef]
- Hu, X.; Liu, X.; Moisan, J.; Wang, Y.; Lesch, C.A.; Spooner, C.; Morgan, R.W.; Zawidzka, E.M.; Mertz, D.; Bousley, D.; et al. Synthetic RORγ Agonists Regulate Multiple Pathways to Enhance Antitumor Immunity. Oncoimmunology 2016, 5, e1254854. [Google Scholar] [CrossRef]
- Zheng, L.; Qin, S.; Si, W.; Wang, A.; Xing, B.; Gao, R.; Ren, X.; Wang, L.; Wu, X.; Zhang, J.; et al. Pan-Cancer Single-Cell Landscape of Tumor-Infiltrating T Cells. Science 2021, 374, abe6474. [Google Scholar] [CrossRef] [PubMed]
- Tretyakova, M.S.; Subbalakshmi, A.R.; Menyailo, M.E.; Jolly, M.K.; Denisov, E.V. Tumor Hybrid Cells: Nature and Biological Significance. Front. Cell Dev. Biol. 2022, 10, 814714. [Google Scholar] [CrossRef]
- Zhu, H.; Peng, B.; Klausen, C.; Yi, Y.; Li, Y.; Xiong, S.; von Dadelszen, P.; Leung, P.C.K. NPFF Increases Fusogenic Proteins Syncytin 1 and Syncytin 2 via GCM1 in First Trimester Primary Human Cytotrophoblast Cells. FASEB J. 2020, 34, 9419–9432. [Google Scholar] [CrossRef] [PubMed]
- Leroy, H.; Han, M.; Woottum, M.; Bracq, L.; Bouchet, J.; Xie, M.; Benichou, S. Virus-Mediated Cell-Cell Fusion. Int. J. Mol. Sci. 2020, 21, 9644. [Google Scholar] [CrossRef]
- Zhang, L.-N.; Huang, Y.-H.; Zhao, L. Fusion of Macrophages Promotes Breast Cancer Cell Proliferation, Migration and Invasion through Activating Epithelial-Mesenchymal Transition and Wnt/β-Catenin Signaling Pathway. Arch. Biochem. Biophys. 2019, 676, 108137. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
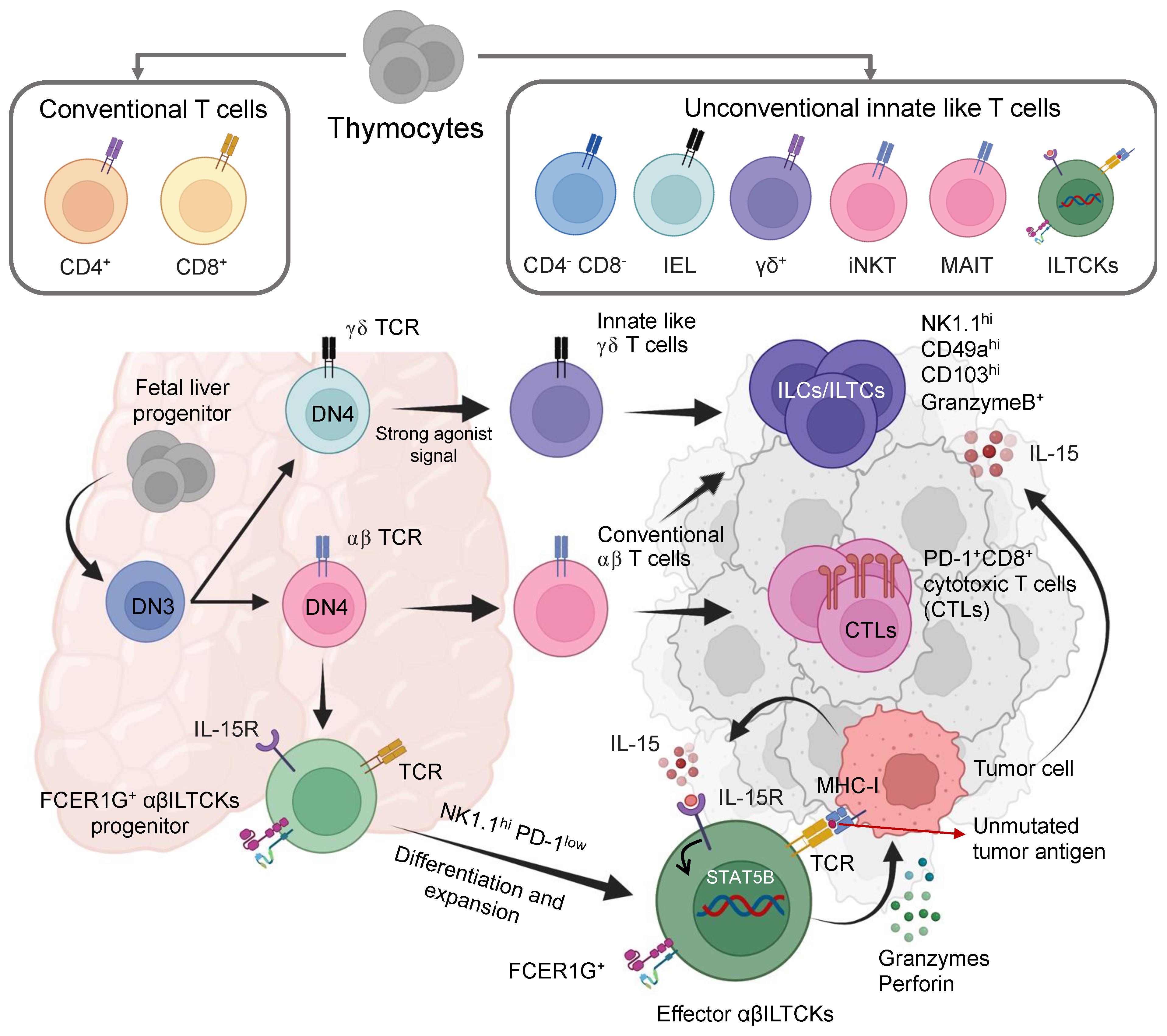
- Molgora, M.; Colonna, M. Innate-like T Cells: A Promising Asset in Anti-Cancer Immunity. Cancer Cell 2022, 40, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Zhang, X.; Krishna, C.; Nixon, B.G.; Dadi, S.; Capistrano, K.J.; Kansler, E.R.; Steele, M.; Han, J.; Shyu, A.; et al. Programme of Self-Reactive Innate-like T Cell-Mediated Cancer Immunity. Nature 2022, 605, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Dadi, S.; Chhangawala, S.; Whitlock, B.M.; Franklin, R.A.; Luo, C.T.; Oh, S.A.; Toure, A.; Pritykin, Y.; Huse, M.; Leslie, C.S.; et al. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell 2016, 164, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Van Rhijn, I.; Godfrey, D.I.; Rossjohn, J.; Moody, D.B. Lipid and Small-Molecule Display by CD1 and MR1. Nat. Rev. Immunol. 2015, 15, 643–654. [Google Scholar] [CrossRef]
