
Mometasone Furoate Inhibits the Progression of Head and Neck Squamous Cell Carcinoma via Regulating Protein Tyrosine Phosphatase Non-Receptor Type 11
Abstract
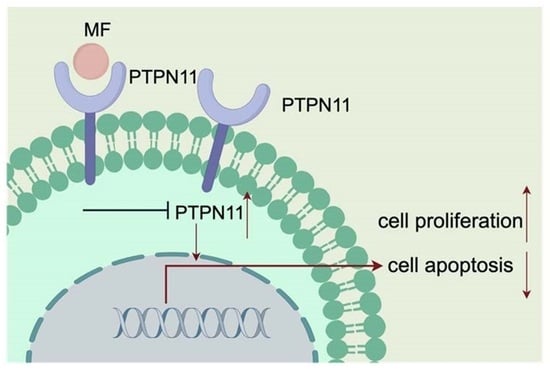
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Counting Kit-8 Assay
2.3. Colony Formation Assay
2.4. Flow Cytometric Analysis
2.5. Western Blotting
2.6. Real-Time PCR Analysis
2.7. Xenograft Study Models
2.8. Hematoxylin–Eosin Staining
2.9. Bioinformatics Analysis
2.10. Molecular Docking
2.11. Construction of PTPN11-Overexpression Plasmid and Cell Lines
2.12. Statistical Analysis
3. Results
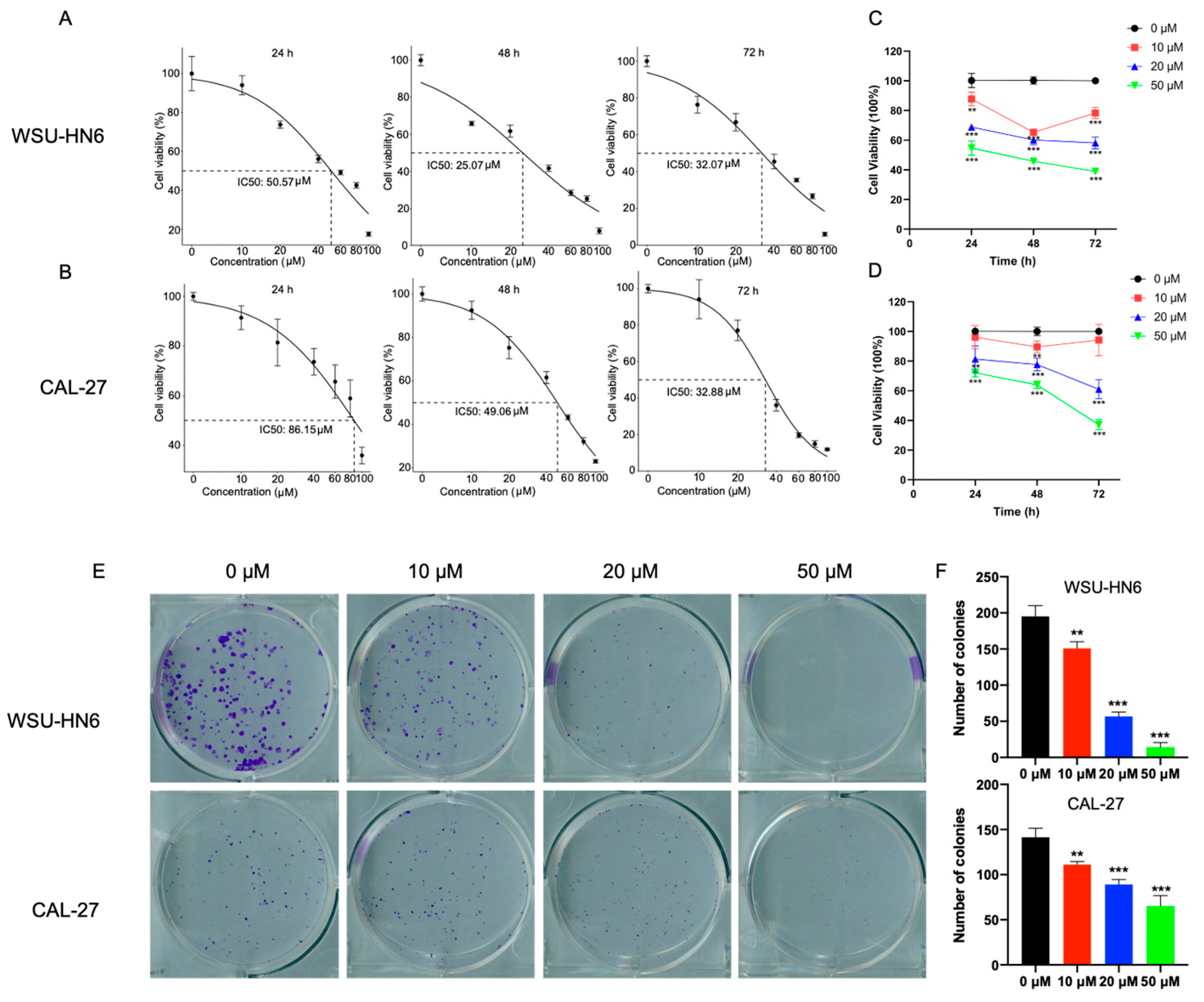
3.1. MF Inhibited the Proliferation of HNSCC Cells
3.2. MF Regulated the Cell Cycle and Induced Apoptosis In Vitro
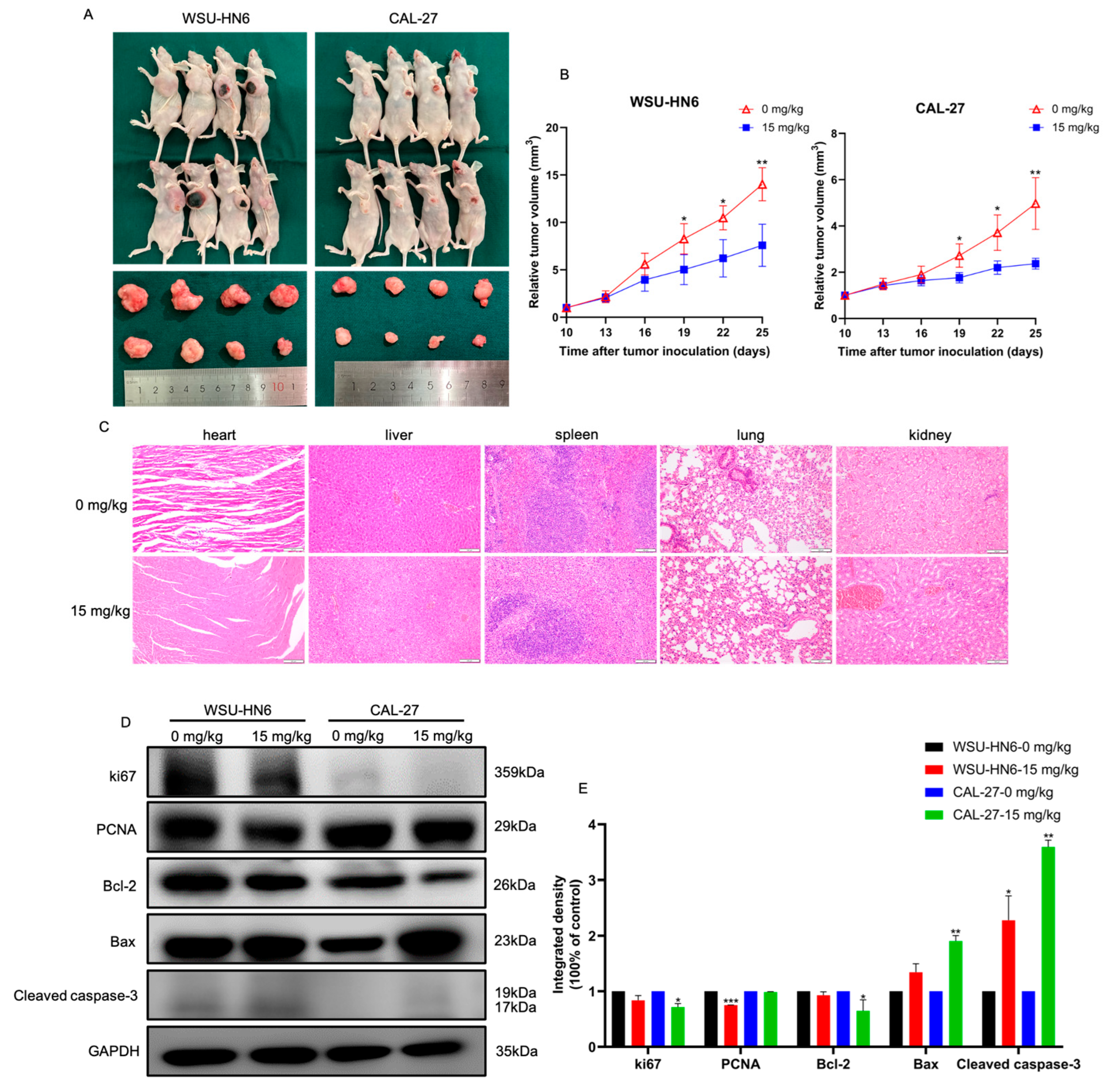
3.3. MF Suppressed Tumor Growth In Vivo
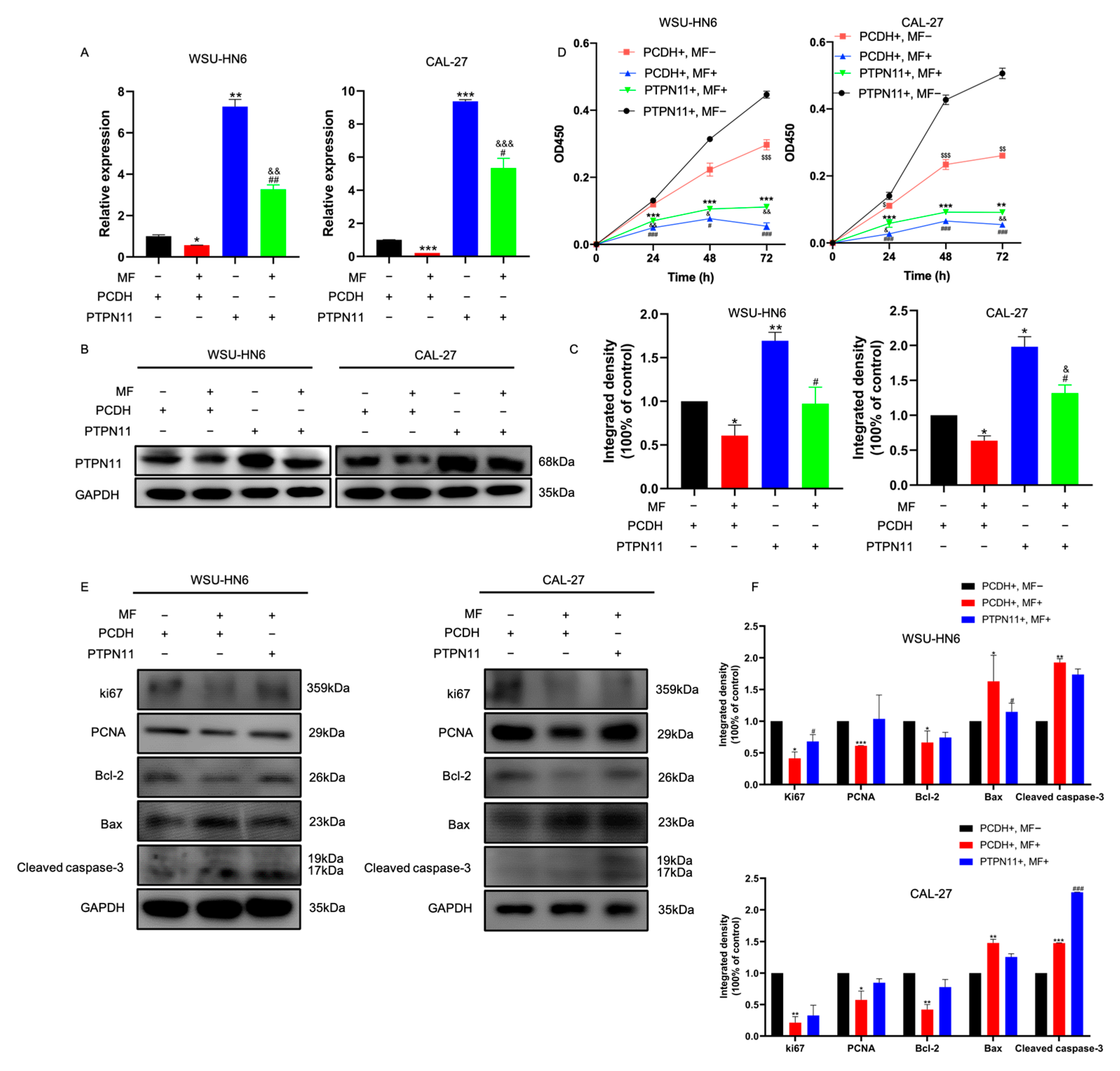
3.4. PTPN11 Was the Core Target of MF against HNSCC
3.5. MF Exerted Anti-Tumor Activity by Targeting PTPN11
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Cuaran, S.; Bouaoud, J.; Karabajakian, A.; Fayette, J.; Saintigny, P. Precision Medicine Approaches to Overcome Resistance to Therapy in Head and Neck Cancers. Front. Oncol. 2021, 11, 614332. [Google Scholar] [CrossRef] [PubMed]
- Tahara, M.; Muro, K.; Hasegawa, Y.; Chung, H.C.; Lin, C.C.; Keam, B.; Takahashi, K.; Cheng, J.D.; Bang, Y.-J. Pembrolizumab in Asia-Pacific patients with advanced head and neck squamous cell carcinoma: Analyses from KEYNOTE-012. Cancer Sci. 2018, 109, 771–776. [Google Scholar] [CrossRef]
- Mayayo-Peralta, I.; Zwart, W.; Prekovic, S. Duality of glucocorticoid action in cancer: Tumor-suppressor or oncogene? Endocr. Relat. Cancer 2021, 28, R157–R171. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cai, G.Q.; Peng, J.P.; Shen, C. Glucocorticoids induce apoptosis and matrix metalloproteinase-13 expression in chondrocytes through the NOX4/ROS/p38 MAPK pathway. J. Steroid. Biochem. Mol. Biol. 2018, 181, 52–62. [Google Scholar] [CrossRef]
