
The Tyrosine Phosphatase SHP2: A New Target for Insulin Resistance?
 , ,
, ,
Abstract
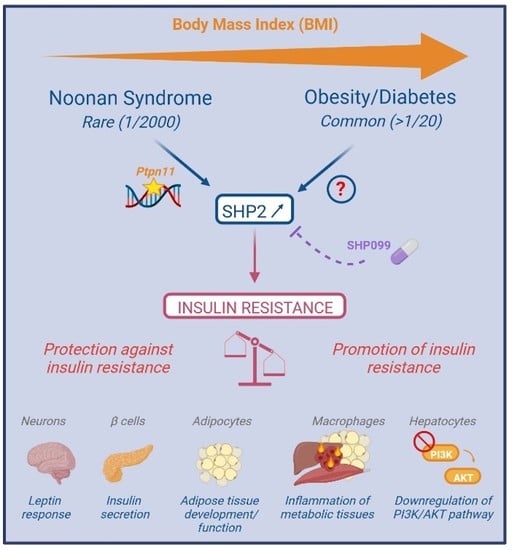
:
1. The Tyrosine Phosphatase SHP2
1.1. Structure, Function and Regulation
1.2. Physiological Roles of SHP2 during Development and Homeostasis, and Pathological Consequences of Its Dysfunction
2. SHP2, in Insulin Resistance: A Fragmented and Confusing View from Cellular and Tissue Specific Models
2.1. General Considerations about Insulin Signaling and Insulin Resistance
2.2. A Direct and Dual Role of SHP2 in Insulin Signaling
2.3. A protective or Causal Role of SHP2 in Insulin Resistance In Vivo?
3. SHP2 in Insulin Resistance: Lessons from Integrated Models
3.1. Genetic Diseases, Susceptibility Gene and SHP2 Dysregulation in Obesity/Diabetes
3.2. SHP2 Targeting in Obesity/Diabetes
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tajan, M.; Serra, D.A.; Valet, P.; Edouard, T.; Yart, A. SHP2 sails from physiology to pathology. Eur. J. Med. Genet. 2015, 58, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, S.; Zhao, M.; Yang, X.; Yu, B. Strategies Targeting Protein Tyrosine Phosphatase SHP2 for Cancer Therapy. J. Med. Chem. 2022, 65, 3066–3079. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.; Geldenhuys, W.J.; Agazie, Y.M. A specific amino acid context in EGFR and HER2 phosphorylation sites enables selective binding to the active site of Src homology phosphatase 2 (SHP2). J. Biol. Chem. 2020, 295, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, R.; Song, J. DephosSite: A machine learning approach for discovering phosphotase-specific dephosphorylation sites. Sci. Rep. 2016, 6, 23510. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Xie, J.; Zhong, Q.; Wang, Y.; Zhang, S.; Luo, F.; Wen, F.; Xie, J.; Zhao, J.; Sun, X.; et al. A novel partially open state of SHP2 points to a “multiple gear” regulation mechanism. J. Biol. Chem. 2021, 296, 100538. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Xie, J.; Kong, W.; Xie, J.; Li, Y.; Du, L.; Zheng, Q.; Sun, L.; Guan, M.; Li, H.; et al. Phase Separation of Disease-Associated SHP2 Mutants Underlies MAPK Hyperactivation. Cell 2020, 183, 490–502.e18. [Google Scholar] [CrossRef]
- Lin, C.-C.; Suen, K.M.; Jeffrey, P.-A.; Wieteska, L.; Lidster, J.A.; Bao, P.; Curd, A.P.; Stainthorp, A.; Seiler, C.; Koss, H.; et al. Receptor tyrosine kinases regulate signal transduction through a liquid-liquid phase separated state. Mol. Cell 2022, 82, 1089–1106.e12. [Google Scholar] [CrossRef]
- Burmeister, B.T.; Wang, L.; Gold, M.G.; Skidgel, R.A.; O’Bryan, J.P.; Carnegie, G.K. Protein Kinase A (PKA) Phosphorylation of Shp2 Protein Inhibits Its Phosphatase Activity and Modulates Ligand Specificity. J. Biol. Chem. 2015, 290, 12058–12067. [Google Scholar] [CrossRef]
