

HAT- and HDAC-Targeted Protein Acetylation in the Occurrence and Treatment of Epilepsy

Abstract
:1. Introduction
2. Protein Acetylation and Deacetylation Modifications
2.1. Classification of HATs
2.2. Classification of HDACs
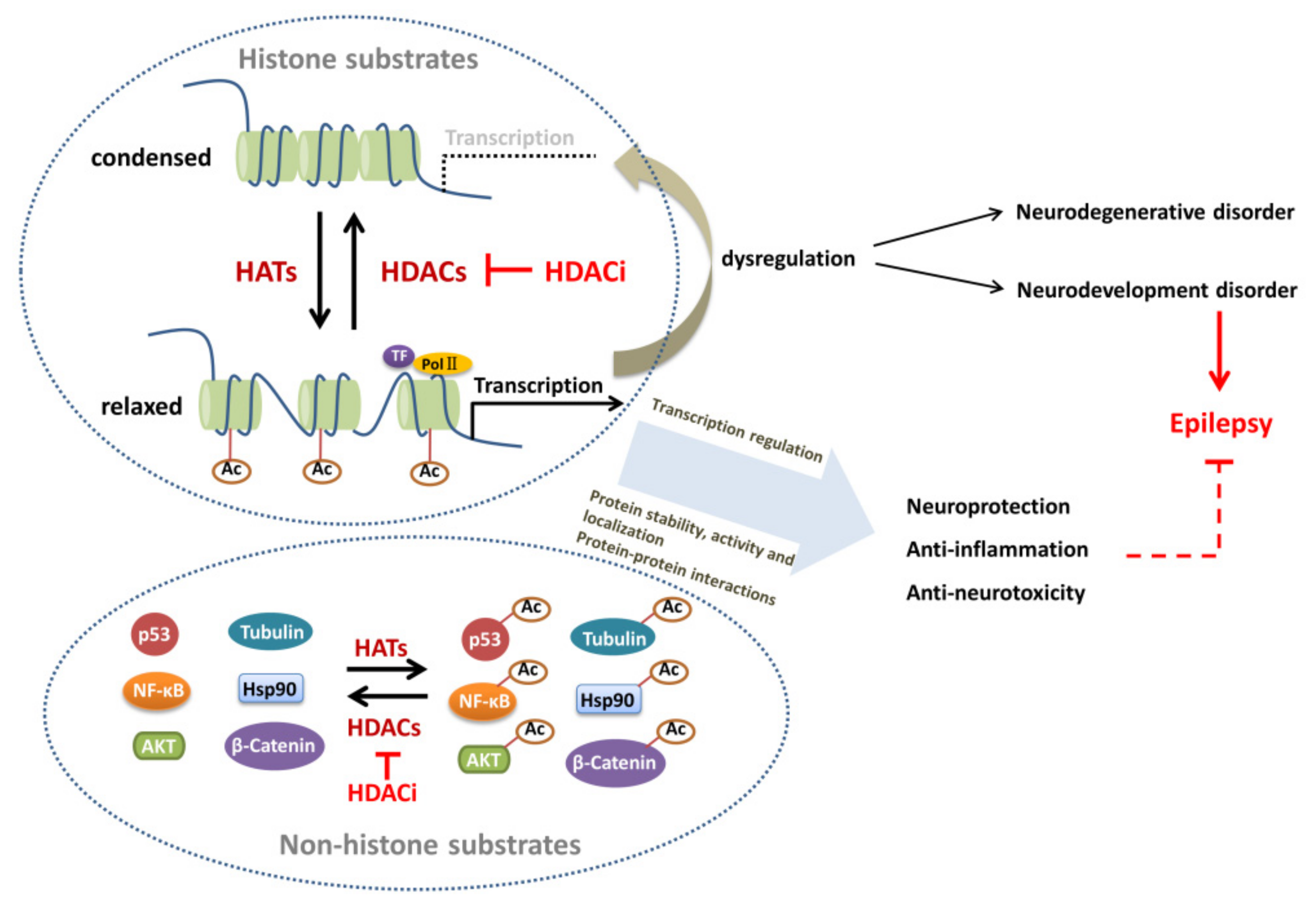
3. Substrates of HATs and HDACs
3.1. Histone Substrates
3.2. Non-Histone Substrates
4. Altered Protein Acetylation in Epileptogenesis
4.1. Alteration and Regulation of Histone Acetylation in Epileptogenesis
4.2. Regulatory Role of Non-Histone Acetylation in Epileptogenesis
4.2.1. TFs
4.2.2. Signaling Pathway Molecules
4.2.3. Chaperones
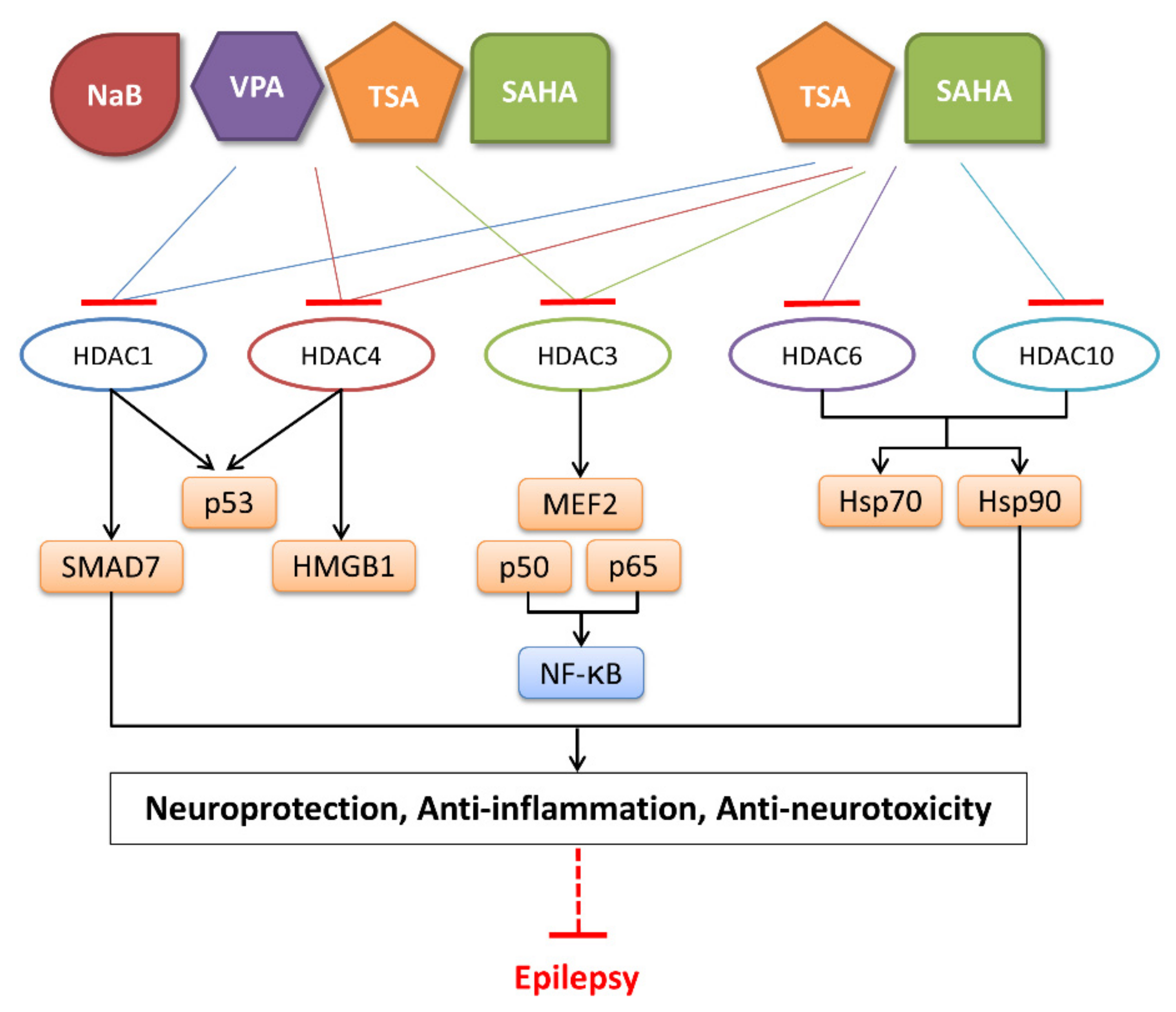
5. Role of HDACi in the Treatment of Epilepsy
5.1. Classification of HDACi
5.2. Role of HDACi in Epilepsy Treatment
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full Name |
| 4-PBA | Sodium Phenylbutyrate |
| Acss1 | Acyl-CoA Synthetase Short-Chain Family Member 1 |
| AKT | Protein Kinase B |
| ARID1A | (A+T)-Rich Interaction Domain 1A |
| ARNT | Aryl Hydrocarbon Receptor Nuclear Translocator |
| ASD | Autism Spectrum Disorder |
| ASS1 | Argininosuccinate Synthase 1 |
| ATAT1 | α-tubulin N-acetyltransferase 1 |
| ATF4 | Activating Transcription Factor 4 |
| Atg3 | Autophagy-Related 3 |
| ATM | Ataxia Telangiectasia Mutated |
| BDNF | Brain-Derived Neurotrophic Factor |
| BET | Bromodomain And Extra-Terminal Domain |
| bHLH | Basic Helix–Loop–Helix |
| BMAL1 | Brain And Muscle Arnt-Like 1 |
| BRPF1 | Bromodomain And Plant Homeodomain Finger-Containing 1 |
| CBP | CREB-binding protein |
| CDT1 | Chromatin Licensing And DNA Replication Factor 1 |
| CoA | Acetyl Coenzyme A |
| Co-IP | Co-Immunoprecipitation |
| CPS1 | Carbamoyl Phosphate Synthetase 1 |
| CREB | cAMP Response Element Binding Protein |
| ELP3 | Complex Subunit 3 |
| ELP3 | Elongator Complex Protein 3 |
| ESCO | Establishment Of Sister Chromatid Cohesion N-Acetyltransferase |
| FKBP51 | FK506-Binding Protein 51 |
| FoxO1 | Forkhead Box Protein O1 |
| G3BP1 | Ras GTPase-Activating Protein-Binding Protein 1 |
| G6PD | Glucose 6-Phosphate Dehydrogenase |
| GABA | γ-aminobutyric Acid |
| GABAAR | γ-aminobutyric Acid Type A Receptor |
| GABRG2 | GABAAR γ2 Subunit |
| GAD | Glutamate Decarboxylase |
| GATA1 | GATA-Binding Factor 1 |
| GCN5 | General Control Of Amino Acid Synthesis Protein 5 |
| GluR2/GRIA2 | Glutamate Receptor 2 |
| GNAT | GCN5-Related N-Acetyltransferase |
| H3K4bhb | β-hydroxybutyrylated Histone H3 Lys4 |
| H3K4cr | Crotonylated Histone H3 Lys4 |
| H3K9ac | Acetylated Histone H3 Lys9 |
| HATs | Histone Acetyltransferases |
| HAWAS | Histone-Acetylome-Wide Association Study |
| HBO1 | Histone Acetyltransferase Binding To ORC1 |
| HDACi | HDAC Inhibitors |
| HDACs | Histone Deacetylases |
| HIF-1α | Hypoxia-Inducible Factor-1α |
| HMGB1 | High-Mobility Group Box-1 |
| hnRNP M | Heterogeneous Nuclear Ribonucleoprotein M |
| Hsp70 | Heat Shock 70 Kda Protein |
| IC50 | Half-Maximal Inhibitory Concentration |
| IDH2 | Isocitrate Dehydrogenase 2 |
| IEGs | Immediate Early Genes |
| ISWI | Imitation Switch |
| KA | Kainic Acid |
| KAT | Lysine Acetyltransferase |
| KATs | Lysine Acetyltransferases |
| KDACs | Lysine Deacetylases |
| LPS | Lipopolysaccharide |
| MCCC | Methylcrotonoyl-CoA Carboxylase |
| MCD | Malonyl-CoA Decarboxylase |
| MEF2 | Myocyte Enhancer Factor 2 |
| MMP2 | Matrix Metalloproteinase 2 |
| MOF | Ortholog Of Drosophila Males-Absent On The First |
| MORF | MOZ-related Factors |
| MOZ | Onocytic Leukemia Zinc Finger Protein |
| MSH2 | Muts Homolog 2 |
| NaB | Sodium Butyrate |
| NAD+ | Nicotinamide Adenine Dinucleotide |
| NCOA | Nuclear Receptor Coactivator |
| NMDA | N-methyl-d-aspartate |
| Nrf2 | Nuclear Factor Erythroid-2 Related Factor 2 |
| Nrg1 | Neuregulin 1 |
| ORC1 | Initiation Recognition Complex 1 |
| p53 | Protein Of 53 kDa |
| p65 | 65kda Polypeptide Of The Transcription Factor NF-κB |
| PCAF | P300/CBP-Related Factors |
| PCNA | Proliferating Cell Nuclear Antigen |
| PGC-1α | Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1 Alpha |
| PKM2 | Pyruvate Kinase M2 |
| PLK4 | Polo-Like Kinase 4 |
| pol II | RNA polymerase II |
| Prx | Peroxiredoxin |
| PTM | Post-translational Modification |
| Ran | Ras-Related Nuclear Protein |
| Runx2 | Runt-Related Transcription Factor 2 |
| Sam68 | Src Associated In Mitosis Of 68 kDa |
| SCFAs | Short-Chain Fatty Acids |
| SDHA | Succinate Dehydrogenase Complex Subunit A |
| SE | Epileptic Seizures |
| SIRT | Sirtuin |
| SMC3 | Structural Maintenance Of Chromosomes 3 |
| Snail | Zinc Finger Protein SNAI1 |
| SNF2 | Sucrose Non-Fermenting Protein 2 |
| SRF | Serum Response Factor |
| STAT1 | Signal Transducer And Activator Of Transcription 1 |
| STRAP | Serine–Threonine Kinase Receptor Associated Protein |
| TAF1 | TATA-Binding Protein (TBP)-Associated Factor 1 |
| TAFII230/250 | Transcription Initiation Factor TFIID 230/250 kDa Subunit |
| TBX5 | T-box Protein 5 |
| TFs | Transcription Factors |
| TGF-β | Transforming Growth Factor-β |
| Tip60 | 60 kDa TAT-interacting Protein |
| TLE | Temporal Lobe Epilepsy |
| TLR4 | Toll-Like Receptor 4 |
| TNF | Tumor Necrosis Factor |
| TPR | Translocated Promoter Region |
| TPX2 | Targeting Protein For Xklp2 |
| TSA | Trichostatin A |
| TSC | Tuberous Sclerosis |
| U3-55k | U3 Small Nucleolar Ribonucleoprotein (snoRNP) Complex Component |
| VPA | Valproic Acid |
References
- Kobow, K.; Blümcke, I. Epigenetics in epilepsy. Neurosci. Lett. 2018, 667, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Attrish, D.; Srivastava, A.; Banerjee, J.; Tripathi, M.; Chandra, P.S.; Dixit, A.B. Non-histone substrates of histone deacetylases as potential therapeutic targets in epilepsy. Expert Opin. Ther. Targets 2021, 25, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Citraro, R.; Leo, A.; Santoro, M.; D’Agostino, G.; Constanti, A.; Russo, E. Role of histone deacetylases (HDACs) in epilepsy and epileptogenesis. Curr. Pharm. Des. 2017, 23, 5546–5562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.J.; Carregari, V.C. Post-translational modifications during brain development. Adv. Exp. Med. Biol. 2022, 1382, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.M.; Henshall, D.C.; Lubin, F.D. The epigenetics of epilepsy and its progression. Neuroscientist 2018, 24, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Van Loo, K.M.J.; Carvill, G.L.; Becker, A.J.; Conboy, K.; Goldman, A.M.; Kobow, K.; Lopes-Cendes, I.; Reid, C.A.; Van Vliet, E.A.; Henshall, D.C. Epigenetic genes and epilepsy—Emerging mechanisms and clinical applications. Nat. Rev. Neurol. 2022, 18, 530–543. [Google Scholar] [CrossRef]