- Toubal, A.; Nel, I.; Lotersztajn, S.; Lehuen, A. Mucosal-Associated Invariant T Cells and Disease. Nat. Rev. Immunol. 2019, 19, 643–657. [Google Scholar] [CrossRef]
- Ponzetta, A.; Carriero, R.; Carnevale, S.; Barbagallo, M.; Molgora, M.; Perucchini, C.; Magrini, E.; Gianni, F.; Kunderfranco, P.; Polentarutti, N.; et al. Neutrophils Driving Unconventional T Cells Mediate Resistance against Murine Sarcomas and Selected Human Tumors. Cell 2019, 178, 346–360.e24. [Google Scholar] [CrossRef]
- Morrish, E.; Ruland, J. Cytotoxic FCER1G+ Innate-like T Cells: New Potential for Tumour Immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 204. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, J.; Bai, X.; Handley, M.; Shan, F. Biological Effects of IL-15 on Immune Cells and Its Potential for the Treatment of Cancer. Int. Immunopharmacol. 2021, 91, 107318. [Google Scholar] [CrossRef]
- Desbois, M.; Béal, C.; Charrier, M.; Besse, B.; Meurice, G.; Cagnard, N.; Jacques, Y.; Béchard, D.; Cassard, L.; Chaput, N. IL-15 Superagonist RLI Has Potent Immunostimulatory Properties on NK Cells: Implications for Antimetastatic Treatment. J. Immunother. Cancer 2020, 8, e000632. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-F.; Shao, J.-H.; Liao, Y.-T.; Wang, L.-N.; Jia, Y.; Dong, P.-J.; Liu, Z.-Z.; He, D.-D.; Li, C.; Zhang, X. Regulation of Short-Chain Fatty Acids in the Immune System. Front. Immunol. 2023, 14, 1186892. [Google Scholar] [CrossRef]
- Hesterberg, R.S.; Cleveland, J.L.; Epling-Burnette, P.K. Role of Polyamines in Immune Cell Functions. Med. Sci. 2018, 6, 22. [Google Scholar] [CrossRef]
- Jung, Y.O.; Cho, M.-L.; Kang, C.-M.; Jhun, J.-Y.; Park, J.-S.; Oh, H.-J.; Min, J.-K.; Park, S.-H.; Kim, H.-Y. Toll-like Receptor 2 and 4 Combination Engagement Upregulate IL-15 Synergistically in Human Rheumatoid Synovial Fibroblasts. Immunol. Lett. 2007, 109, 21–27. [Google Scholar] [CrossRef]
- Dadi, S.; Li, M.O. Tissue-Resident Lymphocytes: Sentinel of the Transformed Tissue. J. Immunother. Cancer 2017, 5, 41. [Google Scholar] [CrossRef]
