
Chloride Ions, Vascular Function and Hypertension

Abstract
:1. Introduction
2. Role of Chloride Ions in Regulation of Vascular Tone and Blood Pressure
3. Alterations in Vascular Chloride Channels and Transporters in Hypertension
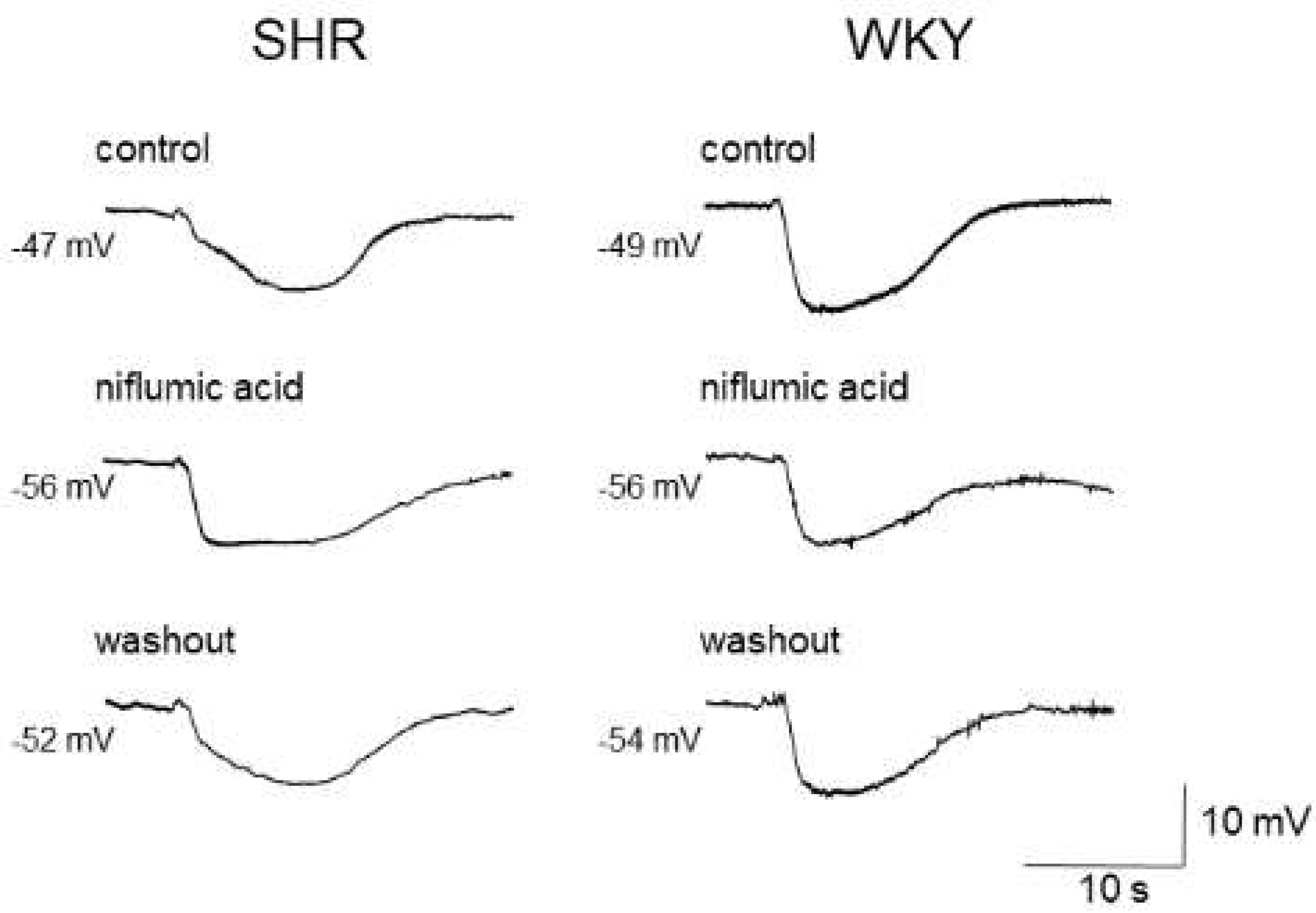
3.1. Ca2+-Activated Chloride Channels (CaCCs) in Vascular Smooth Muscle Cells
3.2. Ca2+-Activated Chloride Channels (CaCCs) in Vascular Endothelial Cells
3.3. Na+–K+–2Cl− Cotransporter1 (NKCC1)
4. Clinical Perspectives
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lawes, C.M.; Hoorn, S.V.; Rodgers, A. Global Burden of Blood-Pressure-Related Disease, 2001. Lancet 2008, 371, 1513–1518. [Google Scholar] [CrossRef]
- Zhou, B.; Perel, P.; Mensah, G.A.; Ezzati, M. Global Epidemiology, Health Burden and Effective Interventions for Elevated Blood Pressure and Hypertension. Nat. Rev. Cardiol. 2021, 18, 785–802. [Google Scholar] [CrossRef]
- Zhou, B.; Carrillo-Larco, R.M.; Danaei, G.; Riley, L.M.; Paciorek, C.J.; Stevens, G.A.; Gregg, E.W.; Bennett, J.E.; Solomon, B.; Singleton, R.K.; et al. Worldwide Trends in Hypertension Prevalence and Progress in Treatment and Control from 1990 to 2019: A Pooled Analysis of 1201 Population-Representative Studies with 104 Million Participants. Lancet 2021, 398, 957–980. [Google Scholar] [CrossRef]
- Umemura, S.; Arima, H.; Arima, S.; Asayama, K.; Dohi, Y.; Hirooka, Y.; Horio, T.; Hoshide, S.; Ikeda, S.; Ishimitsu, T.; et al. The Japanese Society of Hypertension Guidelines for the Management of Hypertension (JSH 2019). Hypertens. Res. 2019, 42, 1235–1481. [Google Scholar] [CrossRef] [PubMed]
- Tsuchihashi, T. Dietary Salt Intake in Japan-Past, Present, and Future. Hypertens. Res. 2022, 45, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Veronesi, M.; Fogacci, F. Dietary Intervention to Improve Blood Pressure Control: Beyond Salt Restriction. High Blood Press. Cardiovasc. Prev. 2021, 28, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Intersalt. Intersalt: An International Study of Electrolyte Excretion and Blood. BMJ 1988, 297, 319–328. [Google Scholar]
- Mozaffarian, D.; Fahimi, S.; Singh, G.M.; Micha, R.; Khatibzadeh, S.; Engell, R.E.; Lim, S.; Danaei, G.; Ezzati, M.; Powles, J. Global Sodium Consumption and Death from Cardiovascular Causes. N. Engl. J. Med. 2014, 371, 624–634. [Google Scholar] [CrossRef]
- Meneton, P.; Jeunemaitre, X.; De Wardener, H.E.; Macgregor, G.A. Links between Dietary Salt Intake, Renal Salt Handling, Blood Pressure, and Cardiovascular Diseases. Physiol. Rev. 2005, 85, 679–715. [Google Scholar] [CrossRef]
- Dahl, L.K.; Love, R.A. Evidence for Relationship between Sodium (Chloride) Intake and Human Essential Hypertension. AMA Arch. Intern. Med. 1954, 94, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Kempner, W. Treatment of Hypertensive Vascular Disease with Rice Diet. Am. J. Med. 1948, 4, 545–577. [Google Scholar] [CrossRef]
- Kurtz, T.W.; Morris, R.C. Dietary Chloride as a Determinant of “Sodium-Dependent” Hypertension. Science 1983, 222, 1139–1141. [Google Scholar] [CrossRef] [PubMed]
- Kotchen, T.A.; Luke, R.G.; Ott, C.E.; Galla, J.H.; Whitescarver, S. Effect of Chloride on Renin and Blood Pressure Responses to Sodium Chloride. Ann. Intern. Med. 1983, 98 Pt 2, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Whitescarver, S.A.; Holtzclaw, B.J.; Downs, J.H.; Ott, C.E.; Sowers, J.R.; Kotchen, T.A. Effect of Dietary Chloride on Salt-Sensitive and Renin-Dependent Hypertension. Hypertension 1986, 8, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Schmidlin, O.; Yi, S.L.; Bollen, A.W.; Morris, R.C. Genetically Determined Chloride-Sensitive Hypertension and Stroke. Proc. Natl. Acad. Sci. USA 1997, 94, 14748–14752. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, T.W.; Al-Bander, H.A.; Morris, R.C. “Salt-Sensitive” Essential Hypertension in Men. Is the Sodium Ion Alone Important? N. Engl. J. Med. 1987, 317, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Luft, F.C.; Zemel, M.B.; Sowers, J.A.; Fine Berg, N.S.; Weinberger, M.H. Sodium Bicarbonate and Sodium Chloride: Effects on Blood Pressure and Electrolyte Homeostasis in Normal and Hypertensive Man. J. Hypertens. 1990, 8, 663–670. [Google Scholar] [CrossRef]