- Sun, J.; Lu, S.; Ouyang, M.; Lin, L.J.; Zhuo, Y.; Liu, B.; Chien, S.; Neel, B.G.; Wang, Y. Antagonism between binding site affinity and conformational dynamics tunes alternative ci.is-interactions within Shp2. Nat. Commun. 2013, 4, 2037. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Masoudi, M.; Takahashi, A.; Fujii, Y.; Hayashi, T.; Kikuchi, I.; Satou, Y.; Taira, M.; Hatakeyama, M. YAP and TAZ, Hippo signaling targets, act as a rheostat for nuclear SHP2 function. Dev. Cell 2013, 26, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, R.; Harizanova, J.; Stockert, R.; Schröder, K.; Bastiaens, P.I.H.; Neel, B.G. Assay to visualize specific protein oxidation reveals spatio-temporal regulation of SHP2. Nat. Commun. 2017, 8, 466. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Hale, A.J.; Lemeer, S.; den Hertog, J. Differential oxidation of protein-tyrosine phosphatases during zebrafish caudal fin regeneration. Sci. Rep. 2017, 7, 8460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, D.; Fan, H.; Wu, R.; Tu, J.; Zhang, F.Q.; Wang, M.; Zheng, H.; Qu, C.-K.; Elf, S.E.; et al. Cellular signals converge at the NOX2-SHP-2 axis to induce reductive carboxylation in cancer cells. Cell Chem. Biol. 2022, 29, 1200–1208.E6. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Chen, W.; Zhu, J.; Wu, G.; Shen, R.; Xi, M.; Sun, H. Therapeutic potential of targeting SHP2 in human developmental disorders and cancers. Eur. J. Med. Chem. 2020, 190, 112117. [Google Scholar] [CrossRef]
- Dance, M.; Montagner, A.; Salles, J.P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Buday, L.; Vas, V. Novel regulation of Ras proteins by direct tyrosine phosphorylation and dephosphorylation. Cancer Metastasis Rev. 2020, 39, 1067–1073. [Google Scholar] [CrossRef]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Tsiaras, W.G.; Araki, T.; Wen, G.; Minichiello, L.; Klein, R.; Neel, B.G. Receptor-specific regulation of phosphatidylinositol 3’-kinase activation by the protein tyrosine phosphatase Shp2. Mol. Cell. Biol. 2002, 22, 4062–4072. [Google Scholar] [CrossRef]
- Guo, W.; Liu, W.; Chen, Z.; Gu, Y.; Peng, S.; Shen, L.; Shen, Y.; Wang, X.; Feng, G.-S.; Sun, Y.; et al. Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis. Nat. Commun. 2017, 8, 2168. [Google Scholar] [CrossRef]
- Lee, I.; Pecinova, A.; Pecina, P.; Neel, B.G.; Araki, T.; Kucherlapati, R.; Roberts, A.E.; Hüttemann, M. A suggested role for mitochondria in Noonan syndrome. Biochim. Biophys. Acta 2010, 1802, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Zang, Q.S.; Martinez, B.; Yao, X.; Maass, D.L.; Ma, L.; Wolf, S.E.; Minei, J.P. Sepsis-induced cardiac mitochondrial dysfunction involves altered mitochondrial-localization of tyrosine kinase Src and tyrosine phosphatase SHP2. PLoS ONE 2012, 7, e43424. [Google Scholar] [CrossRef] [PubMed]
- Chughtai, N.; Schimchowitsch, S.; Lebrun, J.-J.; Ali, S. Prolactin induces SHP-2 association with Stat5, nuclear translocation, and binding to the beta-casein gene promoter in mammary cells. J. Biol. Chem. 2002, 277, 31107–31114. [Google Scholar] [CrossRef] [PubMed]
- Jakob, S.; Schroeder, P.; Lukosz, M.; Buchner, N.; Spyridopoulos, I.; Altschmied, J.; Haendeler, J. Nuclear protein tyrosine phosphatase Shp-2 is one important negative regulator of nuclear export of telomerase reverse transcriptase. J. Biol. Chem. 2008, 283, 33155–33161. [Google Scholar] [CrossRef] [PubMed]