- Burns, A.M.; Gräff, J. Cognitive epigenetic priming: Leveraging histone acetylation for memory amelioration. Curr. Opin. Neurobiol. 2021, 67, 75–84. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Henshall, D.C.; Kobow, K. Epigenetics and epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022731. [Google Scholar] [CrossRef] [Green Version]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Zhao, Y.; Garcia, B.A. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Zhang, X.; Li, H. Beyond histone acetylation—Writing and erasing histone acylations. Curr. Opin. Struct. Biol. 2018, 53, 169–177. [Google Scholar] [CrossRef]
- Imhof, A.; Yang, X.-J.; Ogryzko, V.V.; Nakatani, Y.; Wolffe, A.P.; Ge, H. Acetylation of general transcription factors by histone acetyltransferases. Curr. Biol. 1997, 7, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Ud-Din, A.I.M.S.; Tikhomirova, A.; Roujeinikova, A. Structure and functional diversity of GCN5-related N-acetyltransferases (GNAT). Int. J. Mol. Sci. 2016, 17, 1018. [Google Scholar] [CrossRef] [Green Version]
- Vasileia, S.; Jacques, C. MYST-family histone acetyltransferases: Beyond chromatin. Cell Mol. Life Sci. 2011, 68, 1147–1156. [Google Scholar]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Marshall, C.B.; Ikura, M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: Structural and functional versatility in target recognition. Cell Mol. Life Sci. 2013, 70, 3989–4008. [Google Scholar] [CrossRef]
- Vannam, R.; Sayilgan, J.; Ojeda, S.; Karakyriakou, B.; Hu, E.; Kreuzer, J.; Morris, R.; Lopez, X.I.H.; Rai, S.; Haas, W.; et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem. Biol. 2021, 28, 503–514.e12. [Google Scholar] [CrossRef]
- Wassarman, D.A.; Sauer, F. TAF(II)250: A transcription toolbox. J. Cell Sci. 2001, 114, 2895–2902. [Google Scholar] [CrossRef]
- Even, A.; Morelli, G.; Turchetto, S.; Shilian, M.; Bail, R.L.; Laguesse, S.; Krusy, N.; Brisker, A.; Brandis, A.; Inbar, S.; et al. ATP-citrate lyase promotes axonal transport across species. Nat. Commun. 2021, 12, 5878. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Millard, C.J.; Lin, C.-L.; Gurnett, J.E.; Wu, M.; Lee, K.; Fairall, L.; Schwabe, J.W.; Cole, P.A. Diverse nucleosome Site-Selectivity among histone deacetylase complexes. Elife 2020, 9, 57663. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kawaguchi, Y.; Lai, C.-H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.-P. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef]
- Hu, F.; Zhou, J.; Lu, Y.; Guan, L.; Wei, N.-N.; Tang, Y.-Q.; Wang, K. Inhibition of Hsp70 suppresses neuronal hyperexcitability and attenuates epilepsy by enhancing a-type potassium current. Cell Rep. 2019, 26, 168–181.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.-B.; Bai, J.-Y.; Zhang, H.-B.; Jia, J.; Shi, Q.; Yang, C.; Wang, X.; He, D.; Guo, P. KLF5 inhibits STAT3 activity and tumor metastasis in prostate cancer by suppressing IGF1 transcription cooperatively with HDAC1. Cell Death Dis. 2020, 11, 466. [Google Scholar] [CrossRef]
- Li, S.-Z.; Zeng, F.; Li, J.; Shu, Q.-P.; Zhang, H.-H.; Xu, J.; Ren, J.-W.; Zhang, X.-D.; Song, X.-M.; Du, R.-L. Nemo-like kinase (NLK) primes colorectal cancer progression by releasing the E2F1 complex from HDAC1. Cancer Lett. 2018, 431, 43–53. [Google Scholar] [CrossRef]
- Wagner, F.F.; Zhang, Y.-L.; Fass, D.M.; Joseph, N.; Gale, J.P.; Weïwer, M.; McCarren, P.; Fisher, S.L.; Kaya, T.; Zhao, W.-N.; et al. Kinetically selective inhibitors of histone deacetylase 2 (HDAC2) as cognition enhancers. Chem. Sci. 2015, 6, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Wang, J.; Shiozawa, K.; Jones, K.B.; Zhang, Y.; Prokop, J.W.; Davenport, G.G.; Nihira, N.T.; Hao, Z.; Wong, D.; et al. HDAC2 regulates site-specific acetylation of MDM2 and its ubiquitination signaling in tumor suppression. iScience 2019, 13, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Gao, Z.-W.; Hou, J.; Zhou, Q.; Ma, W.; Dai, Y.-H.; She, W.-D. Nuclear factor erythroid 2-related factor 2-histone deacetylase 2 pathway in the pathogenesis of refractory sudden sensorineural hearing loss and glucocorticoid resistance. ORL 2021, 83, 227–233. [Google Scholar] [CrossRef]
- Gonneaud, A.; Turgeon, N.; Boisvert, F.-M.; Boudreau, F.; Asselin, C. JAK-STAT pathway inhibition partially restores intestinal homeostasis in hdac1- and hdac2-intestinal epithelial cell-deficient mice. Cells 2021, 10, 224. [Google Scholar] [CrossRef]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.-L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef]
- Ziesche, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Muller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-kappaB. Nucleic Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Wu, X.; Guo, J.; Yuan, J. Myocyte-specific enhancer binding factor 2A expression is downregulated during temporal lobe epilepsy. Int. J. Neurosci. 2015, 126, 786–796. [Google Scholar] [CrossRef]
- Grabenstatter, H.; Del Angel, Y.C.; Carlsen, J.; Wempe, M.; White, A.; Cogswell, M.; Russek, S.; Brooks-Kayal, A. The effect of STAT3 inhibition on status epilepticus and subsequent spontaneous seizures in the pilocarpine model of acquired epilepsy. Neurobiol. Dis. 2014, 62, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ke, Q.; Shao, Y.; Zhu, G.; Li, Y.; Geng, N.; Jin, F.; Li, F. GATA1 induces epithelial-mesenchymal transition in breast cancer cells through PAK5 oncogenic signaling. Oncotarget 2015, 6, 4345–4356. [Google Scholar] [CrossRef] [Green Version]
- Castaneda, C.A.; Wolfson, N.A.; Leng, K.R.; Kuo, Y.M.; Andrews, A.J.; Fierke, C.A. HDAC8 substrate selectivity is deter-mined by long- and short-range interactions leading to enhanced reactivity for full-length histone substrates compared with peptides. J. Biol. Chem. 2017, 292, 21568–21577. [Google Scholar] [CrossRef] [Green Version]
