

Kynurenine 3-Monooxygenase Interacts with Huntingtin at the Outer Mitochondrial Membrane
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
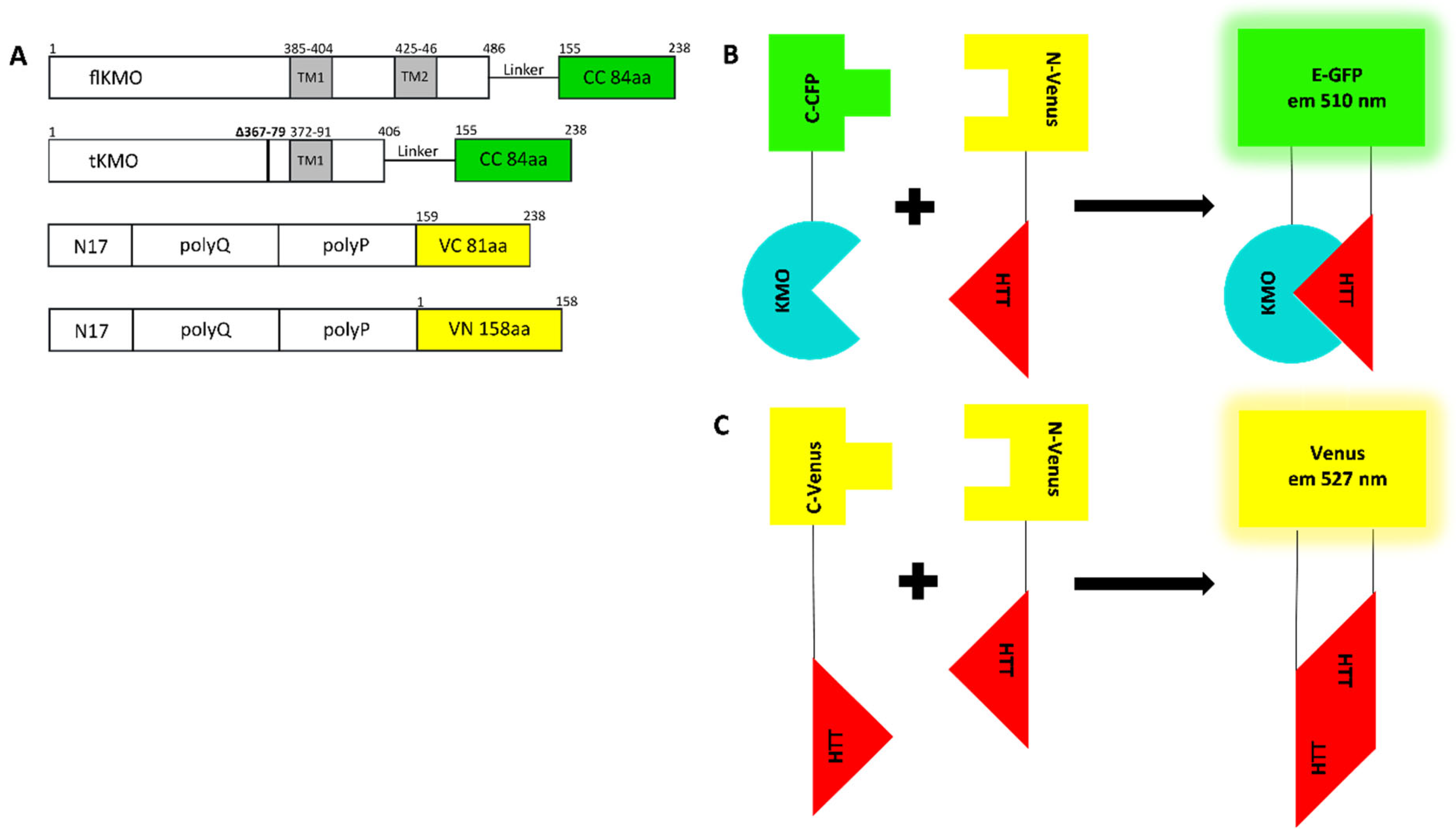
2.1. Construction of KMO and HTT Plasmids
2.2. Mammalian Cell Culture Growth Conditions and Transfection
2.3. Immunocytochemistry (ICC)
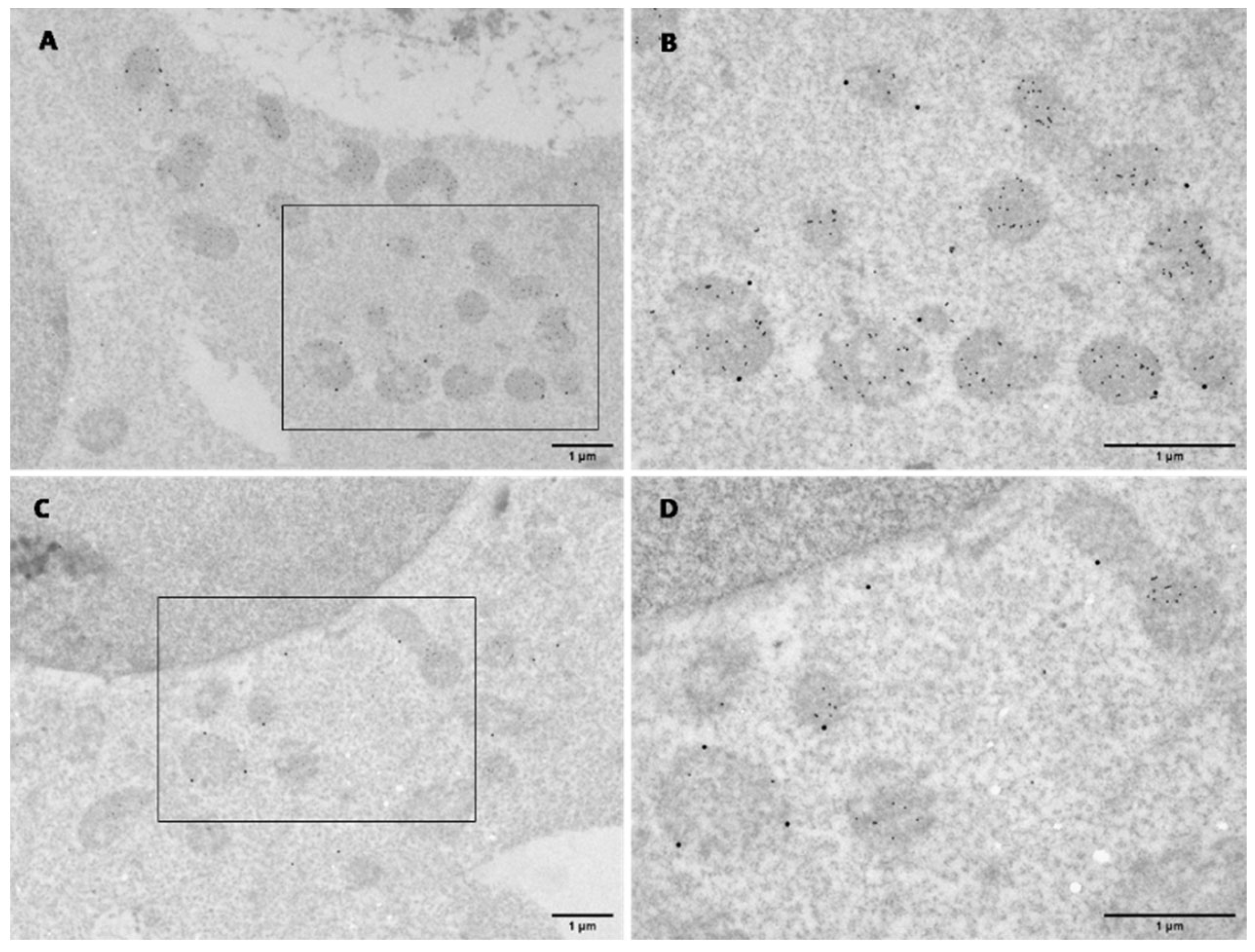
2.4. Transmission Electron Microscopy
2.5. Bimolecular Fluorescence Complementation (BiFC) Assay
2.6. Immunoblotting and Filter Trap Assay
2.7. Kynurenine 3-Monooxygenase Activity
2.8. MYC-Trap Immunoprecipitation
2.9. RFP-Trap Immunoprecipitation
2.10. Statistical Analysis
3. Results
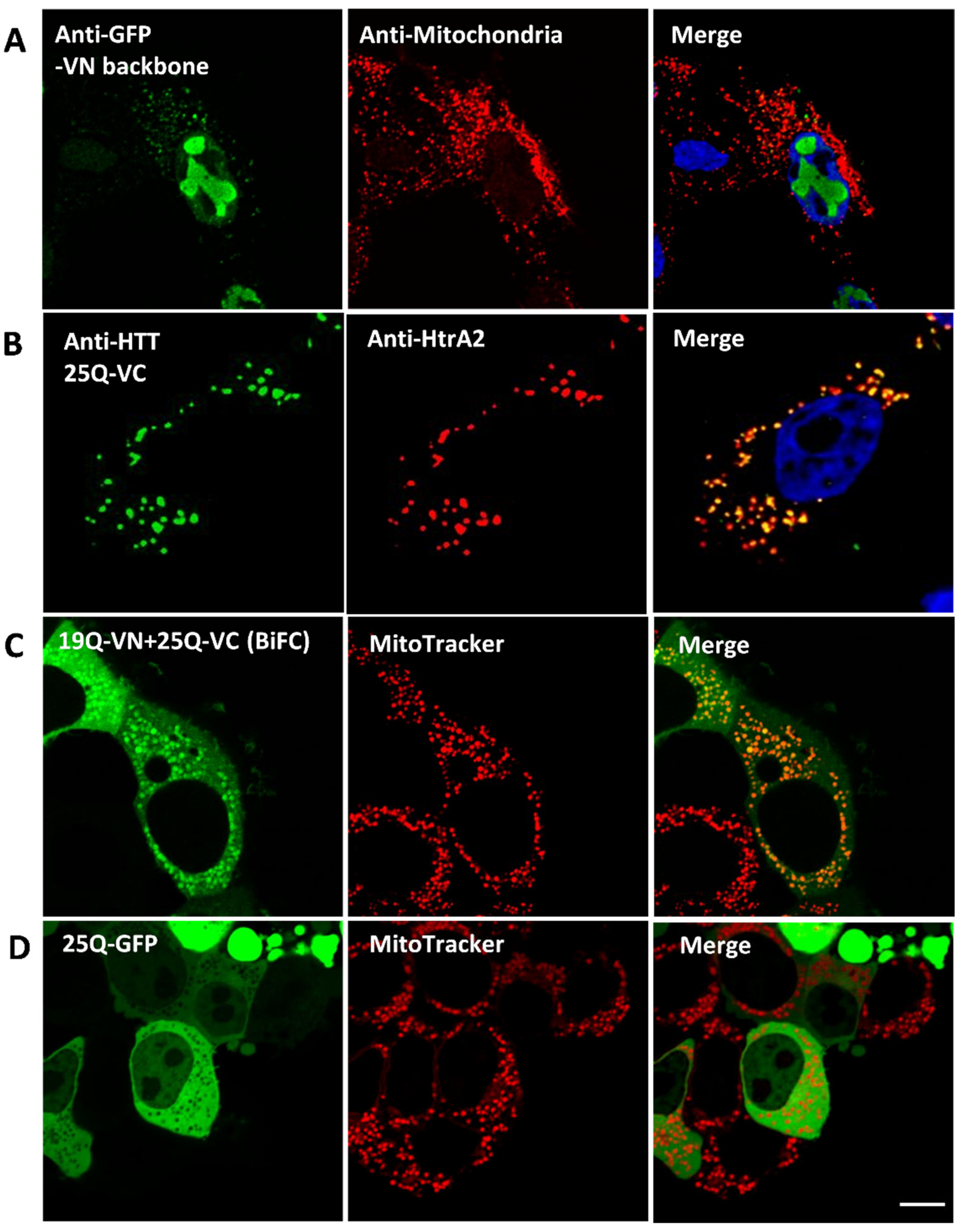
3.1. Human KMO Localises to the Outer Mitochondrial Membrane in HEK293T Cells
3.2. KMO Interacts with Soluble HTT in a PolyQ Dependent Manner at the Mitochondria
3.3. HTT-VN Localises to Mitochondria in HEK293T Cells
3.4. KMO and HTT Co-Localise at the Outer Mitochondrial Membrane
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castellano-Gonzalez, G.; Jacobs, K.R.; Don, E.; Cole, N.J.; Adams, S.; Lim, C.K.; Lovejoy, D.B.; Guillemin, G. Kynurenine 3-Monooxygenase Activity in Human Primary Neurons and Effect on Cellular Bioenergetics Identifies New Neurotoxic Mechanisms. Neurotox. Res. 2019, 35, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Maddison, D.C.; Giorgini, F. The kynurenine pathway and neurodegenerative disease. Semin. Cell. Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahil, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primer 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Bano, D.; Zanetti, F.; Mende, Y.; Nicotera, P. Neurodegenerative processes in Huntington’s disease. Cell. Death Dis. 2011, 2, 228. [Google Scholar] [CrossRef]