- Jakob, S.; Altschmied, J.; Haendeler, J. “Shping 2” different cellular localizations—A potential new player in aging processes. Aging 2009, 1, 664–668. [Google Scholar] [CrossRef]
- He, Z.; Zhang, S.S.; Meng, Q.; Li, S.; Zhu, H.H.; Raquil, M.A.; Alderson, N.; Zhang, H.; Wu, J.; Rui, L.; et al. Shp2 controls female body weight and energy balance by integrating leptin and estrogen signals. Mol. Cell. Biol. 2012, 32, 1867–1878. [Google Scholar] [CrossRef]
- Ran, H.; Kong, S.; Zhang, S.; Cheng, J.; Zhou, C.; He, B.; Xin, Q.; Lydon, J.P.; De Mayo, F.J.; Feng, G.-S.; et al. Nuclear Shp2 directs normal embryo implantation via facilitating the ERα tyrosine phosphorylation by the Src kinase. Proc. Natl. Acad. Sci. USA 2017, 114, 4816–4821. [Google Scholar] [CrossRef]
- Batth, T.S.; Papetti, M.; Pfeiffer, A.; Tollenaere, M.A.X.; Francavilla, C.; Olsen, J.V. Large-Scale Phosphoproteomics Reveals Shp-2 Phosphatase-Dependent Regulators of Pdgf Receptor Signaling. Cell Rep. 2018, 22, 2784–2796. [Google Scholar] [CrossRef]
- Corallino, S.; Iwai, L.K.; Payne, L.S.; Huang, P.H.; Sacco, F.; Cesareni, G.; Castagnoli, L. Alterations in the phosphoproteomic profile of cells expressing a non-functional form of the SHP2 phosphatase. N. Biotechnol. 2016, 33, 524–536. [Google Scholar] [CrossRef]
- Vemulapalli, V.; Chylek, L.A.; Erickson, A.; Pfeiffer, A.; Gabriel, K.-H.; LaRochelle, J.; Subramanian, K.; Cao, R.; Stegmaier, K.; Mohseni, M.; et al. Time-resolved phosphoproteomics reveals scaffolding and catalysis-responsive patterns of SHP2-dependent signaling. eLife 2021, 10, e64251. [Google Scholar] [CrossRef]
- Chaudhari, M.; Thapa, N.; Ismail, H.; Chopade, S.; Caragea, D.; Köhn, M.; Newman, R.H.; Kc, D.B. DTL-DephosSite: Deep Transfer Learning Based Approach to Predict Dephosphorylation Sites. Front. Cell Dev. Biol. 2021, 9, 662983. [Google Scholar] [CrossRef]
- Kan, C.; Yang, F.; Wang, S. SHP2-Mediated Signal Networks in Stem Cell Homeostasis and Dysfunction. Stem Cells Int. 2018, 2018, 8351374. [Google Scholar] [CrossRef] [PubMed]
- Vainonen, J.P.; Momeny, M.; Westermarck, J. Druggable cancer phosphatases. Sci. Transl. Med. 2021, 13, eabe2967. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Ramdas, B.; Wan, C.; Sandusky, G.; Mohseni, M.; Zhang, C.; Kapur, R. SHP2 inhibition reduces leukemogenesis in models of combined genetic and epigenetic mutations. J. Clin. Investig. 2019, 129, 5468–5473. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Everingham, S.; Ramdas, B.; Kapur, R.; Craig, A.W. SHP2 phosphatase promotes mast cell chemotaxis toward stem cell factor via enhancing activation of the Lyn/Vav/Rac signaling axis. J. Immunol. 2014, 192, 4859–4866. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Han, T.; Tang, H.; Huang, K.; Min, J.; Li, J.; Ding, X.; Xu, Z. Shp2 Inhibits Proliferation of Esophageal Squamous Cell Cancer via Dephosphorylation of Stat3. Int. J. Mol. Sci. 2017, 18, 134. [Google Scholar] [CrossRef] [PubMed]
- Bard-Chapeau, E.A.; Li, S.; Ding, J.; Zhang, S.S.; Zhu, H.H.; Princen, F.; Fang, D.D.; Han, T.; Bailly-Maitre, B.; Poli, V.; et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 2011, 19, 629–639. [Google Scholar] [CrossRef]