- Olson, D.E.; Udeshi, N.D.; Wolfson, N.A.; Pitcairn, C.A.; Sullivan, E.D.; Jaffe, J.D.; Svinkina, T.; Natoli, T.; Lu, X.; Paulk, J.; et al. An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem. Biol. 2014, 9, 2210–2216. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.P.; Zhou, L.S.; Zhao, Y.Z.; Wang, S.W.; Chen, L.L.; Liu, L.X.; Ling, Z.Q.; Hu, F.J.; Sun, Y.P.; Zhang, J.Y.; et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014, 33, 1304–1320. [Google Scholar] [CrossRef] [Green Version]
- Provance, O.K.; Lewis-Wambi, J. Deciphering the role of interferon alpha signaling and microenvironment crosstalk in inflammatory breast cancer. Breast Cancer Res. 2019, 21, 59. [Google Scholar] [CrossRef] [Green Version]
- Davis, F.J.; Gupta, M.; Camoretti-Mercado, B.; Schwartz, R.J.; Gupta, M.P. Calcium/calmodulin-dependent protein kinase activates serum response factor transcription activity by its dissociation from histone deacetylase, HDAC4. Implications in cardiac muscle gene regulation during hypertrophy. J. Biol. Chem. 2003, 278, 20047–20058. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 protein regulates HIF1α protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Chen, H.; Liu, X.; Wang, Y.; Fan, A.; Qi, L.; Pan, L.; Bai, W.; Zhang, Y.; Sun, Y. ID1 inhibits foot-and-mouth disease virus replication via targeting of interferon pathways. FEBS J. 2021, 288, 4364–4381. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, A.; Grayson, D.R. DNA methylation and demethylation as targets for antipsychotic therapy. Dialogues Clin. Neurosci. 2014, 16, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Tamamizu-Kato, S.; Shibasaki, F. Histone deacetylase 7 associates with hypoxia-inducible factor 1α and increases transcriptional activity. J. Biol. Chem. 2004, 279, 41966–41974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feller, C.; Forné, I.; Imhof, A.; Becker, P.B. Global and specific responses of the histone acetylome to systematic perturbation. Mol. Cell 2015, 57, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Gal, J.; Chen, J.; Na, D.-Y.; Tichacek, L.; Barnett, K.R.; Zhu, H. The acetylation of lysine-376 of G3BP1 regulates RNA binding and stress granule dynamics. Mol. Cell Biol. 2019, 39, e00052-19. [Google Scholar] [CrossRef]
- Kozyreva, V.K.; McLaughlin, S.L.; Livengood, R.H.; Calkins, R.A.; Kelley, L.C.; Rajulapati, A.; Ice, R.J.; Smolkin, M.B.; Weed, S.A.; Pugacheva, E.N. NEDD9 regulates actin dynamics through cortactin deacetylation in an AURKA/HDAC6-dependent manner. Mol. Cancer Res. 2014, 12, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Scroggins, B.T.; Robzyk, K.; Wang, D.; Marcu, M.G.; Tsutsumi, S.; Beebe, K.; Cotter, R.J.; Felts, S.; Toft, D.; Karnitz, L.; et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 2007, 25, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Mak, A.B.; Nixon, A.M.L.; Kittanakom, S.; Stewart, J.M.; Chen, G.I.; Curak, J.; Gingras, A.-C.; Mazitschek, R.; Neel, B.G.; Stagljar, I.; et al. Regulation of CD133 by HDAC6 promotes β-catenin signaling to suppress cancer cell differentiation. Cell Reports 2012, 2, 951–963. [Google Scholar] [CrossRef] [Green Version]
- Parmigiani, R.B.; Xu, W.S.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Riolo, M.T.; Cooper, Z.; Holloway, M.P.; Cheng, Y.; Bianchi, C.; Yakirevich, E.; Ma, L.; Chin, Y.E.; Altura, R.A. Histone deacetylase 6 (HDAC6) deacetylates survivin for its nuclear export in breast cancer. J. Biol. Chem. 2012, 287, 10885–10893. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Li, Y.; Xiang, S.; Yuan, F.; Yuan, Z.; Telles, E.; Fang, J.; Coppola, D.; Shibata, D.; Lane, W.S.; et al. Histone deacetylase 10 regulates DNA mismatch repair and may involve the deacetylation of muts homolog 2. J. Biol. Chem. 2015, 290, 22795–22804. [Google Scholar] [CrossRef] [Green Version]
- Rempe, R.G.; Hartz, A.M.; Soldner, E.L.; Sokola, B.S.; Alluri, S.R.; Abner, E.L.; Kryscio, R.J.; Pekcec, A.; Schlichtiger, J.; Bauer, B. Matrix metalloproteinase-mediated blood-brain barrier dysfunction in epilepsy. J. Neurosci. 2018, 38, 4301–4315. [Google Scholar] [CrossRef] [Green Version]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Zerr, P.; Palumbo-Zerr, K.; Huang, J.; Tomcik, M.; Sumova, B.; Distler, O.; Schett, G.; Distler, J. OP0267 Sirt1 regulates canonical Tgf-beta signaling to control fibroblast activation and tissue fibrosis. Ann. Rheum. Dis. 2016, 75, 226–233. [Google Scholar] [CrossRef]
- Wang, S.J.; Zhao, X.H.; Chen, W.; Bo, N.; Wang, X.J.; Chi, Z.F.; Wu, W. Sirtuin 1 activation enhances the PGC-1alpha/mitochondrial antioxidant system pathway in status epilepticus. Mol. Med. Rep. 2015, 11, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Tu, W.; Zhang, Q.; Liu, Y.; Han, L.; Wang, Q.; Chen, P.; Zhang, S.; Wang, A.; Zhou, X. Fluoride induces apoptosis via inhibiting SIRT1 activity to activate mitochondrial p53 pathway in human neuroblastoma SH-SY5Y cells. Toxicol. Appl. Pharmacol. 2018, 347, 60–69. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhan, L.; Zhou, Q.-Y.; Zhang, L.-L.; Chen, X.-M.; Hu, X.-M.; Yuan, X.-C. SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy. Eur. J. Pharmacol. 2015, 764, 554–561. [Google Scholar] [CrossRef]
- Zhang, X.; Cao, R.; Niu, J.; Yang, S.; Ma, H.; Zhao, S.; Li, H. Molecular basis for hierarchical histone de-beta-hydroxybutyrylation by SIRT3. Cell Discov. 2019, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Wang, Y.; Li, X.; Li, X.-M.; Liu, Z.; Yang, T.; Wong, C.F.; Zhang, J.; Hao, Q.; Li, X.D. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. eLife 2014, 3, 2999. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wei, W.; Jiang, Y.; Peng, H.; Cai, J.; Mao, C.; Dai, H.; Choy, W.; Bemis, J.E.; Jirousek, M.R.; et al. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 2009, 284, 24394–24405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rardin, M.J.; Newman, J.C.; Held, J.M.; Cusack, M.P.; Sorensen, D.J.; Li, B.; Schilling, B.; Mooney, S.D.; Kahn, C.R.; Verdin, E.; et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 6601–6606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.A.; Huynh, F.K.; Fisher-Wellman, K.; Stuart, J.D.; Peterson, B.S.; Douros, J.D.; Wagner, G.R.; Thompson, J.W.; Madsen, A.S.; Green, M.F.; et al. SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 2017, 25, 838–855.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, G.; German, N.J.; Saha, A.K.; De Boer, V.C.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl COA decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, K.; Xu, W.; Zhao, S.; Ye, D.; Wang, Y.; Xu, Y.; Zhou, L.; Chu, Y.; Zhang, C.; et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1beta production and to prevent DSS-induced colitis in mice. Cell Rep. 2017, 19, 2331–2344. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L. SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 2009, 137, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Etchegaray, J.-P.; Zhong, L.; Li, C.; Henriques, T.; Ablondi, E.; Nakadai, T.; Van Rechem, C.; Ferrer, C.; Ross, K.N.; Choi, J.-E.; et al. The histone deacetylase SIRT6 restrains transcription elongation via promoter-proximal pausing. Mol. Cell 2019, 75, 683–699.e7. [Google Scholar] [CrossRef]