- Gil, J.M.; Rego, A.C. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 2008, 27, 2803–2820. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Sutton, L.; Hayden, M.R. Small changes, big impact: Posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist 2011, 17, 475–492. [Google Scholar] [CrossRef]
- Gipson, T.A.; Neueder, A.; Wexler, N.S.; Bates, G.P.; Housman, D. Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biol. 2013, 10, 1647–1652. [Google Scholar] [CrossRef]
- Sathyasaikumar, K.V.; Stachowski, E.K.; Amori, L.; Guidetti, P.; Muchowski, P.J.; Schwarcz, R. Dysfunctional kynurenine pathway metabolism in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 2010, 113, 1416–1425. [Google Scholar] [CrossRef] [Green Version]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr. Biol. 2011, 21, 961–966. [Google Scholar] [CrossRef]
- Giorgini, F.; Guidetti, P.; Nguyen, Q.; Bennett, S.C.; Muchowski, P.J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 2005, 37, 526–531. [Google Scholar] [CrossRef]
- Zwilling, D.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.-Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef]
- Giorgini, F.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Thomas, M.A.R.; Tararina, M.; Wu, H.Q.; Schwarcz, R.; Muchowski, P.J. Targeted deletion of kynurenine 3-monooxygenase in mice: A new tool for studying kynurenine pathway metabolism in periphery and brain. J. Biol. Chem. 2013, 288, 36554–36566. [Google Scholar] [CrossRef]