- Wagner, D.K.; Clements, M.L.; Reimer, C.B.; Snyder, M.; Nelson, D.L.; Murphy, B.R. Analysis of Immunoglobulin G Antibody Responses after Administration of Live and Inactivated Influenza A Vaccine Indicates That Nasal Wash Immunoglobulin G Is a Transudate from Serum. J. Clin. Microbiol. 1987, 25, 559–562. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TME Components Influence Tumor Progression | ||
|---|---|---|
| Non-Immune Cells | ||
| Type | Mediator | Key Role |
| Cancer-associated fibroblasts (CAFs) | α-Smooth muscle actin, fibroblast activation protein, vimentin, desmin, PDGFR α, and β |
|
| Tumor-associated adipocytes (TAPs) | Secretes adipokines, chemokines, cytokines, CCL2, CCL5, IL-1β, IL-6, TNF-α, VEGF and leptin |
|
| Tumor-associated pericytes (TAPs) | Upregulate MHC class II in response to IFN-γ |
|
| Tumor endothelial cells (TECs) | VWF, P-selectin (CD62P), and angiopoietin-2 (Angpt2) |
|
| Extracellular vesicles (EVs) | IL-6, CXCL-1, CCL2, MDM2, α-SMA, VEGF, NKG2L and PD-L1 |
|
| Immune cells | ||
| Subtype | Phenotype | Key role |
| Tumor-infiltrating Natural Killer cells (TINKs) | CD27hi, CD11bhi |
|
| Invariant NKT (iNKT) | CD1d restricted |
|
| Natural Killer T cells (NKT) | CD56, NKG2, CD94, CD161, NKG2D, NKG2A, NK1, Ly49 |
|
| Neutrophils and Tumor-associated neutrophils (TANs) | CD45, CD16hi, D11c, CXCR2hi, CXCR4low, CD62Lhi, ICAM1, Arginase-1, TGF-β |
|
| Macrophages | CD14, CD11c, CD16, CD64, CD68, CD206, HLA-DR, and CCR5 |
|
| Myeloid-derived suppressor cells (MDSCs) | CD14, CD15, HLA-DR, CD16, CD66b, CD11b and CD33 |
|
| Dendritic cells (DCs) | ITGAX, CX3CR1, FLT3 and CSF1R |
|
| B-lymphocytes | Immature B cells (CD19, CD20, CD24, CD38, CD45R) Mature B cells (IgM and CD19) |
|
| CD4+ T helper cells | CD4+ |
|
| Cytotoxic T lymphocytes (CTLs) | CD8+ |
|
| Regulatory T cells (Tregs) | CD4+, CD25 and FOXP3 |
|
| Small Molecule Inhibitor | TME Component | Cancer Type | Major Impact | References |
|---|---|---|---|---|
| Sorafenib | TAM | Breast cancer |
| [52] |
| Bindarit | Breast and prostate cancer |
| [53] | |
| Trabectedin | Fibrosarcoma, ovarian and lung carcinoma |
| [54,55] | |
| Tasquinimod | Melanoma and prostate cancer |
| [56] | |
| BLZ-945 | Breast and colon cancer |
| [57] | |
| AS1517499 | Breast |
| [58] | |
| Paclitaxel | DC | Lung cancer |
| [59] |