- Hong, J.; Saba, K.; Yi, W.; Guillaume, C.; Bin, H.; Carlos, S.; Jintang, D.; Ray, K.; Eva, G.; Raul, M.; et al. SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature 2013, 496, 110–113. [Google Scholar]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Qin, B.; Wu, F.; Qin, S.; Nowsheen, S.; Shan, S.; Zayas, J.; Pei, H.; Lou, Z.; Wang, L. Regulation of serine-threonine kinase akt activation by NAD(+)-dependent deacetylase SIRT7. Cell Rep. 2017, 18, 1229–1240. [Google Scholar] [CrossRef]
- Sobuz, S.U.; Sato, Y.; Yoshizawa, T.; Karim, F.; Ono, K.; Sawa, T.; Miyamoto, Y.; Oka, M.; Yamagata, K. SIRT7 regulates the nuclear export of NF-kappaB p65 by deacetylating Ran. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1355–1367. [Google Scholar] [CrossRef]
- Chen, S.; Blank, M.F.; Iyer, A.; Huang, B.; Wang, L.; Grummt, I.; Voit, R. SIRT7-dependent deacetylation of the U3-55k protein controls pre-rRNA processing. Nat. Commun. 2016, 7, 10734. [Google Scholar] [CrossRef] [Green Version]
- Ianni, A.; Kumari, P.; Tarighi, S.; Simonet, N.G.; Popescu, D.; Guenther, S.; Hölper, S.; Schmidt, A.; Smolka, C.; Yue, S.; et al. SIRT7-dependent deacetylation of NPM promotes p53 stabilization following UV-induced genotoxic stress. Proc. Natl. Acad. Sci. USA 2021, 118, e2015339118. [Google Scholar] [CrossRef]
- Heim, C.E.; Bosch, M.E.; Yamada, K.J.; Aldrich, A.L.; Chaudhari, S.S.; Klinkebiel, D.; Gries, C.M.; Alqarzaee, A.A.; Li, Y.; Thomas, V.C.; et al. Lactate production by Staphylococcus aureus biofilm inhibits HDAC11 to re-programme the host immune response during persistent infection. Nat. Microbiol. 2020, 5, 1271–1284. [Google Scholar] [CrossRef]
- Voss, A.K.; Thomas, T. Histone lysine and genomic targets of histone acetyltransferases in mammals. Bioessays 2018, 40, e1800078. [Google Scholar] [CrossRef]
- Fournier, M.; Orpinell, M.; Grauffel, C.; Scheer, E.; Garnier, J.-M.; Ye, T.; Chavant, V.; Joint, M.; Esashi, F.; Dejaegere, A.; et al. KAT2A/KAT2B-targeted acetylome reveals a role for PLK4 acetylation in preventing centrosome amplification. Nat. Commun. 2016, 7, 13227. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.; Eberharter, A.; Bonaldi, T.; Chioda, M.; Imhof, A.; Becker, P.B. Site-specific acetylation of ISWI by GCN5. BMC Mol. Biol. 2007, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Saraf, A.; Florens, L.; Washburn, M.; Workman, J.L. Gcn5 regulates the dissociation of SWI/SNF from chromatin by acetylation of Swi2/Snf2. Genes Dev. 2010, 24, 2766–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, K.; Jeng, W.; Chiang, C.; Chai, C.; Chiou, S.; Huang, M.; Yokoyama, K.K.; Wang, S.; Huang, S.; et al. PCAF-mediated acetylation of ISX recruits BRD 4 to promote epithelial-mesenchymal transition. EMBO Rep. 2020, 21, e48795. [Google Scholar] [CrossRef]
- Savoia, M.; Cencioni, C.; Mori, M.; Atlante, S.; Zaccagnini, G.; Devanna, P.; Di Marcotullio, L.; Botta, B.; Martelli, F.; Zeiher, A.M.; et al. P300/CBP-associated factor regulates transcription and function of isocitrate dehydrogenase 2 during muscle differentiation. FASEB J. 2019, 33, 4107–4123. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.A.A.; Nagarajan, P.; Parthun, M.R. Hat1-dependent lysine acetylation targets diverse cellular functions. J. Proteome Res. 2020, 19, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Miśkiewicz, K.; Jose, L.E.; Bento-Abreu, A.; Fislage, M.; Taes, I.; Kasprowicz, J.; Swerts, J.; Sigrist, S.; Versées, W.; Robberecht, W.; et al. ELP3 controls active zone morphology by acetylating the ELKS family member bruchpilot. Neuron 2011, 72, 776–788. [Google Scholar] [CrossRef] [Green Version]
- Sampath, V.; Liu, B.; Tafrov, S.; Srinivasan, M.; Rieger, R.; Chen, E.I.; Sternglanz, R. Biochemical characterization of Hpa2 and Hpa3, two small closely related acetyltransferases from saccharomyces cerevisiae. J. Biol. Chem. 2013, 288, 21506–21513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petkau, N.; Budak, H.; Zhou, X.; Oster, H.; Eichele, G. Acetylation of BMAL1 by TIP60 controls BRD4-P-TEFb recruitment to circadian promoters. eLife 2019, 8, 43235. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Ma, X.; Zhu, Q.; Song, D.; Ding, X.; Li, L.; Jiang, X.; Wang, X.; Tian, R.; Su, H.; et al. Pacer is a mediator of mTORC1 and GSK3-TIP60 signaling in regulation of autophagosome maturation and lipid metabolism. Mol. Cell 2019, 73, 788–802.e7. [Google Scholar] [CrossRef] [Green Version]
- Rokudai, S.; Laptenko, O.; Arnal, S.M.; Taya, Y.; Kitabayashi, I.; Prives, C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl. Acad. Sci. USA 2013, 110, 3895–3900. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.; Pelletier, N.; Xiao, L.; Zhao, S.P.; Wang, K.; Degerny, C.; Tahmasebi, S.; Cayrou, C.; Doyon, Y.; Goh, S.-L.; et al. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell Biol. 2008, 28, 6828–6843. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, N.; Champagne, N.; Stifani, S.; Yang, X.-J. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene 2002, 21, 2729–2740. [Google Scholar] [CrossRef] [Green Version]
- Miotto, B.; Struhl, K. HBO1 histone acetylase is a coactivator of the replication licensing factor Cdt1. Genes Dev. 2008, 22, 2633–2638. [Google Scholar] [CrossRef] [Green Version]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.W.; Reva, B.; et al. Proteogenomic characterization reveals therapeutic vul-nerabilities in lung adenocarcinoma. Cell 2020, 182, 200–225.e35. [Google Scholar] [CrossRef]