- Liu, J.J.; Xin, B.; Du, L.; Chen, L.; Long, Y.; Feng, G.-S. Pharmaceutical Shp2 inhibition suppresses primary and metastasized liver tumors by provoking hepatic innate immunity. Hepatology 2022. Online Version of Record before inclusion in an issue. [Google Scholar] [CrossRef]
- Chen, W.S.; Liang, Y.; Zong, M.; Liu, J.J.; Kaneko, K.; Hanley, K.L.; Zhang, K.; Feng, G.-S. Single-cell transcriptomics reveals opposing roles of Shp2 in Myc-driven liver tumor cells and microenvironment. Cell Rep. 2021, 37, 109974. [Google Scholar] [CrossRef]
- Tajan, M.; Paccoud, R.; Branka, S.; Edouard, T.; Yart, A. The RASopathy Family: Consequences of Germline Activation of the RAS/MAPK Pathway. Endocr. Rev. 2018, 39, 676–700. [Google Scholar] [CrossRef]
- Kim, H.K.; Feng, G.S.; Chen, D.; King, P.D.; Kamiya, N. Targeted disruption of Shp2 in chondrocytes leads to metachondromatosis with multiple cartilaginous protrusions. J. Bone Miner. Res. 2014, 29, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Do Carmo, J.M.; da Silva, A.A.; Ebaady, S.E.; Sessums, P.O.; Abraham, R.S.; Elmquist, J.K.; Lowell, B.B.; Hall, J.E. Shp2 signaling in POMC neurons is important for leptin’s actions on blood pressure, energy balance, and glucose regulation. Am. J. Physiol. 2014, 307, R1438–R1447. [Google Scholar] [CrossRef]
- Zhang, E.E.; Chapeau, E.; Hagihara, K.; Feng, G.S. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 16064–16069. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhu, H.H.; Bauler, T.J.; Wang, J.; Ciaraldi, T.; Alderson, N.; Li, S.; Raquil, M.A.; Ji, K.; Wang, S.; et al. Nonreceptor tyrosine phosphatase Shp2 promotes adipogenesis through inhibition of p38 MAP kinase. Proc. Natl. Acad. Sci. USA 2012, 110, E79–E88. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Delibegovic, M.; Matsuo, I.; Nagata, N.; Liu, S.; Bettaieb, A.; Xi, Y.; Araki, K.; Yang, W.; Kahn, B.B.; et al. Altered glucose homeostasis in mice with liver-specific deletion of Src homology phosphatase 2. J. Biol. Chem. 2010, 285, 39750–39758. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.S.; Hao, E.; Yu, J.; Liu, W.; Wang, J.; Levine, F.; Feng, G.S. Coordinated regulation by Shp2 tyrosine phosphatase of signaling events controlling insulin biosynthesis in pancreatic beta-cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7531–7536. [Google Scholar] [CrossRef] [PubMed]
- Princen, F.; Bard, E.; Sheikh, F.; Zhang, S.S.; Wang, J.; Zago, W.M.; Wu, D.; Diaz Trelles, R.; Bailly-Maitre, B.; Kahn, C.R.; et al. Deletion of Shp2 Tyrosine Phosphatase in Muscle Leads to Dilated Cardiomyopathy, Insulin Resistance and Premature Death. Mol. Cell. Biol. 2009, 29, 378–388. [Google Scholar] [CrossRef]
- Nagata, N.; Matsuo, K.; Bettaieb, A.; Bakke, J.; Matsuo, I.; Graham, J.; Xi, Y.; Liu, S.; Tomilov, A.; Tomilova, N.; et al. Hepatic SRC homology phosphatase 2 regulates energy balance in mice. Endocrinology 2012, 153, 3158–3169. [Google Scholar] [CrossRef]