- Shore, A.C.; Markandu, N.D.; MacGregor, G.A. A Randomized Crossover Study to Compare the Blood Pressure Response to Sodium Loading with and without Chloride in Patients with Essential Hypertension. J. Hypertens. 1988, 6, 613–617. [Google Scholar] [CrossRef] [PubMed]
- McCallum, L.; Lip, S.; Padmanabhan, S. The Hidden Hand of Chloride in Hypertension. Pflug. Arch. 2015, 467, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Chipperfield, A.R.; Harper, A.A. Chloride in Smooth Muscle. Prog. Biophys. Mol. Biol. 2000, 74, 175–221. [Google Scholar] [CrossRef]
- Bulley, S.; Jaggar, J.H. Cl− Channels in Smooth Muscle Cells. Pflug. Arch. 2014, 466, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.J.; Harder, D.R.; Pamnani, M.B.; Haddy, F.J. In Vivo Membrane Potentials of Smooth Muscle Cells in the Caudal Artery of the Rat. Am. J. Physiol. 1985, 249 Pt 1, C78–C83. [Google Scholar] [CrossRef]
- Aickin, C.C.; Brading, A.F. Measurement of Intracellular Chloride in Guinea-pig Vas Deferens by Ion Analysis, 36chloride and Micro-electrodes. J. Physiol. 1982, 326, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Large, W.A.; Wang, Q. Characteristics and Physiological Role of the Ca(2+)-Activated Cl− Conductance in Smooth Muscle. Am. J. Physiol. 1996, 271 Pt 1, C435–C454. [Google Scholar] [CrossRef]
- Goto, K.; Fujii, K.; Onaka, U.; Abe, I.; Fujishima, M. Effects of Adrenomedullin and PAMP on Membrane Potential and Neurotransmission. Peptides 2000, 21, 257–263. [Google Scholar] [CrossRef]
- Brock, M.A.; Cunnane, T.C. Effects of Ca2+ Concentration and Ca2+ Channel Blockers on Noradrenaline Release and Purinergic Neuroeffector Transmission in Rat Tail Artery. Br. J. Pharmacol. 1999, 126, 11–18. [Google Scholar] [CrossRef]
- Burnstock, G.; Ralevic, V. New Insights into the Local Regulation of Blood Flow by Perivascular Nerves and Endothelium. Br. J. Plast. Surg. 1994, 47, 527–543. [Google Scholar] [CrossRef]
- Hill, C.E.; Phillips, J.K.; Sandow, S.L. Heterogeneous Control of Blood Flow amongst Different Vascular Beds. Med. Res. Rev. 2001, 21, 1–60. [Google Scholar] [CrossRef]
- Plane, F.; Garland, C.J. Electrophysiology of Cerebral Blood Vessels. Pharmacol. Ther. 1992, 56, 341–358. [Google Scholar] [CrossRef]
- Gould, D.J.; Hill, C.E. Alpha-Adrenoceptor Activation of a Chloride Conductance in Rat Iris Arterioles. Am. J. Physiol. 1996, 271 Pt 2, H2469–H2476. [Google Scholar] [CrossRef]
- Kitamura, K.; Yamazaki, J. Chloride Channels and Their Functional Roles in Smooth Muscle Tone in the Vasculature. Jpn. J. Pharmacol. 2001, 85, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, N.; Ledoux, J.; Saleh, S.; Sanguinetti, A.; Angermann, J.; O’Driscoll, K.; Britton, F.; Perrino, B.A.; Greenwood, I.A. Regulation of Calcium-Activated Chloride Channels in Smooth Muscle Cells: A Complex Picture Is Emerging. Can. J. Physiol. Pharmacol. 2005, 83, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Hill, M.A. Signaling Mechanisms Underlying the Vascular Myogenic Response. Physiol. Rev. 1999, 79, 387–423. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Conway, M.A.; Knot, H.J.; Brayden, J.E. Chloride Channel Blockers Inhibit Myogenic Tone in Rat Cerebral Arteries. J. Physiol. 1997, 502 Pt 2, 259–264. [Google Scholar] [CrossRef]