- Karoutas, A.; Szymanski, W.; Rausch, T.; Guhathakurta, S.; Rog-Zielinska, E.A.; Peyronnet, R.; Seyfferth, J.; Chen, H.-R.; De Leeuw, R.; Herquel, B.; et al. The NSL complex maintains nuclear architecture stability via lamin A/C acetylation. Nature 2019, 21, 1248–1260. [Google Scholar] [CrossRef]
- Yi, C.; Ma, M.; Ran, L.; Zheng, J.; Tong, J.; Zhu, J.; Ma, C.; Sun, Y.; Zhang, S.; Feng, W.; et al. Function and molecular mechanism of acetylation in autophagy regulation. Science 2012, 336, 474–477. [Google Scholar] [CrossRef]
- Weinert, B.T.; Narita, T.; Satpathy, S.; Srinivasan, B.; Hansen, B.K.; Schölz, C.; Hamilton, W.B.; Zucconi, B.E.; Wang, W.W.; Liu, W.R.; et al. Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell 2018, 174, 231–244.e12. [Google Scholar] [CrossRef] [Green Version]
- Siam, A.; Baker, M.; Amit, L.; Regev, G.; Rabner, A.; Najar, R.A.; Bentata, M.; Dahan, S.; Cohen, K.; Araten, S.; et al. Regulation of alternative splicing by p300-mediated acetylation of splicing factors. RNA 2019, 25, 813–824. [Google Scholar] [CrossRef]
- Chang, R.; Zhang, Y.; Zhang, P.; Zhou, Q. Snail acetylation by histone acetyltransferase p300 in lung cancer. Thorac. Cancer 2017, 8, 131–137. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, J.; Xue, F.; Zhang, C.; Mei, Z.; Wang, Y.; Bi, M.; Shan, D.; Meredith, A.; Li, H.; et al. Pokemon (FBI-1) interacts with Smad4 to repress TGF-beta-induced transcriptional responses. Biochim. Biophys. Acta 2015, 1849, 270–281. [Google Scholar] [CrossRef]
- Boyes, J.; Byfield, P.; Nakatani, Y.; Ogryzko, V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 1998, 396, 594–598. [Google Scholar] [CrossRef]
- Lin, C.-C.; Kitagawa, M.; Tang, X.; Hou, M.-H.; Wu, J.; Qu, D.C.; Srinivas, V.; Liu, X.; Thompson, J.W.; Mathey-Prevot, B.; et al. CoA synthase regulates mitotic fidelity via CBP-mediated acetylation. Nat. Commun. 2018, 9, 1039. [Google Scholar] [CrossRef]
- Hung, H.-L.; Lau, J.; Kim, A.Y.; Weiss, M.J.; Blobel, G.A. CREB-binding protein acetylates hematopoietic transcription factor gata-1 at functionally important sites. Mol. Cell Biol. 1999, 19, 3496–3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senf, S.M.; Sandesara, P.B.; Reed, S.A.; Judge, A.R. P300 acetyltransferase activity differentially regulates the localization and activity of the FOXO homologues in skeletal muscle. Am. J. Physiol. Physiol. 2011, 300, C1490–C1501. [Google Scholar] [CrossRef] [PubMed]
- Kalebic, N.; Sorrentino, S.; Perlas, E.; Bolasco, G.; Martinez, C.; Heppenstall, P.A. αTAT1 is the major α-tubulin acetyltransferase in mice. Nat. Commun. 2013, 4, 1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamino, M.; Ishibashi, M.; Nakato, R.; Akiyama, K.; Tanaka, H.; Kato, Y.; Negishi, L.; Hirota, T.; Sutani, T.; Bando, M.; et al. Esco1 acetylates cohesin via a mechanism different from that of Esco2. Curr. Biol. 2015, 25, 1694–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Mo, Y.; Zha, H.; Qu, Z.; Xie, P.; Zhu, Z.-J.; Xu, Y.; Xiong, Y.; Guan, K.-L. CLOCK acetylates ASS1 to drive circadian rhythm of ureagenesis. Mol. Cell 2017, 68, 198–209.e6. [Google Scholar] [CrossRef] [Green Version]
- Guelman, S.; Kozuka, K.; Mao, Y.; Pham, V.; Solloway, M.J.; Wang, J.; Wu, J.; Lill, J.R.; Zha, J. The double-histone-acetyltransferase complex ATAC is essential for mammalian development. Mol. Cell Biol. 2009, 29, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Geneviève, P.D.; Dilshad, H.K.; James, R.D. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Banerjee, J.; Dubey, V.; Tripathi, M.; Chandra, P.S.; Sharma, M.C.; Lalwani, S.; Siraj, F.; Doddamani, R.; Dixit, A.B. Role of altered expression, activity and sub-cellular distribution of various histone deacetylases (HDACs) in mesial temporal lobe epilepsy with hippocampal sclerosis. Cell Mol. Neurobiol. 2022, 42, 1049–1064. [Google Scholar] [CrossRef]
- Baltan, S.; Bachleda, A.; Morrison, R.S.; Murphy, S.P. Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Transl. Stroke Res. 2011, 2, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Broide, R.S.; Redwine, J.M.; Aftahi, N.; Young, W.; Bloom, F.E.; Winrow, C.J. Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 2007, 31, 47–58. [Google Scholar] [CrossRef]
- Cho, Y.; Cavalli, V. HDAC signaling in neuronal development and axon regeneration. Curr. Opin. Neurobiol. 2014, 27, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Kettmann, R.; Dequiedt, F. Class IIa histone deacetylases: Regulating the regulators. Oncogene 2007, 26, 5450–5467. [Google Scholar] [CrossRef]
- Morrison, B.E.; Majdzadeh, N.; D’Mello, S.R. Histone deacetylases: Focus on the nervous system. Cell Mol. Life Sci. 2007, 64, 2258–2269. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Hu, Q.; Kaufman, A.; D’Ercole, A.J.; Ye, P. Developmental expression of histone deacetylase 11 in the murine brain. J. Neurosci. Res. 2007, 86, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Mariño-Ramírez, L.; Kann, M.G.; Shoemaker, B.A.; Landsman, D. Histone structure and nucleosome stability. Expert Rev. Proteom. 2005, 2, 719–729. [Google Scholar] [CrossRef]
- Happel, N.; Doenecke, D. Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene 2009, 431, 1–12. [Google Scholar] [CrossRef]
- Roque, A.; Ponte, I.; Suau, P. Interplay between histone H1 structure and function. Biochim. Et Biophys. Acta 2016, 1859, 444–454. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2018, 20, 156–174. [Google Scholar] [CrossRef]
- Diallo, I.; Seve, M.; Cunin, V.; Minassian, F.; Poisson, J.-F.; Michelland, S.; Bourgoin-Voillard, S. Current trends in protein acetylation analysis. Expert Rev. Proteom. 2019, 16, 139–159. [Google Scholar] [CrossRef]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Chen, Z.; Cen, Z.; Ye, Y.; Li, S.; Lu, X.; Shao, Q.; Wang, D.; Ji, J.; Ji, Q. A comprehensive mouse brain acetylome-the cellular-specific distribution of acetylated brain proteins. Front Cell Neurosci. 2022, 16, 980815. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Poschmann, J.; Del Rosario, R.C.-H.; Parikshak, N.N.; Hajan, H.S.; Kumar, V.; Ramasamy, R.; Belgard, T.G.; Elanggovan, B.; Wong, C.C.Y.; et al. Histone acetylome-wide association study of autism spectrum disorder. Cell 2016, 167, 1385–1397.e11. [Google Scholar] [CrossRef] [Green Version]