- Cari, L.; De Rosa, F.; Nocentini, G.; Riccardi, C. Context-Dependent Effect of Glucocorticoids on the Proliferation, Differentiation, and Apoptosis of Regulatory T Cells: A Review of the Empirical Evidence and Clinical Applications. Int. J. Mol. Sci. 2019, 20, 1142. [Google Scholar] [CrossRef]
- Liu, X.; Suo, H.; Zhou, S.; Hou, Z.; Bu, M.; Liu, X.; Xu, W. Afatinib induces pro-survival autophagy and increases sensitivity to apoptosis in stem-like HNSCC cells. Cell Death Dis. 2021, 12, 728. [Google Scholar] [CrossRef]
- Raudenska, M.; Balvan, J.; Masarik, M. Cell death in head and neck cancer pathogenesis and treatment. Cell Death Dis. 2021, 12, 192. [Google Scholar] [CrossRef]
- Wang, X.; Shi, J.; Gong, D. Mometasone furoate inhibits growth of acute leukemia cells in childhood by regulating PI3K signaling pathway. Hematology 2018, 23, 478–485. [Google Scholar] [CrossRef]
- Xu, K.; Qin, X.S.; Zhang, Y.; Yang, M.Y.; Zheng, H.S.; Li, L.; Yang, X.; Xu, Q.; Li, Y.; Xu, P.; et al. Lycium ruthenicum Murr. anthocyanins inhibit hyperproliferation of synovial fibroblasts from rheumatoid patients and the mechanism study powered by network pharmacology. Phytomedicine 2023, 118, 154949. [Google Scholar] [CrossRef]
- Tang, B.H.; Dong, Y. Network pharmacology and bioinformatics analysis on the underlying mechanisms of baicalein against oral squamous cell carcinoma. J. Gene Med. 2023, 25, e3490. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gao, S.; Elhassan, R.M.; Hou, X.B.; Fang, H. Strategies to overcome drug resistance using SHP2 inhibitors. Acta Pharm. Sin. B 2021, 11, 3908–3924. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Zhao, M.; Zhang, H.Q.; Yu, B. Double-edged roles of protein tyrosine phosphatase SHP2 in cancer and its inhibitors in clinical trials. Pharmacol. Ther. 2022, 230, 107966. [Google Scholar] [CrossRef] [PubMed]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.Y.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Li, S.; Wang, X.; Li, Q.; Li, C. Role of SHP2/PTPN11 in the occurrence and prognosis of cancer: A systematic review and meta-analysis. Oncol. Lett. 2023, 25, 19. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.E.; Elamin, Y.Y.; Carr, A.; Gately, K.; Rafee, S.; Cremona, M.; Hanrahan, E.; Smyth, R.; Ryan, D.; Morgan, R.K.; et al. Protein Tyrosine Phosphatase Non-Receptor 11 (PTPN11/Shp2) as a Driver Oncogene and a Novel Therapeutic Target in Non-Small Cell Lung Cancer (NSCLC). Int. J. Mol. Sci. 2023, 24, 10545. [Google Scholar] [CrossRef]
- Hoffmann, L.; Coras, R.; Kobow, K.; López-Rivera, J.A.; Lal, D.; Leu, C.; Najm, I.; Nürnberg, P.; Herms, J.; Harter, P.N.; et al. Ganglioglioma with adverse clinical outcome and atypical histopathological features were defined by alterations in PTPN11/KRAS/NF1 and other RAS-/MAP-Kinase pathway genes. Acta Neuropathol. 2023, 145, 815–827. [Google Scholar] [CrossRef]
- Wang, H.C.; Chiang, W.F.; Huang, H.H.; Shen, Y.Y.; Chiang, H.C. Src-homology 2 domain-containing tyrosine phosphatase 2 promotes oral cancer invasion and metastasis. BMC Cancer 2014, 14, 442. [Google Scholar] [CrossRef]
- Leibowitz, M.S.; Srivastava, R.M.; Andrade Filho, P.A.; Egloff, A.M.; Wang, L.; Seethala, R.R.; Ferrone, S.; Ferris, R.L. SHP2 is overexpressed and inhibits pSTAT1-mediated APM component expression, T-cell attracting chemokine secretion, and CTL recognition in head and neck cancer cells. Clin. Cancer Res. 2013, 19, 798–808. [Google Scholar] [CrossRef]
- Yuan, X.; Bu, H.; Zhou, J.; Yang, C.Y.; Zhang, H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem. 2020, 63, 11368–11396. [Google Scholar] [CrossRef]