- Doughty, J.M.; Langton, P.D. Measurement of Chloride Flux Associated with the Myogenic Response in Rat Cerebral Arteries. J. Physiol. 2001, 534 Pt 3, 753–761. [Google Scholar] [CrossRef]
- Doughty, J.M.; Miller, A.L.; Langton, P.D. Non-Specificity of Chloride Channel Blockers in Rat Cerebral Arteries: Block of the L-Type Calcium Channel. J. Physiol. 1998, 507 Pt 2, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Welsh, D.G.; Nelson, M.T.; Eckman, D.M.; Brayden, J.E. Swelling-Activated Cation Channels Mediate Depolarization of Rat Cerebrovascular Smooth Muscle by Hyposmolarity and Intravascular Pressure. J. Physiol. 2000, 527 Pt 1, 139–148. [Google Scholar] [CrossRef]
- Byrne, N.G.; Large, W.A. Membrane Mechanism Associated with Muscarinic Receptor Activation in Single Cells Freshly Dispersed from the Rat Anococcygeus Muscle. Br. J. Pharmacol. 1987, 92, 371–379. [Google Scholar] [CrossRef]
- Matchkov, V.V. Mechanisms of Cellular Synchronization in the Vascular Wall. Mechanisms of Vasomotion. Dan. Med. Bull. 2010, 57, B4191. [Google Scholar]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.M.; et al. TMEM16A Confers Receptor-Activated Calcium-Dependent Chloride Conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J.V. TMEM16A, a Membrane Protein Associated with Calcium-Dependent Chloride Channel Activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression Cloning of TMEM16A as a Calcium-Activated Chloride Channel Subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dam, V.S.; Boedtkjer, D.M.B.; Aalkjaer, C.; Matchkov, V. The Bestrophin- and TMEM16A-Associated Ca(2+)- Activated Cl(–) Channels in Vascular Smooth Muscles. Channels 2014, 8, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Hawn, M.B.; Akin, E.; Hartzell, H.C.; Greenwood, I.A.; Leblanc, N. Molecular Mechanisms of Activation and Regulation of ANO1-Encoded Ca2+-Activated Cl− Channels. Channels 2021, 15, 569–603. [Google Scholar] [CrossRef]
- Wray, S.; Prendergast, C.; Arrowsmith, S. Calcium-Activated Chloride Channels in Myometrial and Vascular Smooth Muscle. Front. Physiol. 2021, 12, 1805. [Google Scholar] [CrossRef]
- Bulley, S.; Neeb, Z.P.; Burris, S.K.; Bannister, J.P.; Thomas-Gatewood, C.M.; Jangsangthong, W.; Jaggar, J.H. TMEM16A/ANO1 Channels Contribute to the Myogenic Response in Cerebral Arteries. Circ. Res. 2012, 111, 1027–1036. [Google Scholar] [CrossRef]
- Yip, K.P.; Balasubramanian, L.; Kan, C.; Wang, L.; Liu, R.; Ribeiro-Silva, L.; Sham, J.S.K. Intraluminal Pressure Triggers Myogenic Response via Activation of Calcium Spark and Calcium-Activated Chloride Channel in Rat Renal Afferent Arteriole. Am. J. Physiol. Renal Physiol. 2018, 315, F1592–F1600. [Google Scholar] [CrossRef]
- Heinze, C.; Seniuk, A.; Sokolov, M.V.; Huebner, A.K.; Klementowicz, A.E.; Szijártó, I.A.; Schleifenbaum, J.; Vitzthum, H.; Gollasch, M.; Ehmke, H.; et al. Disruption of Vascular Ca2+-Activated Chloride Currents Lowers Blood Pressure. J. Clin. Investig. 2014, 124, 675–686. [Google Scholar] [CrossRef]
- Mulvany, M.J.; Halpern, W. Contractile Properties of Small Arterial Resistance Vessels in Spontaneously Hypertensive and Normotensive Rats. Circ. Res. 1977, 41, 19–26. [Google Scholar] [CrossRef]
- Pintérová, M.; Kuneš, J.; Zicha, J. Altered Neural and Vascular Mechanisms in Hypertension. Physiol. Res. 2011, 60, 381–402. [Google Scholar] [CrossRef]
- Goto, K.; Ohtsubo, T.; Kitazono, T. Endothelium-Dependent Hyperpolarization (EDH) in Hypertension: The Role of Endothelial Ion Channels. Int. J. Mol. Sci. 2018, 19, 315. [Google Scholar] [CrossRef] [PubMed]