- Gano, L.B.; Liang, L.-P.; Ryan, K.; Michel, C.R.; Gomez, J.; Vassilopoulos, A.; Reisdorph, N.; Fritz, K.S.; Patel, M. Altered mitochondrial acetylation profiles in a kainic acid model of temporal lobe epilepsy. Free. Radic. Biol. Med. 2018, 123, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Nadia, M.T.; Arvind, K.; Eric, J.N. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J. Neurosci. 2004, 24, 5603–5610. [Google Scholar]
- Taniura, H.; Sng, J.C.G.; Yoneda, Y. Histone modifications in status epilepticus induced by kainate. Histol. Histopathol. 2006, 21, 785–791. [Google Scholar] [CrossRef]
- Jagirdar, R.; Drexel, M.; Kirchmair, E.; Tasan, R.O.; Sperk, G. Rapid changes in expression of class I and IV histone deacetylases during epileptogenesis in mouse models of temporal lobe epilepsy. Exp. Neurol. 2015, 273, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Jagirdar, R.; Drexel, M.; Bukovac, A.; Tasan, R.O.; Sperk, G. Expression of class II histone deacetylases in two mouse models of temporal lobe epilepsy. J. Neurochem. 2016, 136, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Doherty, J.J.; Dingledine, R. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J. Neurosci. 2002, 22, 8422–8428. [Google Scholar] [CrossRef] [Green Version]
- Sng, J.C.G.; Taniura, H.; Yoneda, Y. Histone modifications in kainate-induced status epilepticus. Eur. J. Neurosci. 2006, 23, 1269–1282. [Google Scholar] [CrossRef]
- Sng, J.C.G.; Taniura, H.; Yoneda, Y. Inhibition of histone deacetylation by trichostatin A intensifies the transcriptions of neuronal c-fos and c-jun genes after kainate stimulation. Neurosci. Lett. 2005, 386, 150–155. [Google Scholar] [CrossRef]
- Wu, G.; Yu, J.; Wang, L.; Ren, S.; Zhang, Y. PKC/CREB pathway mediates the expressions of GABAA receptor subunits in cultured hippocampal neurons after low-Mg(2+) solution treatment. Epilepsy Res. 2018, 140, 155–161. [Google Scholar] [CrossRef]
- Mizielinska, S.; Greenwood, S.; Connolly, C. The role of GABAA receptor biogenesis, structure and function in epilepsy. Biochem. Soc. Trans. 2006, 34, 863–867. [Google Scholar] [CrossRef]
- Wang, J.G.; Cai, Q.; Zheng, J.; Dong, Y.S.; Li, J.J.; Li, J.C.; Hao, G.Z.; Wang, C.; Wang, J.L. Epigenetic suppression of GADs expression is involved in temporal lobe epilepsy and pilocarpine-induced mice epilepsy. Neurochem. Res. 2016, 41, 1751–1760. [Google Scholar] [CrossRef]
- Park, H.G.; Yu, H.S.; Park, S.; Ahn, Y.M.; Kim, Y.S.; Kim, S.H. Repeated treatment with electroconvulsive seizures induces HDAC2 expression and down-regulation of NMDA receptor-related genes through histone deacetylation in the rat frontal cortex. Int. J. Neuropsychopharmacol. 2014, 17, 1487–1500. [Google Scholar] [CrossRef] [Green Version]
- Jordan, J.; Galindo, M.F.; Prehn, J.; Weichselbaum, R.R.; Beckett, M.; Ghadge, G.D.; Roos, R.P.; Leiden, J.M.; Miller, R.J. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J. Neurosci. 1997, 17, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Morrison, R.S.; Kinoshita, Y. The role of p53 in neuronal cell death. Cell Death Differ. 2000, 7, 868–879. [Google Scholar] [CrossRef] [Green Version]
- Engel, T.; Murphy, B.M.; Schindler, C.K.; Henshall, D.C. Elevated p53 and lower MDM2 expression in hippocampus from patients with intractable temporal lobe epilepsy. Epilepsy Res. 2007, 77, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Sakhi, S.; Sun, N.; Wing, L.L.; Mehta, P.; Schreiber, S.S. Nuclear accumulation of p53 protein following kainic acid-induced seizures. Neuroreport 1996, 7, 493–496. [Google Scholar] [CrossRef]
- Jie, X.; Xiaomin, X.; Qimei, Z. Inhibited p38/p53 signaling pathway protects on neuron injury in epilepsy rats. J. Practical. Med. 2021, 37, 1666–1669+1673. [Google Scholar]
- Basile, V.; Mantovani, R.; Imbriano, C. DNA damage promotes histone deacetylase 4 nuclear localization and repression of G2/M promoters, via p53 C-terminal lysines. J. Biol. Chem. 2006, 281, 2347–2357. [Google Scholar] [CrossRef] [Green Version]
- Redondo, P.M.; Vaquero, A. The diversity of histone versus nonhistone sirtuin substrates. Genes Cancer 2013, 4, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, Z.; Zhang, Y.; Wang, G.; Wei, M.; Hu, Y.; Ma, S.; Jiang, Y.; Che, N.; Wang, X.; et al. Targeting of mi-croRNA-199a-5p protects against pilocarpine-induced status epilepticus and seizure damage via SIRT1-p53 cascade. Epilepsia 2016, 57, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Engel, T.; Tanaka, K.; Jimenez-Mateos, E.M.; Caballero-Caballero, A.; Prehn, J.H.; Henshall, D.C. Loss of p53 results in pro-tracted electrographic seizures and development of an aggravated epileptic phenotype following status epilepticus. Cell Death Dis. 2010, 1, e79. [Google Scholar] [CrossRef] [Green Version]
- Grégoire, S.; Xiao, L.; Nie, J.; Zhang, X.; Xu, M.; Li, J.; Wong, J.; Seto, E.; Yang, X.-J. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol. Cell Biol. 2007, 27, 1280–1295. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhao, Y. Progress on the roles of MEF2C in neuropsychiatric diseases. Mol. Brain 2022, 15, 8. [Google Scholar] [CrossRef]
- Zhang, W.; Du, Y.; Zou, Y.; Luo, J.; Lü, Y.; Yu, W. Smad anchor for receptor activation and phospho-smad3 were upregulated in patients with temporal lobe epilepsy. J. Mol. Neurosci. 2019, 68, 91–98. [Google Scholar] [CrossRef]
- Yu, W.; Du, Y.; Zou, Y.; Wang, X.; Stephani, U.; Lü, Y. Smad anchor for receptor activation contributes to seizures in temporal lobe epilepsy. Synapse 2017, 71, e21957. [Google Scholar] [CrossRef]
- Wei, L.; Rui, S.; Wenxiu, Y.; Nan, Z.; Yingshi, D.; Yan, Z.; Weihua, Y. Synchronous alteration pattern between serine-threonine kinase receptor-associated protein and Smad7 in pilocarpine-induced rats of epilepsy. Synapse 2014, 68, 275–282. [Google Scholar]
- Simonsson, M.; Heldin, C.-H.; Ericsson, J.; Gronroos, E. The balance between acetylation and deacetylation controls smad7 stability. J. Biol. Chem. 2005, 280, 21797–21803. [Google Scholar] [CrossRef] [Green Version]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, F.; Zhai, F.; Liang, S. Role of HMGB1/TLR4 and IL-1beta/IL-1R1 signaling pathways in epilepsy. Front Neurol. 2022, 13, 904225. [Google Scholar] [CrossRef]