- Qiu, L.; Liu, H.; Wang, S.; Dai, X.H.; Shang, J.W.; Lian, X.L.; Wang, G.H.; Zhang, J. FKBP11 promotes cell proliferation and tumorigenesis via p53-related pathways in oral squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2021, 559, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Huang, S.; Li, W.; Zhao, H.; Zhang, T.; Zhang, D. Upregulation of Src homology phosphotyrosyl phosphatase 2 (Shp2) expression in oral cancer and knockdown of Shp2 expression inhibit tumor cell viability and invasion in vitro. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2014, 117, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.F.; Hall, A.; Walker, K.; Sherborne, A.; Tute, R.M.D.; Newnham, N.; Roberts, S.; Ingleson, E.; Bowles, K.; Garg, M.; et al. Daratumumab, Cyclophosphamide, Bortezomib, Lenalidomide, and Dexamethasone as Induction and Extended Consolidation Improves Outcome in Ultra-High-Risk Multiple Myeloma. J. Clin. Oncol. 2023, 41, 3945–3955. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Fleshner, N.; Chiuri, V.E.; Bruwaene, S.V.; Hafron, J.; McNeel, D.G.; Porre, P.D.; Maul, R.S.; Daksh, M.; Zhong, X.; et al. Niraparib with Abiraterone Acetate and Prednisone for Metastatic Castration-Resistant Prostate Cancer: Phase II QUEST Study Results. Oncologist 2023, 28, e309–e312. [Google Scholar] [CrossRef]
- Ahmed, A.; Reinhold, C.; Breunig, E.; Phan, T.S.; Dieteich, L.; Kostadinova, F.; Urwyler, C.; Merk, V.M.; Noti, M.; da Silva, I.T.; et al. Immune escape of colorectal tumours via local LRH-1/Cyp11b1-mediated synthesis of immunosuppressive glucocorticoids. Mol. Oncol. 2023, 17, 1545–1566. [Google Scholar] [CrossRef]
- Cairat, M.; Rahmoun, M.A.; Gunter, M.J.; Heudel, P.E.; Severi, G.; Dossus, L.; Fournier, A. Use of systemic glucocorticoids and risk of breast cancer in a prospective cohort of postmenopausal women. BMC Med. 2021, 19, 186. [Google Scholar] [CrossRef]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 15, e493–e503. [Google Scholar] [CrossRef]