- Stekiel, W.J.; Contney, S.J.; Lombard, J.H. Small Vessel Membrane Potential, Sympathetic Input, and Electrogenic Pump Rate in SHR. Am. J. Physiol. 1986, 250 Pt 1, C547–C556. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Tominaga, M.; Ohmori, S.; Kobayashi, K.; Koga, T.; Takata, Y.; Fujishima, M. Decreased Endothelium-Dependent Hyperpolarization to Acetylcholine in Smooth Muscle of the Mesenteric Artery of Spontaneously Hypertensive Rats. Circ. Res. 1992, 70, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Fujii, K.; Abe, I. Impaired β-Adrenergic Hyperpolarization in Arteries from Prehypertensive Spontaneously Hypertensive Rats. Hypertension 2001, 37, 609–613. [Google Scholar] [CrossRef]
- Wang, Z.; Chai, Q.; Liu, Z.; Liu, D.; Chen, L. Chloride Channel Activity of Vascular Smooth Muscle in the Spontaneous Hypertensive Rats. Chin. J. Physiol. 2004, 47, 129–135. [Google Scholar] [PubMed]
- Wang, B.; Li, C.; Huai, R.; Qu, Z. Overexpression of ANO1/TMEM16A, an Arterial Ca2+-Activated Cl− Channel, Contributes to Spontaneous Hypertension. J. Mol. Cell. Cardiol. 2015, 82, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Askew Page, H.R.; Dalsgaard, T.; Baldwin, S.N.; Jepps, T.A.; Povstyan, O.; Olesen, S.P.; Greenwood, I.A. TMEM16A Is Implicated in the Regulation of Coronary Flow and Is Altered in Hypertension. Br. J. Pharmacol. 2019, 176, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Cil, O.; Chen, X.; Askew Page, H.R.; Baldwin, S.N.; Jordan, M.C.; Myat Thwe, P.; Anderson, M.O.; Haggie, P.M.; Greenwood, I.A.; Roos, K.P.; et al. A Small Molecule Inhibitor of the Chloride Channel TMEM16A Blocks Vascular Smooth Muscle Contraction and Lowers Blood Pressure in Spontaneously Hypertensive Rats. Kidney Int. 2021, 100, 311–320. [Google Scholar] [CrossRef]
- Li, R.S.; Wang, Y.; Chen, H.S.; Jiang, F.Y.; Tu, Q.; Li, W.J.; Yin, R.X. TMEM16A Contributes to Angiotensin II-Induced Cerebral Vasoconstriction via the RhoA/ROCK Signaling Pathway. Mol. Med. Rep. 2016, 13, 3691–3699. [Google Scholar] [CrossRef]
- Wang, M.; Yang, H.; Zheng, L.Y.; Zhang, Z.; Tang, Y.B.; Wang, G.L.; Du, Y.H.; Lv, X.F.; Liu, J.; Zhou, J.G.; et al. Downregulation of TMEM16A Calcium-Activated Chloride Channel Contributes to Cerebrovascular Remodeling during Hypertension by Promoting Basilar Smooth Muscle Cell Proliferation. Circulation 2012, 125, 697–707. [Google Scholar] [CrossRef]
- Shiono, K.; Sokabe, H. Renin-Angiotensin System in Spontaneously Hypertensive Rats. Am. J. Physiol. 1976, 231, 1295–1299. [Google Scholar] [CrossRef]
- Okamura, T.; Miyazaki, M.; Inagami, T.; Toda, N. Vascular Renin-Angiotensin System in Two-Kidney, One Clip Hypertensive Rats. Hypertension 1986, 8, 560–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.H.; Zheng, B.; Yang, Z.; He, M.; Yue, L.Y.; Zhang, R.N.; Zhang, M.; Zhang, W.; Zhang, X.; Wen, J.K. TMEM16A and Myocardin Form a Positive Feedback Loop That Is Disrupted by KLF5 during Ang II-Induced Vascular Remodeling. Hypertension 2015, 66, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Abel, P.W.; Hermsmeyer, K. Sympathetic Cross-Innervation of SHR and Genetic Controls Suggests a Trophic Influence on Vascular Muscle Membranes. Circ. Res. 1981, 49, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Kostyunina, D.S.; Zhang, L.; Shvetsova, A.A.; Selivanova, E.K.; Tarasova, O.S.; Matchkov, V.V.; Gaynullina, D.K. Trophic Sympathetic Influence Weakens Pro-Contractile Role of Cl− Channels in Rat Arteries during Postnatal Maturation. Sci. Rep. 2020, 10, 20002. [Google Scholar] [CrossRef] [PubMed]
- Liskova, S.; Petrova, M.; Karen, P.; Behuliak, M.; Zicha, J. Contribution of Ca2+-Dependent Cl− Channels to Norepinephrine-Induced Contraction of Femoral Artery Is Replaced by Increasing EDCF Contribution during Ageing. Biomed Res. Int. 2014, 2014, 289361. [Google Scholar] [CrossRef]