- Park, E.J.; Kim, Y.M.; Kim, H.J.; Chang, K.C. Degradation of histone deacetylase 4 via the TLR 4/ JAK / STAT 1 signaling pathway promotes the acetylation of high mobility group box 1 ( HMGB 1) in lipopolysaccharide-activated macrophages. FEBS Open Bio. 2018, 8, 1119–1126. [Google Scholar] [CrossRef] [Green Version]
- Lubin, F.D.; Ren, Y.; Xu, X.; Anderson, A.E. Nuclear factor-kappa B regulates seizure threshold and gene transcription fol-lowing convulsant stimulation. J. Neurochem. 2007, 103, 1381–1395. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef]
- Karin, M.R.; Süheda, E.; Susanne, W.; Bernhard, L.; Michael, O.H. SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 123, 4251–4258. [Google Scholar]
- Xu, B.; Lang, L.-M.; Lian, S.; Guo, J.-R.; Wang, J.-F.; Liu, J.; Yang, H.-M.; Li, S.-Z. Neuroinflammation induced by secretion of acetylated HMGB1 from activated microglia in hippocampi of mice following chronic cold exposure. Brain Res. 2020, 1726, 146495. [Google Scholar] [CrossRef]
- Yang, T.; Hsu, C.; Liao, W.; Chuang, J.S. Heat shock protein 70 expression in epilepsy suggests stress rather than protection. Acta Neuropathol. 2008, 115, 219–230. [Google Scholar] [CrossRef]
- Johnson, C.A.; White, D.A.; Lavender, J.S.; O’Neill, L.P.; Turner, B.M. Human class I histone deacetylase complexes show enhanced catalytic activity in the presence of ATP and co-immunoprecipitate with the ATP-dependent chaperone protein Hsp70. J. Biol. Chem. 2002, 277, 9590–9597. [Google Scholar] [CrossRef] [Green Version]
- Oehme, I.; Linke, J.-P.; Böck, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longze, S.; Xueqin, W.; Jing, L.; Xinze, S.; Liwen, W.; Yan, S.; Qi, X. Pharmacologic inhibition of Hsp90 to prevent GLT-1 degradation as an effective therapy for epilepsy. J. Exp. Med. 2017, 214, 547–563. [Google Scholar]
- Marks, P.A. Histone deacetylase inhibitors: A chemical genetics approach to understanding cellular functions. Biochim. Biophys. Acta 2010, 1799, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Atalaya, J.P.; Ito, S.; Valor, L.M.; Benito, E.; Barco, A. Genomic targets, and histone acetylation and gene expression profiling of neural HDAC inhibition. Nucleic Acids Res. 2013, 41, 8072–8084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Hsing, C.-H.; Hung, S.-K.; Chen, Y.-C.; Wei, T.-S.; Sun, D.-P.; Wang, J.-J.; Yeh, C.-H. Histone deacetylase inhibitor trichostatin a ameliorated endotoxin-induced neuroinflammation and cognitive dysfunction. Mediat. Inflamm. 2015, 2015, 163140. [Google Scholar] [CrossRef] [Green Version]
- Sara, E.; Boris, Y.; Eyal, S.; Yoram, A.; Miriam, S.; Meir, B. The activity of antiepileptic drugs as histone deacetylase inhibitors. Epilepsia 2004, 45, 737–744. [Google Scholar]
- Younus, I.; Reddy, D.S. Epigenetic interventions for epileptogenesis: A new frontier for curing epilepsy. Pharmacol. Ther. 2017, 177, 108–122. [Google Scholar] [CrossRef]
- Eleuteri, S.; Monti, B.; Brignani, S.; Contestabile, A. Chronic dietary administration of valproic acid protects neurons of the rat nucleus basalis magnocellularis from ibotenic acid neurotoxicity. Neurotox. Res. 2009, 15, 127–132. [Google Scholar] [CrossRef]
- Jessberger, S.; Nakashima, K.; Clemenson, G.D., Jr.; Mejia, E.; Mathews, E.; Ure, K.; Ogawa, S.; Sinton, C.M.; Gage, F.H.; Hsieh, J. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci. 2007, 27, 5967–5975. [Google Scholar] [CrossRef] [Green Version]
- Basu, T.; O’Riordan, K.J.; Schoenike, B.A.; Khan, N.N.; Wallace, E.P.; Rodriguez, G.; Maganti, R.K.; Roopra, A. Histone deacetylase inhibitors restore normal hippocampal synaptic plasticity and seizure threshold in a mouse model of tuberous sclerosis complex. Sci. Rep. 2019, 9, 5266. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.D.; Clossen, B.L.; Reddy, D.S. Epigenetic histone deacetylation inhibition prevents the development and persistence of temporal lobe epilepsy. J. Pharmacol. Exp. Ther. 2018, 364, 97–109. [Google Scholar] [CrossRef]
- Maejima, H.; Kitahara, M.; Takamatsu, Y.; Mani, H.; Inoue, T. Effects of exercise and pharmacological inhibition of histone deacetylases (HDACs) on epigenetic regulations and gene expressions crucial for neuronal plasticity in the motor cortex. Brain Res. 2021, 1751, 147191. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; De Caro, C.; Nesci, V.; Gallo Cantafio, M.E.; Amodio, N.; Mattace Raso, G.; Lama, A.; Russo, R.; Calignano, A.; et al. Effects of histone deacetylase inhibitors on the development of epilepsy and psychiatric comorbidity in WAG/Rij Rats. Mol. Neurobiol. 2020, 57, 408–421. [Google Scholar] [CrossRef]
- Deutsch, S.I.; Rosse, R.B.; Long, K.D.; Gaskins, B.L.; Burket, J.A.; Mastropaolo, J. Sodium butyrate, an epigenetic interventional strategy, attenuates a stress-induced alteration of MK-801’s pharmacologic action. Eur. Neuropsychopharmacol. 2008, 18, 565–568. [Google Scholar] [CrossRef]
- Wang, J.; Huang, J.; Yao, S.; Wu, J.-H.; Li, H.-B.; Gao, F.; Wang, Y.; Huang, G.-B.; You, Q.-L.; Li, J.; et al. The ketogenic diet increases neuregulin 1 expression via elevating histone acetylation and its anti-seizure effect requires ErbB4 kinase activity. Cell Biosci. 2021, 11, 93. [Google Scholar] [CrossRef]
- Qing-Peng, H.; Ding-An, M. Histone deacetylase inhibitor SAHA attenuates post-seizure hippocampal microglia TLR4/MYD88 signaling and inhibits TLR4 gene expression via histone acetylation. BMC Neurosci. 2016, 17, 22. [Google Scholar]
- Shen, D.; Chen, J.; Liu, D.; Shen, M.; Wang, X.; Wu, Y.; Ke, S.; Macdonald, R.L.; Zhang, Q. The GABRG2 F343L allele causes spontaneous seizures in a novel transgenic zebrafish model that can be treated with suberanilohydroxamic acid (SAHA). Ann. Transl. Med. 2020, 8, 1560. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Class/Family | Members | Localization | Histone Substrates | Non-Histone Substrates | ||
|---|---|---|---|---|---|---|
| HDACs | Class I | HDAC1 | Nucleus | H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac [22] | p53 (K382ac) [23], Hsp70 [24], STAT3 [25], E2F-1 [26] | |
| HDAC2 | Nucleus | H3K9ac, H4K12ac [27] | p53 (K320ac) [28], Nrf2 [29], Bcl-6, STAT3 [30] | |||