- Sen, B.; Saigal, B.; Parikh, N.; Gallick, G.; Johnson, F.M. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009, 69, 1958–1965. [Google Scholar] [CrossRef]
- Stabile, L.P.; Egloff, A.M.; Gibson, M.K.; Gooding, W.E.; Ohr, J.; Zhou, P.; Rothenberger, N.J.; Wang, J.; Geiger, J.L.; Flaherty, J.T.; et al. IL6 is associated with response to dasatinib and cetuximab: Phase II clinical trial with mechanistic correlatives in cetuximab-resistant head and neck cancer. Oral. Oncol. 2017, 69, 38–45. [Google Scholar] [CrossRef]
- Wu, Q.J.; Li, W.; Zhao, J.; Sun, W.; Yang, Q.Q.; Chen, C.; Xia, P.; Zhu, J.; Huang, G.; Yong, C.; et al. Apigenin ameliorates doxorubicin-induced renal injury via inhibition of oxidative stress and inflammation. Biomed. Pharmacother. 2021, 137, 111308. [Google Scholar] [CrossRef]
- Albouy, B.; Tourani, J.M.; Allain, P.; Rolland, F.; Staerman, F.; Eschwege, P.; Pfister, C. Preliminary results of the Prostacox phase II trial in hormonal refractory prostate cancer. BJU Int. 2007, 100, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Feng, G.; Dai, T.; Long, F.; Tang, J.; Pu, Y.; Zheng, X.; Cao, S.; Xu, S.; Du, X. Randomized, self-controlled, prospective assessment of the efficacy of mometasone furoate local application in reducing acute radiation dermatitis in patients with head and neck squamous cell carcinomas. Medicine 2019, 98, e18230. [Google Scholar] [CrossRef] [PubMed]
- Hindley, A.; Zain, Z.; Wood, L.; Whitehead, A.; Sanneh, A.; Barber, D.; Hornsby, R. Mometasone furoate cream reduces acute radiation dermatitis in patients receiving breast radiation therapy: Results of a randomized trial. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.; Bhadri, V.A.; Beck, D.; Thoms, J.A.; Yakob, N.A.; Wong, J.W.; Knezevic, K.; Pimanda, J.E.; Lock, R.B. Opposing regulation of BIM and BCL2 controls glucocorticoid-induced apoptosis of pediatric acute lymphoblastic leukemia cells. Blood 2015, 125, 273–283. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 2011, 15, 1013–1031. [Google Scholar] [CrossRef]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Tateishi, K.; Tsubaki, M.; Takeda, T.; Yamatomo, Y.; Imano, M.; Satou, T.; Nishida, S. FTI-277 and GGTI-289 induce apoptosis via inhibition of the Ras/ERK and Ras/mTOR pathway in head and neck carcinoma HEp-2 and HSC-3 cells. J. BUON 2021, 26, 606–612. [Google Scholar]
- Solomon, L.W.; Frustino, J.L.; Loree, T.R.; Brecher, M.L.; Alberico, R.A.; Sullivan, M. Ewing sarcoma of the mandibular condyle: Multidisciplinary management optimizes outcome. Head Neck 2008, 30, 405–410. [Google Scholar] [CrossRef]
- Sun, Y.; Meyers, B.A.; Czako, B.; Leonard, P.; Mseeh, F.; Harris, A.L.; Wu, Q.; Johnson, S.; Parker, C.A.; Cross, J.B.; et al. Allosteric SHP2 Inhibitor, IACS-13909, Overcomes EGFR-Dependent and EGFR-Independent Resistance Mechanisms toward Osimertinib. Cancer Res. 2020, 80, 4840–4853. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence |
|---|---|
| EGFR | F: GGTGAGTGGCTTGTCTGGAA |
| EGFR | R: CCTTACGCCCTTCACTGTGT |
| GBR2 | F: AAGCTACTGCAGACGACGAG |
| GBR2 | R: CTTGGCTCTGGGGATTTTGC |
| IGF1R | F: AGGCTGGGGCTCTTGTTTAC |
| IGF1R | R: CCTCTCTCGAGTTCGCCTG |
| SRC | F: TTCTGCTGTTGACTGGCTGT |
| SRC | R: TGAGGATGGTCAGGTTGTGC |
| PTPN11 | F: CGTCATGCGTGTTAGGAACG |
| PTPN11 | R: TCTCTCCGTATTCCCCTGGA |
| MAPK1 | F: TCCTTTGAGCCGTTTGGAGG |
| MAPK1 | R: AGTACATACTGCCGCAGGTC |
| Primer | Oligonucleotides Sequence |
|---|---|
| PTPN11-OE-F | GGGGGAGGAGGGGGATCCGGAATGACATCGCGGAGATGGT |
| PTPN11-OE-R | GATCCTTCGCGGCCGCGATCCTCATCTGAAACTTTTCTGC |
| ID | Description | Gene Ratio | p Value | p. Adjust | q Value |
|---|---|---|---|---|---|