- Jensen, A.B.; Joergensen, H.B.; Dam, V.S.; Kamaev, D.; Boedtkjer, D.; Füchtbauer, E.M.; Aalkjaer, C.; Matchkov, V.V. Variable Contribution of TMEM16A to Tone in Murine Arterial Vasculature. Basic Clin. Pharmacol. Toxicol. 2018, 123, 30–41. [Google Scholar] [CrossRef]
- Dam, V.S.; Boedtkjer, D.M.B.; Nyvad, J.; Aalkjaer, C.; Matchkov, V. TMEM16A Knockdown Abrogates Two Different Ca(2+)-Activated Cl(−) Currents and Contractility of Smooth Muscle in Rat Mesenteric Small Arteries. Pflug. Arch. 2014, 466, 1391–1409. [Google Scholar] [CrossRef]
- Wang, Q.; Dennis Leo, M.; Narayanan, D.; Kuruvilla, K.P.; Jaggar, J.H. Local Coupling of TRPC6 to ANO1/TMEM16A Channels in Smooth Muscle Cells Amplifies Vasoconstriction in Cerebral Arteries. Am. J. Physiol. Cell Physiol. 2016, 310, C1001–C1009. [Google Scholar] [CrossRef]
- Ohya, Y.; Abe, I.; Fujii, K.; Takata, Y.; Fujishima, M. Voltage-Dependent Ca2+ Channels in Resistance Arteries from Spontaneously Hypertensive Rats. Circ. Res. 1993, 73, 1090–1099. [Google Scholar] [CrossRef]
- Pesic, A.; Madden, J.A.; Pesic, M.; Rusch, N.J. High Blood Pressure Upregulates Arterial L-Type Ca2+ Channels: Is Membrane Depolarization the Signal? Circ. Res. 2004, 94, e97–e104. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.M.; Kim, A.; Lee, Y.J.; Lim, W.; Noh, Y.H.; Kim, E.J.; Kim, J.; Kim, T.K.; Park, S.W.; Kim, B.; et al. Enhancement of Receptor-Operated Cation Current and TRPC6 Expression in Arterial Smooth Muscle Cells of Deoxycorticosterone Acetate-Salt Hypertensive Rats. J. Hypertens. 2007, 25, 809–817. [Google Scholar] [CrossRef]
- Suh, B.C.; Hille, B. PIP2 Is a Necessary Cofactor for Ion Channel Function: How and Why? Annu. Rev. Biophys. 2008, 37, 175–195. [Google Scholar] [CrossRef]
- Ta, C.M.; Acheson, K.E.; Rorsman, N.J.G.; Jongkind, R.C.; Tammaro, P. Contrasting Effects of Phosphatidylinositol 4,5-Bisphosphate on Cloned TMEM16A and TMEM16B Channels. Br. J. Pharmacol. 2017, 174, 2984–2999. [Google Scholar] [CrossRef] [PubMed]
- Tembo, M.; Wozniak, K.L.; Bainbridge, R.E.; Carlson, A.E. Phosphatidylinositol 4,5-Bisphosphate (PIP 2) and Ca2+ Are Both Required to Open the Cl− Channel TMEM16A. J. Biol. Chem. 2019, 294, 12556–12564. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Chen, J. Specific PIP 2 Binding Promotes Calcium Activation of TMEM16A Chloride Channels. Commun. Biol. 2021, 4, 259. [Google Scholar] [CrossRef]
- Pritchard, H.A.T.; Leblanc, N.; Albert, A.P.; Greenwood, I.A. Inhibitory Role of Phosphatidylinositol 4,5-Bisphosphate on TMEM16A-Encoded Calcium-Activated Chloride Channels in Rat Pulmonary Artery. Br. J. Pharmacol. 2014, 171, 4311–4321. [Google Scholar] [CrossRef]
- Ek, T.P.; Campbell, M.D.; Deth, R.C. Reduction of Norepinephrine-Induced Tonic Contraction and Phosphoinositide Turnover in Arteries of Spontaneously Hypertensive Rats. A Possible Role for Protein Kinase C. Am. J. Hypertens. 1989, 2, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Shah, S.; Liu, Y.; Zhang, H.; Lees, M.; Fu, Z.; Lippiat, J.D.; Beech, D.J.; Sivaprasadarao, A.; Baldwin, S.A.; et al. Activation of the Cl− Channel ANO1 by Localized Calcium Signals in Nociceptive Sensory Neurons Requires Coupling with the IP3 Receptor. Sci. Signal. 2013, 6, ra73. [Google Scholar] [CrossRef]
- Abou-Saleh, H.; Pathan, A.R.; Daalis, A.; Hubrack, S.; Abou-Jassoum, H.; Al-Naeimi, H.; Rusch, N.J.; Machaca, K. Inositol 1,4,5-Trisphosphate (IP3) Receptor up-Regulation in Hypertension Is Associated with Sensitization of Ca2+ Release and Vascular Smooth Muscle Contractility. J. Biol. Chem. 2013, 288, 32941–32951. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Droogmans, G. Ion Channels and Their Functional Role in Vascular Endothelium. Physiol. Rev. 2001, 81, 1415–1459. [Google Scholar] [CrossRef]