- Beaumont, V.; Mrzljak, L.; Dijkman, U.; Freije, R.; Heins, M.; Rassoulpour, A.; Tombaugh, G.; Gelman, S.; Bradaia, A.; Steidl, E.; et al. The novel KMO inhibitor CHDI-340246 leads to a restoration of electrophysiological alterations in mouse models of Huntington’s disease. Exp. Neurol. 2016, 282, 99–118. [Google Scholar] [CrossRef]
- Bondulich, M.K.; Fan, Y.; Song, Y.; Giorgini, F.; Bates, G.P. Ablation of kynurenine 3-monooxygenase rescues plasma inflammatory cytokine levels in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 2021, 11, 5484. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.M.; Lowe, A.J.; Tortelli, R.; Heins, M.; Flik, G.; Johnson, E.B.; Vita, E.D.; Scahill, R.I.; Giorgini, F.; et al. Kynurenine pathway metabolites in cerebrospinal fluid and blood as potential biomarkers in Huntington’s disease. J. Neurochem. 2021, 158, 539–553. [Google Scholar] [CrossRef]
- Garrison, A.M.; Parrott, J.M.; Tunon, A.; Delgado, J.; Redus, L.; O’Connor, J.C. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef]
- Valadao, P.A.C.; Santos, K.B.S.; Vieira, T.H.F.E.; Cordeiro, T.M.E.; Teixeira, A.L.; Guatimosim, C.; Miranda, A.S. Inflammation in Huntington’s disease: A few new twists on an old tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar]
- DiFiglia, M.; Sapp, E.; Chase, K.; Schwarz, C.; Meloni, A.; Young, C.; Martin, E.; Vonsattel, J.P.; Carraway, R.; Reeves, S.A.; et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.H.; Love, S.J.; Schilling, G.; Li, S.H.; Li, X.J.; Bao, J.; Wangster, M.V.; Kotzuk, J.A.; Steiner, J.P.; Lo, P.; et al. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. 1995, 14, 1065–1074. [Google Scholar] [CrossRef]
- Choo, Y.S.; Johnson, G.V.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Ferrante, R.J.; Li, X.J.; Hersch, S.M. The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): Comparison with huntingtin in rat and human. J. Neurosci. 1998, 18, 7674–7686. [Google Scholar] [CrossRef]