- De Rocca Serra-Nedelec, A.; Edouard, T.; Treguer, K.; Tajan, M.; Araki, T.; Dance, M.; Mus, M.; Montagner, A.; Tauber, M.; Salles, J.P.; et al. Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc. Natl. Acad. Sci. USA 2012, 109, 4257–4262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, W.; Zhao, Q.; Chen, J.; Wang, T.; Ji, J. The role of the protein tyrosine phosphatase SHP2 in ossification. Dev. Dyn. 2022, 251, 748–758. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Wang, Y.; Yang, Y.; Sun, D.; Li, H.; Chen, L. Targeting SHP2 as a therapeutic strategy for inflammatory diseases. Eur. J. Med. Chem. 2021, 214, 113264. [Google Scholar] [CrossRef]
- Mélique, S.; Yang, C.; Lesourne, R. Negative times negative equals positive, THEMIS sets the rule on thymic selection and peripheral T cell responses. Biomed. J. 2022, 45, 334–346. [Google Scholar] [CrossRef]
- Niogret, C.; Birchmeier, W.; Guarda, G. SHP-2 in Lymphocytes’ Cytokine and Inhibitory Receptor Signaling. Front. Immunol. 2019, 10, 2468. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-J.; Wang, C.-W.; Tao, L.; Yan, Y.-H.; Zhang, M.-J.; Liu, Z.-X.; Li, Y.-X.; Zhao, H.-Q.; Li, X.-M.; He, X.-D.; et al. Insulin signaling regulates longevity through protein phosphorylation in Caenorhabditis elegans. Nat. Commun. 2021, 12, 4568. [Google Scholar] [CrossRef] [PubMed]
- Séité, S.; Harrison, M.C.; Sillam-Dussès, D.; Lupoli, R.; Van Dooren, T.J.M.; Robert, A.; Poissonnier, L.-A.; Lemainque, A.; Renault, D.; Acket, S.; et al. Lifespan prolonging mechanisms and insulin upregulation without fat accumulation in long-lived reproductives of a higher termite. Commun. Biol. 2022, 5, 44. [Google Scholar] [CrossRef]
- Ruzzi, L.R.; Schilman, P.E.; San Martin, A.; Lew, S.E.; Gelb, B.D.; Pagani, M.R. The Phosphatase CSW Controls Life Span by Insulin Signaling and Metabolism Throughout Adult Life in Drosophila. Front. Genet. 2020, 11, 364. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef]
- Vinayagam, A.; Kulkarni, M.M.; Sopko, R.; Sun, X.; Hu, Y.; Nand, A.; Villalta, C.; Moghimi, A.; Yang, X.; Mohr, S.E.; et al. An Integrative Analysis of the InR/PI3K/Akt Network Identifies the Dynamic Response to Insulin Signaling. Cell Rep. 2016, 16, 3062–3074. [Google Scholar] [CrossRef]
- Tajan, M.; Batut, A.; Cadoudal, T.; Deleruyelle, S.; Le Gonidec, S.; Saint Laurent, C.; Vomscheid, M.; Wanecq, E.; Treguer, K.; De Rocca Serra-Nédélec, A.; et al. LEOPARD syndrome-associated SHP2 mutation confers leanness and protection from diet-induced obesity. Proc. Natl. Acad. Sci. USA 2014, 11, E4494–E4503. [Google Scholar] [CrossRef] [Green Version]
- Myers, M.G., Jr.; Mendez, R.; Shi, P.; Pierce, J.H.; Rhoads, R.; White, M.F. The COOH-terminal tyrosine phosphorylation sites on IRS-1 bind SHP-2 and negatively regulate insulin signaling. J. Biol. Chem. 1998, 273, 26908–26914. [Google Scholar] [CrossRef]
- Yue, X.; Han, T.; Hao, W.; Wang, M.; Fu, Y. SHP2 knockdown ameliorates liver insulin resistance by activating IRS-2 phosphorylation through the AKT and ERK1/2 signaling pathways. FEBS Open Bio 2020, 10, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Wang, X.; Wu, Y.; Xu, Y.; Zhuo, S.; Qi, M.; Ji, W.; Zhan, L. Polarity Protein AF6 Controls Hepatic Glucose Homeostasis and Insulin Sensitivity by Modulating IRS1/AKT Insulin Pathway in an SHP2-Dependent Manner. Diabetes 2019, 68, 1577–1590. [Google Scholar] [CrossRef]