| HDAC3 | Nucleus Cytoplasm | H3K9ac, H3K14ac, H4K5ac, H4K12ac [31] | p65 (K310ac, K314ac, K315ac) [32], MEF2 [33], STAT1, STAT3 [34], GATA1 [35] | |||
| HDAC8 | Nucleus Cytoplasm | H3K9ac, H3K14ac, H3K56ac [36] | ARID1A (K1808ac) [37], cortactin [38] | |||
| Class II | Class IIa | HDAC4 | Nucleus | p53, STAT1 [39], SRF [40], HIF-1α [41], ATF4 [40], FoxO1 [42] | ||
| HDAC5 | Nucleus | reelin [43] | ||||
| HDAC7 | Nucleus | HIF-1α [44] | ||||
| HDAC9 | Nucleus | |||||
| Class IIb | HDAC6 | Cytoplasm | H3K14ac, H4K5ac, H4K12ac, H4K16ac [45] | G3BP1 (K376ac) [46], cortactin (K124ac) [47], Hsp90 (K294ac) [48], β-Catenin [49], Prx I, Prx II [50], Survivin [51], AKT (K37ac, K163ac) [51] | ||
| HDAC10 | Cytoplasm | MSH2 [52], MMP2, MMP9 [53], Hsp70 [24] | ||||
| Class III | SITR1 | Nucleus | H3K9ac, H3K14ac, H3K56ac, H4K16ac [54] | BMAL1 [55], TGF-β [56], PGC-1α [57], p53 (K379ac) [58] | ||
| SIRT2 | Nucleus Cytoplasm | H3K56ac, H4K16ac [54] | G6PD [38], α-tubulin [59] | |||
| SIRT3 | Nucleus Mitochondria | H4K16ac [54], H3K4bhb, H3K9bhb, H3K18bhb, H3K23bhb, H3K27bhb, H4K16bhb [60], H3K4cr [61] | ACSS1 (K642ac) [62], SDHA (K179ac) [63] | |||
| SIRT4 | Mitochondria | MCCC [64], MCD [65] | ||||
| SIRT5 | Mitochondria | PKM2 (K311ac) [66], CPS1 [67] | ||||
| SIRT6 | Nucleus Endoplasmic Reticulum Mitochondria | H3K9ac, H3K56ac [68] | TNF (K19ac, K20ac) [69] | |||
| SIRT7 | Nucleus Cytoplasm | H3K18ac [70] | FKBP51 (K28ac, K155ac) [71], Ran (K37ac) [72], U3-55k (K12ac, K25ac) [73], nucleophosmin (K27ac, K54ac) [74] | |||
| Class IV | HDAC11 | Nucleus | IL-10 [75] | |||
| HATs | GNAT | GCN5 (KAT2A) | Nucleus | H3K9 [76] | PLK4 (K45, K46) [77], ISWI (K753) [78], SNF2 (K1493, K1497) [79], TBX5 [77] | |
| PCAF (KAT2B) | Nucleus | H3K9 [76] | ISX (K69) [80], IDH2 (K180) [81] | |||
| HAT1 (KAT1) | Nucleus Cytoplasm Mitochondria | H4K5, H4K12 [45] | TPR (K531) [82] | |||
| ELP3 | Nucleus Cytoplasm | Bruchpilot [83] | ||||
| HPA2 | Cytoplasm | Polyamines, small basic proteins [84] | ||||
| HPA3 | Cytoplasm | Polyamines, D-amino acids [84] | ||||
| MYST | Tip60 (KAT5) | Nucleus Cytoplasm | H2AK5, H2AK15, H3K14, H4K5, H4K8, H4K12, H4K16 [76] | BMAL1 (K538) [85], ATM (K3016) [86], Ran, Pacer [87] | ||
| MOZ (KAT6A) | Nucleus | H3K9, H3K14, H3K23 [76] | p53 (K120, K382) [88], BRPF1 [89] | |||
| MORF (KAT6B) | Nucleus | H3K14, H3K23 [76] | BRPF1 [89], Runx2 [90] | |||
| HBO1 (KAT7) | Nucleus | H3K9, H3K14, H4K5, H4K8, H4K12 [76] | CDT1 [91] | |||
| MOF (KAT8) | Nucleus Mitochondria | H4K5, H4K8, H4K16 [92] | Lamin A/C (K311) [93] | |||
| SAS2 | Nucleus | |||||
| SAS3 | Nucleus | |||||
| ESA1 | Nucleus | Atg3 [94] | ||||
| p300/CBP | p300 (KAT3B) | Nucleus Cytoplasm | H2AK4, H2AK5, H2AK7, H2AK9, H2AK11, H2AK13, H2BK5, H2BK11, H2BK12, H2BK15, H2BK16, H2BK20, H2BK21, H2BK23, H2BK24, H3K18, H3K27, H3K36, H4K5 [95] | Sam68, hnRNP M [96], Snail, Smad4, PCNA [97], FoxO1 [98], GATA1 [99], NCOA1, NCOA2, NCOA3, ARNT, ARNT2 [95] | ||
| CBP (KAT3A) | Nucleus Cytoplasm | H2AK4, H2AK5, H2AK7, H2AK9, H2AK11, H2AK13, H2BK5, H2BK11, H2BK12, H2BK15, H2BK16, H2BK20, H2BK21, H2BK23, H2BK24, H3K18, H3K27, H3K36, H4K5 [95] | TPX2 (K75, K476, and K582) [100], GATA1 [101], NCOA1, NCOA2, NCOA3, ARNT, ARNT2 [95], Snail, FoxO1, PCNA [102] | |||
| TAFII230/250 | TAFII250 | Nucleus | TAF1 | |||
| Others | ATAT1 | Nucleus Cytoplasm | α-Tubulin (K40) [103] | |||
| ESCO1 | Nucleus Cytoplasm | SMC3 (K105, K106) [104] | ||||
| ESCO2 | Nucleus Cytoplasm | SMC3 (K105, K106) [104] | ||||
| CLOCK (KAT13D) | Nucleus Cytoplasm | ASS1 (K165, K176) [105] | ||||
| ATAC2 (KAT14) | Nucleus Cytoplasm | H3K9, H4K5, H4K12, H4K16 [106] | ||||
| Classification | Compounds | Structure | IC50 | HDAC Targets | Therapeutic Potential |
|---|---|---|---|---|---|
| Hydroxamic acids | Trichostatin A (TSA) |  | 1.8 nM | Class I and II | Inhibition of breast cancer cell line proliferation |
| Vorinostat (SAHA) |  | ~10 nM | Class I and II | Inhibition of tumor growth | |
| Panobinostat (LBH589) |  | 5 nM | Class I and II | Both autophagy and apoptosis can be induced, effectively disrupting the latency of HIV in vivo. | |
| Belinostat (PXD101) |  | 27 nM | Class I and II | Inhibition of tumor cell growth | |
| Givinostat (ITF2357) |  | 10 nM (HD2), 16 nM (HD1-A), 7.5 nM (HD1-B) | Class I and II | Improving islet cell survival and action on multiple myeloma cell lines | |
| Abexinostat (PCI-24781) |  | Class I and II | Anti-neoplasmic activity | ||
| Dacinostat (LAQ824) |  | 32 nM | Class I and II | Inhibition of cancer cell growth | |
| Benzamides | Entinostat (MS-275) |  | 0.51 μM (HDAC1), 1.7 μM (HDAC3) | HDAC1, HDAC2, HDAC3 | Induction of autophagy and apoptosis in tumor cells |
| Mocetinostat (MGCD0103) |  | 0.15 μM (HDAC1), 0.29 μM (HDAC2), 1.66 μM (HDAC3), 0.59 μM (HDAC11) | HDAC1, HDAC2, HDAC3, HDAC11 | Anticancer activity | |
| Cyclic peptides | Romidepsin (FK228, Depsipeptide) |  | 36 nM (HDAC1), 47 nM (HDAC2) | HDAC1, HDAC2 | Inhibition of growth and induced apoptosis in neuroblastoma tumor cells |
| Apicidin |  | 0.7 nM | HDAC1, HDAC4, HDAC8 | Inhibition of tumor cell proliferation | |
| Cycloheximide (NSC-185) |  | 532.5 nM (protein synthesis), 2880 nM (RNA synthesis) | HDACs associated with protein synthesis | Inhibition of iron death and autophagy | |
| SCFAs | Valproic Acid (VPA) |  | 0.4 mM | Class I and IIa | Treatment of epilepsy |
| Sodium Butyrate (NaB) |  | Class I and IIa | Inhibition of cell cycle progression in cancer cells, promotion of differentiation, and induction of apoptosis and autophagy | ||
| Sodium Phenylbutyrate (4-PBA) |  | Class I and IIa | Induction of apoptosis in prostate cancer cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Yun, F.; Sui, J.; Liang, W.; Shen, D.; Zhang, Q. HAT- and HDAC-Targeted Protein Acetylation in the Occurrence and Treatment of Epilepsy. Biomedicines 2023, 11, 88. https://doi.org/10.3390/biomedicines11010088
Wang J, Yun F, Sui J, Liang W, Shen D, Zhang Q. HAT- and HDAC-Targeted Protein Acetylation in the Occurrence and Treatment of Epilepsy. Biomedicines. 2023; 11(1):88. https://doi.org/10.3390/biomedicines11010088
Chicago/Turabian StyleWang, Jie, Feng Yun, Jiahui Sui, Wenpeng Liang, Dingding Shen, and Qi Zhang. 2023. "HAT- and HDAC-Targeted Protein Acetylation in the Occurrence and Treatment of Epilepsy" Biomedicines 11, no. 1: 88. https://doi.org/10.3390/biomedicines11010088