- Yablonska, S.; Ganesan, V.; Ferrando, L.M.; Kim, J.; Pyzel, A.; Baranova, O.V.; Khattar, N.K.; Larkin, T.M.; Baranov, S.V.; Chen, N.; et al. Mutant huntingtin disrupts mitochondrial proteostasis by interacting with TIM23. Proc. Nat. Acad. Sci. USA. 2019, 116, 16593–16602. [Google Scholar] [CrossRef]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef]
- Mason, R.P.; Casu, M.; Butler, N.; Breda, C.; Campesan, S.; Clapp, J.; Green, E.W.; Dhulkhed, D.; Kyriacou, C.P.; Giorgini, F. Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat. Genet. 2013, 45, 1249–1254. [Google Scholar] [CrossRef]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef]
- Hirai, K.; Kuroyanagi, H.; Tatebayashi, Y.; Hayashi, Y.; Hirabayashi-Takahashi, K.; Saito, K.; Haga, S.; Uemura, T.; Izumi, S. Dual role of the carboxyl-terminal region of pig liver L-kynurenine 3-monooxygenase: Mitochondrial-targeting signal and enzymatic activity. J. Biochem. 2010, 148, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Hirai, K. L-kynurenine 3-monooxygenase from mitochondrial outer membrane of pig liver: Purification, some properties, and monoclonal antibodies directed to the enzyme. J. Biochem. 1998, 123, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Kerppola, T.K. Visualization of molecular interactions by fluorescence complementation. Nat. Rev. Mol. Cell. Biol. 2006, 7, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.; Tenreiro, S.; Miller-Fleming, L.; Outeiro, T.F. Visualization of cell-to-cell transmission of mutant huntingtin oligomers. PLoS Curr. 2011, 3, 1210. [Google Scholar] [CrossRef] [PubMed]