- Choi, E.; Kikuchi, S.; Gao, H.; Brodzik, K.; Nassour, I.; Yopp, A.; Singal, A.G.; Zhu, H.; Yu, H. Mitotic regulators and the SHP2-MAPK pathway promote IR endocytosis and feedback regulation of insulin signaling. Nat. Commun. 2019, 10, 1473. [Google Scholar] [CrossRef]
- Hall, C.; Yu, H.; Choi, E. Insulin receptor endocytosis in the pathophysiology of insulin resistance. Exp. Mol. Med. 2020, 52, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Saxton, T.M.; Henkemeyer, M.; Gasca, S.; Shen, R.; Rossi, D.J.; Shalaby, F.; Feng, G.S.; Pawson, T. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J. 1997, 16, 2352–2364. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, H.; Hasegawa, M.; Sugai, S.; Obata, T.; Ugi, S.; Morino, K.; Egawa, K.; Fujita, T.; Sakamoto, T.; Nishio, Y.; et al. Expression of a dominant negative SHP-2 in transgenic mice induces insulin resistance. J. Biol. Chem. 1999, 274, 30236–30243. [Google Scholar] [CrossRef]
- Krajewska, M.; Banares, S.; Zhang, E.E.; Huang, X.; Scadeng, M.; Jhala, U.S.; Feng, G.S.; Krajewski, S. Development of diabesity in mice with neuronal deletion of Shp2 tyrosine phosphatase. Am. J. Pathol. 2008, 172, 1312–1324. [Google Scholar] [CrossRef]
- Bettaieb, A.; Matsuo, K.; Matsuo, I.; Nagata, N.; Chahed, S.; Liu, S.; Haj, F.G. Adipose-specific deletion of Src homology phosphatase 2 does not significantly alter systemic glucose homeostasis. Metabolism 2011, 286, 9225–9235. [Google Scholar] [CrossRef]
- Qi, X.; Sun, Z.; Li, X.; Jiao, Y.; Chen, S.; Song, P.; Qian, Z.; Qian, J.; Qiu, X.; Tang, L. Shp2 suppresses fat accumulation in white adipose tissue by activating Wnt/β-catenin signaling following vertical sleeve gastrectomy in obese rats with type-2 diabetes. Exp. Ther. Med. 2022, 23, 302. [Google Scholar] [CrossRef]
- Liu, W.; Yin, Y.; Wang, M.; Fan, T.; Zhu, Y.; Shen, L.; Peng, S.; Gao, J.; Deng, G.; Meng, X.; et al. Disrupting phosphatase SHP2 in macrophages protects mice from high-fat diet-induced hepatic steatosis and insulin resistance by elevating IL-18 levels. J. Biol. Chem. 2020, 31, 10842–10856. [Google Scholar] [CrossRef]
- Ranza, E.; Guimier, A.; Verloes, A.; Capri, Y.; Marques, C.; Auclair, M.; Mathieu-Dramard, M.; Morin, G.; Thevenon, J.; Faivre, L.; et al. Overlapping phenotypes between SHORT and Noonan syndromes in patients with PTPN11 pathogenic variants. Clin. Genet. 2020, 98, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Noronha, R.M.; Villares, S.M.F.; Torres, N.; Quedas, E.P.S.; Homma, T.K.; Albuquerque, E.V.A.; Moraes, M.B.; Funari, M.F.A.; Bertola, D.R.; Jorge, A.A.L.; et al. Noonan syndrome patients beyond the obvious phenotype: A potential unfavorable metabolic profile. Am. J. Med. Genet. A 2021, 185, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Paccoud, R.; Saint-Laurent, C.; Piccolo, E.; Tajan, M.; Dortignac, A.; Pereira, O.; Le Gonidec, S.; Baba, I.; Gélineau, A.; Askia, H.; et al. SHP2 drives inflammation-triggered insulin resistance by reshaping tissue macrophage populations. Sci. Transl. Med. 2021, 13, 591. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Zhang, H.; Zhang, Y.; Zheng, M.; Liu, R.; Zhao, Y.; Zhang, X.; Cheng, H.; Cao, Q.; Ke, Y. Phosphatase Shp2 exacerbates intestinal inflammation by disrupting macrophage responsiveness to interleukin-10. J. Exp. Med. 2019, 216, 337–349. [Google Scholar] [CrossRef]