- Ma, M.M.; Gao, M.; Guo, K.M.; Wang, M.; Li, X.Y.; Zeng, X.L.; Sun, L.; Lv, X.F.; Du, Y.H.; Wang, G.L.; et al. TMEM16A Contributes to Endothelial Dysfunction by Facilitating Nox2 NADPH Oxidase-Derived Reactive Oxygen Species Generation in Hypertension. Hypertension 2017, 69, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yasumoto, M.; Suzuki, Y.; Asai, K.; Imaizumi, Y.; Yamamura, H. TMEM16A Ca2+-Activated Cl− Channel Regulates the Proliferation and Migration of Brain Capillary Endothelial Cells. Mol. Pharmacol. 2020, 98, 61–71. [Google Scholar] [CrossRef]
- Skofic Maurer, D.; Zabini, D.; Nagaraj, C.; Sharma, N.; Lengyel, M.; Nagy, B.M.; Frank, S.; Klepetko, W.; Gschwandtner, E.; Enyedi, P.; et al. Endothelial Dysfunction Following Enhanced TMEM16A Activity in Human Pulmonary Arteries. Cells 2020, 9, 1984. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Suzuki, H. Effects of Increased Intracellular Cl− Concentration on Membrane Responses to Acetylcholine in the Isolated Endothelium of Guinea Pig Mesenteric Arteries. J. Physiol. Sci. 2007, 57, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Edwards, F.R.; Hill, C.E. Depolarization Evoked by Acetylcholine in Mesenteric Arteries of Hypertensive Rats Attenuates Endothelium-Dependent Hyperpolarizing Factor. J. Hypertens. 2007, 25, 345–359. [Google Scholar] [CrossRef]
- Goto, K.; Rummery, N.M.; Grayson, T.H.; Hill, C.E. Attenuation of Conducted Vasodilation in Rat Mesenteric Arteries during Hypertension: Role of Inwardly Rectifying Potassium Channels. J. Physiol. 2004, 561, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Hirst, G.D.S.; Bramich, N.J.; Teramoto, N.; Suzuki, H.; Edwards, F.R. Regenerative Component of Slow Waves in the Guinea-Pig Gastric Antrum Involves a Delayed Increase in [Ca2+]i and Cl− Channels. J. Physiol. 2002, 540 Pt 3, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Hill, C.E. Incidence of Myoendothelial Gap Junctions in the Proximal and Distal Mesenteric Arteries of the Rat Is Suggestive of a Role in Endothelium-Derived Hyperpolarizing Factor–Mediated Responses. Circ. Res. 2000, 86, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Fujii, K.; Kansui, Y.; Abe, I.; Iida, M. Critical Role of Gap Junctions in Endothelium-Dependent Hyperpolarization in Rat Mesenteric Arteries. Clin. Exp. Pharmacol. Physiol. 2002, 29, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Mather, S.; Dora, K.A.; Sandow, S.L.; Winter, P.; Garland, C.J. Rapid Endothelial Cell-Selective Loading of Connexin 40 Antibody Blocks Endothelium-Derived Hyperpolarizing Factor Dilation in Rat Small Mesenteric Arteries. Circ. Res. 2005, 97, 399–407. [Google Scholar] [CrossRef]
- Doughty, J.M.; Boyle, J.P.; Langton, P.D. Blockade of Chloride Channels Reveals Relaxations of Rat Small Mesenteric Arteries to Raised Potassium. Br. J. Pharmacol. 2001, 132, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Kwan, Y.W.; Chan, S.W.; Lee, S.M.Y.; Leung, G.P.H. Potentiation of EDHF-Mediated Relaxation by Chloride Channel Blockers. Acta Pharmacol. Sin. 2010, 31, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.W.; Flagella, M.; Sutliff, R.L.; Lorenz, J.N.; Nieman, M.L.; Weber, C.S.; Paul, R.J.; Shull, G.E. Decreased Blood Pressure and Vascular Smooth Muscle Tone in Mice Lacking Basolateral Na+-K+-2Cl− Cotransporter. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1846–H1855. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Roy, K.; Vidossich, P.; Cancedda, L.; De Vivo, M.; Forbush, B.; Cao, E. Structural Basis for Inhibition of the Cation-Chloride Cotransporter NKCC1 by the Diuretic Drug Bumetanide. Nat. Commun. 2022, 13, 2747. [Google Scholar] [CrossRef]