- Kwan, W.; Trager, U.; Davalos, D.; Chou, A.; Bouchard, J.; Andre, R.; Miller, A.; Weiss, A.; Giorgini, F.; Cheah, C.; et al. Mutant huntingtin impairs immune cell migration in Huntington disease. J. Clin. Investig. 2012, 122, 4737–4747. [Google Scholar] [CrossRef]
- Repici, M.; Straatman, K.R.; Balduccio, N.; Enguita, F.J.; Outeiro, T.F.; Giorgini, F. Parkinson’s disease-associated mutations in DJ-1 modulate its dimerization in living cells. J. Mol. Med. 2013, 91, 599–611. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224 Pt 3, 213–232. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Parrott, J.M.; O’Connor, J.C. Kynurenine 3-Monooxygenase: An Influential Mediator of Neuropathology. Front. Psychiatry 2015, 6, 116. [Google Scholar] [CrossRef]
- Shyu, Y.J.; Liu, H.; Deng, X.; Hu, C.D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotech 2006, 40, 61–66. [Google Scholar] [CrossRef]
- Gafni, J.; Ellerby, L.M. Calpain activation in Huntington’s disease. J. Neurosci. 2002, 22, 4842–4849. [Google Scholar] [CrossRef]
- Kegel, K.B.; Schewkunow, V.; Sapp, E.; Masso, N.; Wanker, E.E.; DiFiglia, M.; Goldmann, W.H. Polyglutamine expansion in huntingtin increases its insertion into lipid bilayers. Biochem. Biophys. Res. Commun. 2009, 387, 472–475. [Google Scholar] [CrossRef]
- Kegel, K.B.; Sapp, E.; Alexander, J.; Valencia, A.; Reeves, P.; Li, X.; Masso, N.; Sobin, L.; Aronin, N.; Difiglia, M. Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J. Neurochem. 2009, 110, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Kegel, K.B.; Sapp, E.; Yoder, J.; Cuiffo, B.; Sobin, L.; Kim, Y.J.; Qin, Z.-H.; Hayden, M.R.; Aronin, N.; Scott, D.L.; et al. Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol Chem. 2005, 280, 36464–36473. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.; Auer, M.; Binnie, M.; Zheng, X.; Pham, N.T.; Iredale, J.P.; Webster, S.P.; Mole, D.J. Overexpression of human kynurenine-3-monooxygenase protects against 3-hydroxykynurenine-mediated apoptosis through bidirectional nonlinear feedback. Cell Death Dis. 2016, 7, e2197. [Google Scholar] [CrossRef] [Green Version]
- Maddison, D.C.; Alfonso-Nunez, M.; Swaih, A.M.; Breda, C.; Campesan, S.; Allcock, N.; Straatman-Iwanowska, A.; Kyriacou, C.P.; Giorgini, F. A novel role for kynurenine 3-monooxygenase in mitochondrial dynamics. PLoS Genet. 2020, 16, e1009129. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swaih, A.M.; Breda, C.; Sathyasaikumar, K.V.; Allcock, N.; Collier, M.E.W.; Mason, R.P.; Feasby, A.; Herrera, F.; Outeiro, T.F.; Schwarcz, R.; et al. Kynurenine 3-Monooxygenase Interacts with Huntingtin at the Outer Mitochondrial Membrane. Biomedicines 2022, 10, 2294. https://doi.org/10.3390/biomedicines10092294
Swaih AM, Breda C, Sathyasaikumar KV, Allcock N, Collier MEW, Mason RP, Feasby A, Herrera F, Outeiro TF, Schwarcz R, et al. Kynurenine 3-Monooxygenase Interacts with Huntingtin at the Outer Mitochondrial Membrane. Biomedicines. 2022; 10(9):2294. https://doi.org/10.3390/biomedicines10092294
Chicago/Turabian StyleSwaih, Aisha M., Carlo Breda, Korrapati V. Sathyasaikumar, Natalie Allcock, Mary E. W. Collier, Robert P. Mason, Adam Feasby, Federico Herrera, Tiago F. Outeiro, Robert Schwarcz, and et al. 2022. "Kynurenine 3-Monooxygenase Interacts with Huntingtin at the Outer Mitochondrial Membrane" Biomedicines 10, no. 9: 2294. https://doi.org/10.3390/biomedicines10092294