- Hsu, A.Y.; Gurol, T.; Sobreira, T.J.P.; Zhang, S.; Moore, N.; Cai, C.; Zhang, Z.Y.; Deng, Q. Development and Characterization of an Endotoxemia Model in Zebra Fish. Front. Immunol. 2018, 9, 607. [Google Scholar] [CrossRef]
- Wang, J.; Mizui, M.; Zeng, L.F.; Bronson, R.; Finnell, M.; Terhorst, C.; Kyttaris, V.C.; Tsokos, G.C.; Zhang, Z.Y.; Kontaridis, M.I. Inhibition of SHP2 ameliorates the pathogenesis of systemic lupus erythematosus. J. Clin. Investig. 2016, 126, 2077–2092. [Google Scholar] [CrossRef]
- Tao, B.; Jin, W.; Xu, J.; Liang, Z.; Yao, J.; Zhang, Y.; Wang, K.; Cheng, H.; Zhang, X.; Ke, Y. Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J. Immunol. 2014, 193, 2801–2811. [Google Scholar] [CrossRef]
- Xu, J.; Tao, B.; Guo, X.; Zhou, S.; Li, Y.; Zhang, Y.; Zhou, Z.; Cheng, H.; Zhang, X.; Ke, Y. Macrophage-Restricted Shp2 Tyrosine Phosphatase Acts as a Rheostat for MMP12 through TGF-beta Activation in the Prevention of Age-Related Emphysema in Mice. J. Immunol. 2017, 199, 2323–2332. [Google Scholar] [CrossRef]
- Zhao, L.; Xia, J.; Li, T.; Zhou, H.; Ouyang, W.; Hong, Z.; Ke, Y.; Qian, J.; Xu, F. Shp2 Deficiency Impairs the Inflammatory Response Against Haemophilus influenzae by Regulating Macrophage Polarization. J. Infect. Dis. 2016, 214, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Kraja, A.T.; Chasman, D.I.; North, K.E.; Reiner, A.P.P.; Yanek, L.R.; Kilpelainen, T.O.; Smith, J.A.; Dehghan, A.; Dupuis, J.; Johnson, A.D.; et al. Pleiotropic genes for metabolic syndrome and inflammation. Mol. Genet. Metab. 2014, 112, 317–338. [Google Scholar] [CrossRef]
- Ahmad, F.; Goldstein, B.J. Increased abundance of specific skeletal muscle protein-tyrosine phosphatases in a genetic model of insulin-resistant obesity and diabetes mellitus. Metab. Clin. Exp. 1995, 44, 1175–1184. [Google Scholar] [CrossRef]
- Bonini, J.A.; Colca, J.R.; Dailey, C.; White, M.; Hofmann, C. Compensatory alterations for insulin signal transduction and glucose transport in insulin-resistant diabetes. Am. J. Physiol. 1995, 269, E759–E765. [Google Scholar] [CrossRef] [PubMed]
- Banno, R.; Zimmer, D.; De Jonghe, B.C.; Atienza, M.; Rak, K.; Yang, W.; Bence, K.K. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J. Clin. Investig. 2010, 120, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Hou, Y.; Cheng, R.; Zheng, L.; Alvarez, A.A.; Hu, B.; Cheng, S.-Y.; Zhang, W.; Li, Y.; Feng, H. Targeting PDGFRα-activated glioblastoma through specific inhibition of SHP-2-mediated signaling. Neuro-Oncology 2019, 21, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh Grewal, A.; Grover, R.; Sharma, N.; Chopra, B.; Kumar Dhingra, A.; Arora, S.; Redhu, S.; Lather, V. Recent updates on development of protein-tyrosine phosphatase 1B inhibitors for treatment of diabetes, obesity and related disorders. Bioorg. Chem. 2022, 121, 105626. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Targeted Organism/Tissue/Cell | Approach | Impact on Glucose Metabolism and Major Associated Phenotype | References |