- Garg, P.; Martin, C.F.; Elms, S.C.; Gordon, F.J.; Wall, S.M.; Garland, C.J.; Sutliff, R.L.; O’Neill, W.C. Effect of the Na-K-2Cl Cotransporter NKCC1 on Systemic Blood Pressure and Smooth Muscle Tone. Am. J. Physiol. Heart Circ. Physiol. 2007, 292. [Google Scholar] [CrossRef]
- Orlov, S.N.; Resink, T.J.; Bernhardt, J.; Bühler, F.R. Na(+)-K+ Pump and Na(+)-K+ Co-Transport in Cultured Vascular Smooth Muscle Cells from Spontaneously Hypertensive and Normotensive Rats: Baseline Activity and Regulation. J. Hypertens. 1992, 10, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, S.; Denny, T.N.; Aviv, A. 22Na+ and 86Rb+ Transport in Vascular Smooth Muscle of SHR, Wistar Kyoto, and Wistar Rats. J. Cardiovasc. Pharmacol. 1988, 11, 722–729. [Google Scholar] [CrossRef]
- Tokushige, A.; Kino, M.; Tamura, H.; Hopp, L.; Searle, B.M.; Aviv, A. Bumetanide-Sensitive Sodium-22 Transport in Vascular Smooth Muscle Cell of the Spontaneously Hypertensive Rat. Hypertension 1986, 8, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Canessa, M.; Salazar, G.; Werner, E.; Vallega, G.; Gonzalez, A. Cell Growth and Na-K-Cl Cotransport Responses of Vascular Smooth Muscle Cells of Milan Rats. Hypertension 1994, 23 Pt 2, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.P.L.; Chipperfield, A.R.; Harper, A.A. Accumulation of Intracellular Chloride by (Na-K-Cl) Co-Transport in Rat Arterial Smooth Muscle Is Enhanced in Deoxycorticosterone Acetate (DOCA)/Salt Hypertension. J. Mol. Cell. Cardiol. 1993, 25, 233–237. [Google Scholar] [CrossRef]
- Lee, H.A.; Baek, I.; Seok, Y.M.; Yang, E.; Cho, H.M.; Lee, D.Y.; Hong, S.H.; Kim, I.K. Promoter Hypomethylation Upregulates Na+-K+-2Cl− Cotransporter 1 in Spontaneously Hypertensive Rats. Biochem. Biophys. Res. Commun. 2010, 396, 252–257. [Google Scholar] [CrossRef]
- Cho, H.M.; Lee, H.A.; Kim, H.Y.; Han, H.S.; Kim, I.K. Expression of Na+-K+-2Cl Cotransporter 1 Is Epigenetically Regulated during Postnatal Development of Hypertension. Am. J. Hypertens. 2011, 24, 1286–1293. [Google Scholar] [CrossRef]
- Cho, H.M.; Lee, D.Y.; Kim, H.Y.; Lee, H.A.; Seok, Y.M.; Kim, I.K. Upregulation of the Na(+)-K(+)-2Cl(−) Cotransporter 1 via Histone Modification in the Aortas of Angiotensin II-Induced Hypertensive Rats. Hypertens. Res. 2012, 35, 819–824. [Google Scholar] [CrossRef]
- Bergaya, S.; Faure, S.; Baudrie, V.; Rio, M.; Escoubet, B.; Bonnin, P.; Henrion, D.; Loirand, G.; Achard, J.M.; Jeunemaitre, X.; et al. WNK1 Regulates Vasoconstriction and Blood Pressure Response to α 1-Adrenergic Stimulation in Mice. Hypertension 2011, 58, 439–445. [Google Scholar] [CrossRef]
- Yang, S.S.; Lo, Y.F.; Wu, C.C.; Lin, S.W.; Yeh, C.J.; Chu, P.; Sytwu, H.K.; Uchida, S.; Sasaki, S.; Lin, S.H. SPAK-Knockout Mice Manifest Gitelman Syndrome and Impaired Vasoconstriction. J. Am. Soc. Nephrol. 2010, 21, 1868–1877. [Google Scholar] [CrossRef]
- Zeniya, M.; Sohara, E.; Kita, S.; Iwamoto, T.; Susa, K.; Mori, T.; Oi, K.; Chiga, M.; Takahashi, D.; Yang, S.; et al. Dietary Salt Intake Regulates WNK3-SPAK-NKCC1 Phosphorylation Cascade in Mouse Aorta through Angiotensin II. Hypertension 2013, 62, 872–878. [Google Scholar] [CrossRef]
- Murthy, M.; Kurz, T.; O’Shaughnessy, K.M. WNK Signalling Pathways in Blood Pressure Regulation. Cell. Mol. Life Sci. 2017, 74, 1261–1280. [Google Scholar] [CrossRef]
- Jin, H.-S.; Jung, D. Gender-Specific Association of the ANO1 Genetic Variations with Hypertension. Biomed. Sci. Lett. 2015, 21, 144–151. [Google Scholar] [CrossRef]