| Sildenafil | MDSC | Multiple myeloma and head and neck cancer |
| [60] |
| Sunitinib | Metastatic renal cell carcinoma and pancreatic neuroendocrine tumor |
| [61,62] | |
| GW2580 | Prostate cancer |
| [63] | |
| Axitinib | Metastatic renal cell carcinoma |
| [64] | |
| IC87114 | TIL | Colon cancer and melanoma |
| [65] |
| Trametinib | Colon cancer |
| [66] | |
| SB415286 | Melanoma and lymphoma |
| [67] | |
| RKN5755 | CAF | Breast cancer |
| [68] |
| WRG-28 | Breast cancer |
| [69] | |
| Scriptaid | Melanoma |
| [70] | |
| Navitoclax | Cholangiocarcinoma |
| [71] | |
| Sunitinib | EC | Colon, Renal cell carcinoma Epidermoid carcinoma |
| [72,73] |
| Dasatinib | Colon and prostate cancer |
| [74] | |
| TNP470 | Breast cancer Glioblastoma Melanoma |
| [75] | |
| DIMP53-I | Colon cancer |
| [76] | |
| Pazopanib | Metastatic renal cell carcinoma Multiple myeloma |
| [73,77] | |
| CC5079 | Colon cancer |
| [78] | |
| CX-4945 | Pancreatic cancer Breast cancer |
| [79] | |
| LLL12 | Osteosarcoma |
| [80] | |
| Biochanin A | Angiogenic glioma |
| [81] | |
| PD173074 | CAF and EC | Head and neck squamous cell carcinoma |
| [82] |
| Combination Study | ||||
| Cyclophosphamide + OX40 | TIL | Melanoma |
| [83] |
| TW-37 + radiotherapy | EC | Head and Neck cancer |
| [84] |
| BEZ235 + Verteporfin | Prostate cancer |
| [85] | |
| SOM230 + Gemcitabine | CAF | Pancreatic cancer |
| [86] |
| PT-100 + Oxaliplatin | Colon cancer |
| [87] | |
| AC1MMYR2 + Taxol | Breast cancer |
| [88] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Barik, D.; Arukha, A.P.; Prasad, S.; Mohapatra, I.; Singh, A.; Singh, G. Small Molecule Targeting Immune Cells: A Novel Approach for Cancer Treatment. Biomedicines 2023, 11, 2621. https://doi.org/10.3390/biomedicines11102621
Singh S, Barik D, Arukha AP, Prasad S, Mohapatra I, Singh A, Singh G. Small Molecule Targeting Immune Cells: A Novel Approach for Cancer Treatment. Biomedicines. 2023; 11(10):2621. https://doi.org/10.3390/biomedicines11102621
Chicago/Turabian StyleSingh, Shilpi, Debashis Barik, Ananta Prasad Arukha, Sujata Prasad, Iteeshree Mohapatra, Amar Singh, and Gatikrushna Singh. 2023. "Small Molecule Targeting Immune Cells: A Novel Approach for Cancer Treatment" Biomedicines 11, no. 10: 2621. https://doi.org/10.3390/biomedicines11102621