| hsa05205 | Proteoglycans in cancer | 26/150 | 4.1534 × 10−15 | 1.109 × 10−12 | 5.2901 × 10−13 |
| hsa01522 | Endocrine resistance | 18/150 | 1.4301 × 10−13 | 1.9092 × 10−11 | 9.1077 × 10−12 |
| hsa05215 | Prostate cancer | 17/150 | 1.5834 × 10−12 | 1.4092 × 10−10 | 6.7226 × 10−11 |
| hsa01521 | EGFR tyrosine kinase inhibitor resistance | 15/150 | 1.0373 × 10−11 | 6.9241 × 10−10 | 3.3031 × 10−10 |
| hsa04068 | FoxO signaling pathway | 18/150 | 2.4517 × 10−11 | 1.3092 × 10−9 | 6.2455 × 10−10 |
| hsa04659 | Th17 cell differentiation | 16/150 | 9.1379 × 10−11 | 4.0664 × 10−9 | 1.9398 × 10−9 |
| hsa04917 | Prolactin signaling pathway | 13/150 | 3.5466 × 10−10 | 1.3528 × 10−8 | 6.4532 × 10−9 |
| hsa04926 | Relaxin signaling pathway | 16/150 | 1.5842 × 10−9 | 5.2872 × 10−8 | 2.5222 × 10−8 |
| hsa05145 | Toxoplasmosis | 15/150 | 1.7878 × 10−9 | 5.3037 × 10−8 | 2.5301 × 10−8 |
| hsa04933 | AGE-RAGE signaling pathway in diabetic complications | 14/150 | 3.5417 × 10−9 | 9.3914 × 10−8 | 4.48 × 10−8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, L.; Gao, Q.; Tao, A.; Jiang, J.; Li, C. Mometasone Furoate Inhibits the Progression of Head and Neck Squamous Cell Carcinoma via Regulating Protein Tyrosine Phosphatase Non-Receptor Type 11. Biomedicines 2023, 11, 2597. https://doi.org/10.3390/biomedicines11102597
Qiu L, Gao Q, Tao A, Jiang J, Li C. Mometasone Furoate Inhibits the Progression of Head and Neck Squamous Cell Carcinoma via Regulating Protein Tyrosine Phosphatase Non-Receptor Type 11. Biomedicines. 2023; 11(10):2597. https://doi.org/10.3390/biomedicines11102597
Chicago/Turabian StyleQiu, Lin, Qian Gao, Anqi Tao, Jiuhui Jiang, and Cuiying Li. 2023. "Mometasone Furoate Inhibits the Progression of Head and Neck Squamous Cell Carcinoma via Regulating Protein Tyrosine Phosphatase Non-Receptor Type 11" Biomedicines 11, no. 10: 2597. https://doi.org/10.3390/biomedicines11102597