- Thongprayoon, C.; Cheungpasitporn, W.; Hansrivijit, P.; Thirunavukkarasu, S.; Chewcharat, A.; Medaura, J.; Mao, M.A.; Kashani, K. Association of Serum Chloride Level Alterations with In-Hospital Mortality. Postgrad. Med. J. 2020, 96, 731–736. [Google Scholar] [CrossRef]
- Takahashi, A.; Maeda, K.; Sasaki, K.; Doi, S.; Nakashima, A.; Doi, T.; Masaki, T. Relationships of Hyperchloremia with Hypertension and Proteinuria in Patients with Chronic Kidney Disease. Clin. Exp. Nephrol. 2022, 26, 880–885. [Google Scholar] [CrossRef] [PubMed]
- McCallum, L.; Jeemon, P.; Hastie, C.E.; Patel, R.K.; Williamson, C.; Redzuan, A.M.; Dawson, J.; Sloan, W.; Muir, S.; Morrison, D.; et al. Serum Chloride Is an Independent Predictor of Mortality in Hypertensive Patients. Hypertension 2013, 62, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellino, M.C.; Massari, F.; Albanese, M.; Ursi, R.; Angelini, G.; Lisi, F.; Amato, L.; Scicchitano, P.; Guida, P.; Brunetti, N.D.; et al. Baseline and Incident Hypochloremia in Chronic Heart Failure Outpatients: Clinical Correlates and Prognostic Role. Eur. J. Intern. Med. 2021, 84, 32–37. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animals | Alterations in Vascular Smooth Muscle CaCCs during Hypertension | Ref. |
|---|---|---|
| SHRs | Increased TMEM16A expression and function in aorta, carotid arteries, hindlimb arteries and mesenteric arteries | [56] |
| Increased TMEM16A expression and function in coronary arteries | [57] | |
| Increased TMEM16A expression and function in renal arterioles | [47] | |
| Knockdown of TMEM16A by siRNA transfection lowered blood pressure | [56] | |
| Inhibition of TMEM16A activity by T16Ainh-A01 lowered blood pressure | [56] | |
| Treatment of mesenteric resistance arteries with TMinh-23 blocked vasoconstriction | [58] | |
| Inhibition of TMEM16A activity by TMinh-23 lowered blood pressure | [58] | |
| 2K2C renal hypertensive rats | Reduced TMEM16A expression and function in basilar arteries during the development of hypertension | [59,60] |
| Animals | Alterations in Endothelial CaCCs during Hypertension | Ref. |
|---|---|---|
| SHRs | Increased CaCC function in endothelium of mesenteric arteries | [86] |
| Increased CaCC function, reduced EDH in mesenteric arteries | [86] | |
| Ang Ⅱ-induced hypertensive mice | Increased TMEM16A expression in endothelium of aorta | [82] |
| Endothelial-specific TMEM16A knockout ameliorated endothelial function and lowered blood pressure | [82] | |
| Endothelial-specific TMEM16A overexpression deteriorated endothelial function and elevated blood pressure | [82] |
| Animals | Alterations in Vascular Smooth Muscle NKCC1 during Hypertension | Ref. |
|---|---|---|
| SHRs | Increased NKCC1 function in aorta and carotid arteries | [97,98,99] |
| Epigenetic upregulation of aorta NKCC1 due to Nkcc1 gene promoter hypomethylation | [102] | |
| Nkcc1 gene promoter hypomethylation resulted from the decreased activity of DNA methyltransferase 3B | [103] | |
| Milan hypertensive rats | Increased NKCC1 function in thoracic aorta | [100] |
| DOCA salt hypertensive rats | Increased NKCC1 function in saphenous branch of femoral arteries | [101] |
| Ang Ⅱ-induced hypertensive rats | Epigenetic upregulation of aorta NKCC1 due to histone modifications | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goto, K.; Kitazono, T. Chloride Ions, Vascular Function and Hypertension. Biomedicines 2022, 10, 2316. https://doi.org/10.3390/biomedicines10092316
Goto K, Kitazono T. Chloride Ions, Vascular Function and Hypertension. Biomedicines. 2022; 10(9):2316. https://doi.org/10.3390/biomedicines10092316
Chicago/Turabian StyleGoto, Kenichi, and Takanari Kitazono. 2022. "Chloride Ions, Vascular Function and Hypertension" Biomedicines 10, no. 9: 2316. https://doi.org/10.3390/biomedicines10092316