|---|---|---|---|
| Cultured hepatocytes | SHP2 knockdown or inhibition | Improved insulin signaling | [59,63,64] |
| C. elegans | Ptp2−/− | Modulation of insulin signaling Increased lifespan | [53] |
| Drosophila | Csw−/− | Modulation of insulin signaling Increased lifespan | [55] |
| Mouse/ubiquitous | Ptpn11−/− Ptpn11+/− | Undetermined (embryonic lethality) No obvious phenotype | [66] |
| Mouse/ubiquitous | Ptpn11T468M/+ | Insulin hypersensitivity Improved glucose tolerance Resistance to HFD-induced insulin resistance | [61] |
| Mouse/ubiquitous | Ptpn11D61G/+ | Insulin resistance Glucose intolerance Inflammation | [74] |
| Mouse/transgenic (liver, muscle, adipose tissue) | SHP2∆PTP | Insulin resistance | [67] |
| HFD-fed mouse/systemic | SHP099 treatment | Improved glucose tolerance | [71,74] |
| Mouse/muscle specific | Ptpn11fl/fl × MHC-cre or MCK-cre | Insulin resistance Glucose intolerance Altered myofibers (number and size) Dilated cardiomyopathy | [46] |
| Mouse/neuron specific | Ptpn11fl/fl × CRE3 | Insulin resistance Diabetes Obesity Hyperphagia Leptin resistance Nephropathy | [41,68] |
| Mouse/transgenic (forebrain neuron, CamKII-driven) | SHP2D61A | Improved insulin sensitivity Improved glucose homeostasis | [25] |
| Mouse/POMC neuron specific | Ptpn11fl/fl × POMC-cre | Altered glucose metabolism Obesity Leptin resistance Reduced energy expenditure | [41,84] |
| Mouse/liver specific | Ptpn11fl/fl × Alb-cre | Improved insulin sensitivity Improved glucose tolerance Resistance to obesity Increased energy expenditure | [44,47] |
| Mouse/adipose tissue specific | Ptpn11fl/fl × Adipoq-cre | No phenotype | [69] |
| Ptpn11fl/fl × aP2-cre | Not assessed Severe lipodystrophy Altered adipogenesis Premature death | [43] | |
| Mouse/pancreas specific | Ptpn11fl/fl × Pdx1-cre | Glucose intolerance Reduced insulin secretion | [45] |
| Mouse/macrophage specific | Ptpn11fl/fl × Lyz2-cre | Resistance to HFD-induced insulin resistance | [71] |
| Human/ubiquitous | NS-Ptpn11 | Glucose intolerance Noonan syndrome | [72,73,74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saint-Laurent, C.; Mazeyrie, L.; Tajan, M.; Paccoud, R.; Castan-Laurell, I.; Valet, P.; Edouard, T.; Pradère, J.-P.; Dray, C.; Yart, A. The Tyrosine Phosphatase SHP2: A New Target for Insulin Resistance? Biomedicines 2022, 10, 2139. https://doi.org/10.3390/biomedicines10092139
Saint-Laurent C, Mazeyrie L, Tajan M, Paccoud R, Castan-Laurell I, Valet P, Edouard T, Pradère J-P, Dray C, Yart A. The Tyrosine Phosphatase SHP2: A New Target for Insulin Resistance? Biomedicines. 2022; 10(9):2139. https://doi.org/10.3390/biomedicines10092139
Chicago/Turabian StyleSaint-Laurent, Céline, Laurène Mazeyrie, Mylène Tajan, Romain Paccoud, Isabelle Castan-Laurell, Philippe Valet, Thomas Edouard, Jean-Philippe Pradère, Cédric Dray, and Armelle Yart. 2022. "The Tyrosine Phosphatase SHP2: A New Target for Insulin Resistance?" Biomedicines 10, no. 9: 2139. https://doi.org/10.3390/biomedicines10092139