
Membranolytic Mechanism of Amphiphilic Antimicrobial β-Stranded [KL]n Peptides

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis
2.3. Circular Dichroism Spectroscopy (CD)
2.3.1. Sample Preparation
2.3.2. Measurements
2.4. Oriented CD
2.5. MIC (Minimum Inhibitory Concentration) Assay
2.6. Hemolysis Assay
2.7. Vesicle Leakage Assay
2.8. Leakage of FITC-Dextrans
2.9. Solid-State NMR
NMR Data Analysis
3. Results
3.1. Peptide Synthesis
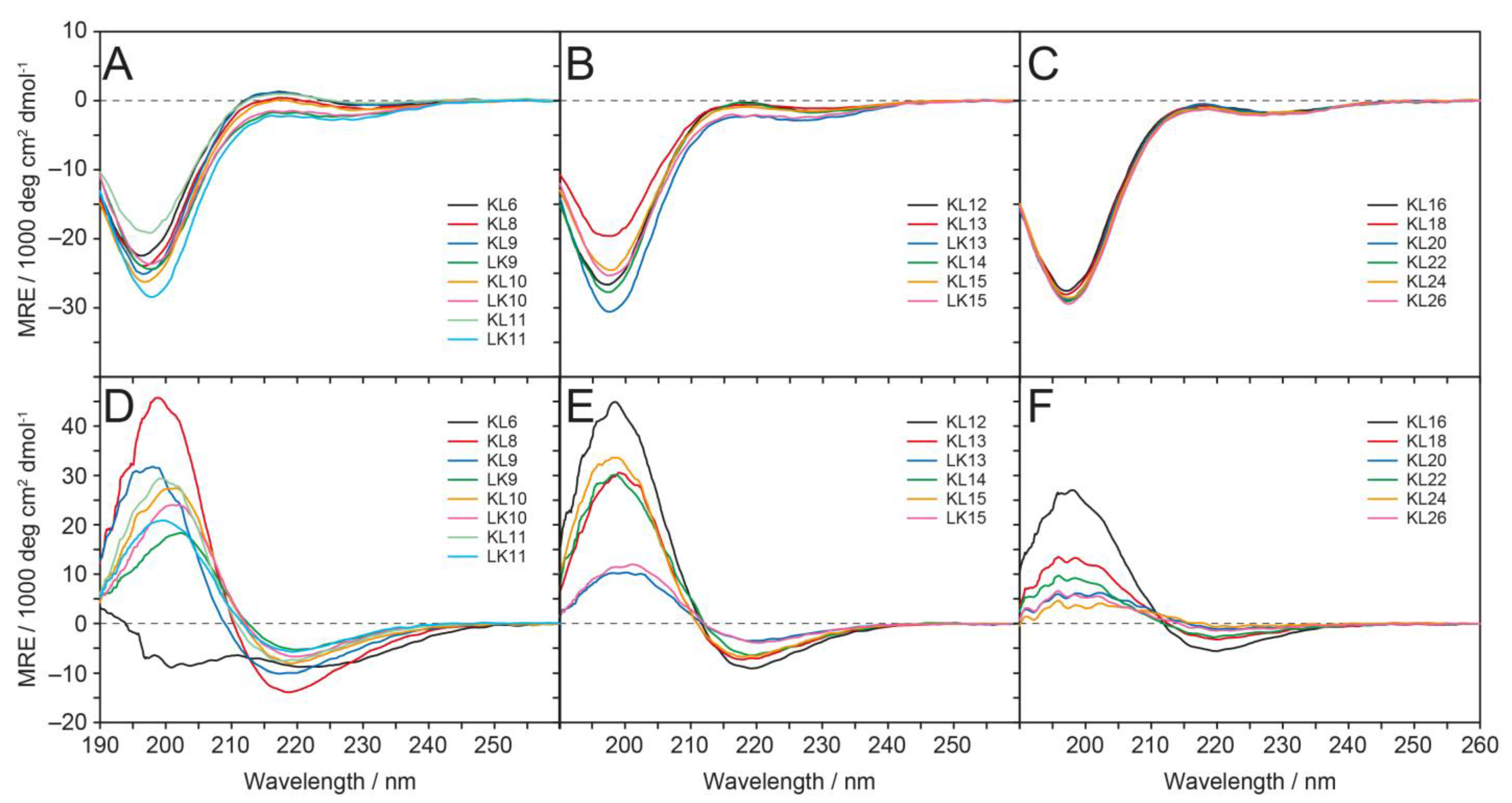
3.2. Circular Dichroism (CD)
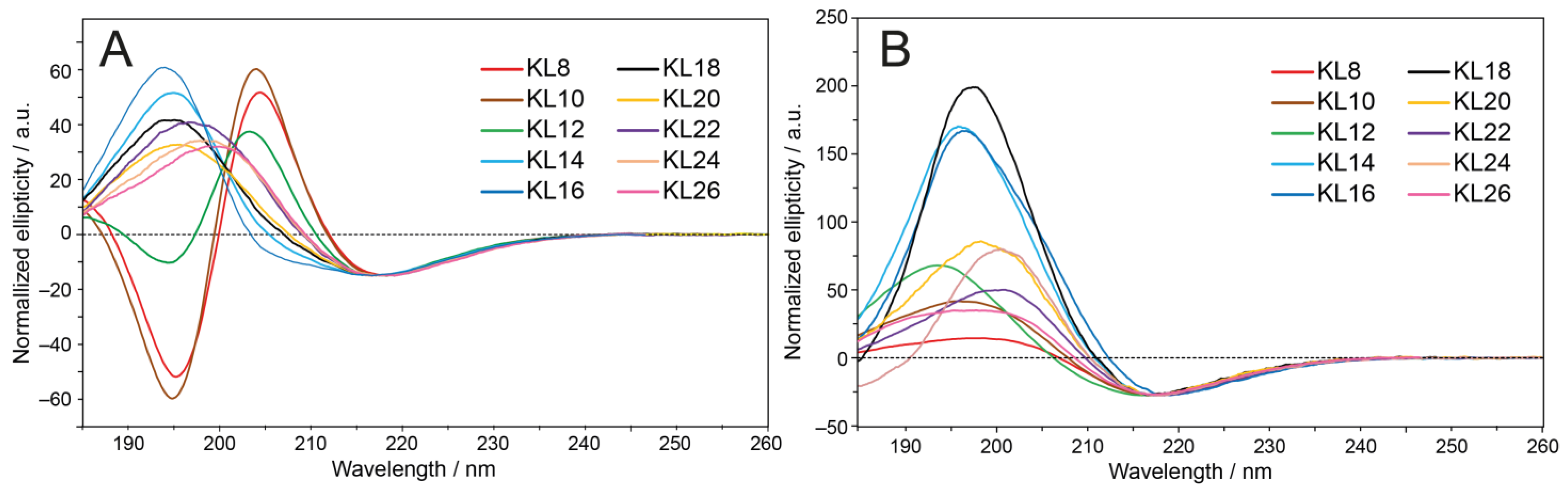
3.3. Oriented CD
3.4. Antimicrobial Activity
3.5. Hemolysis
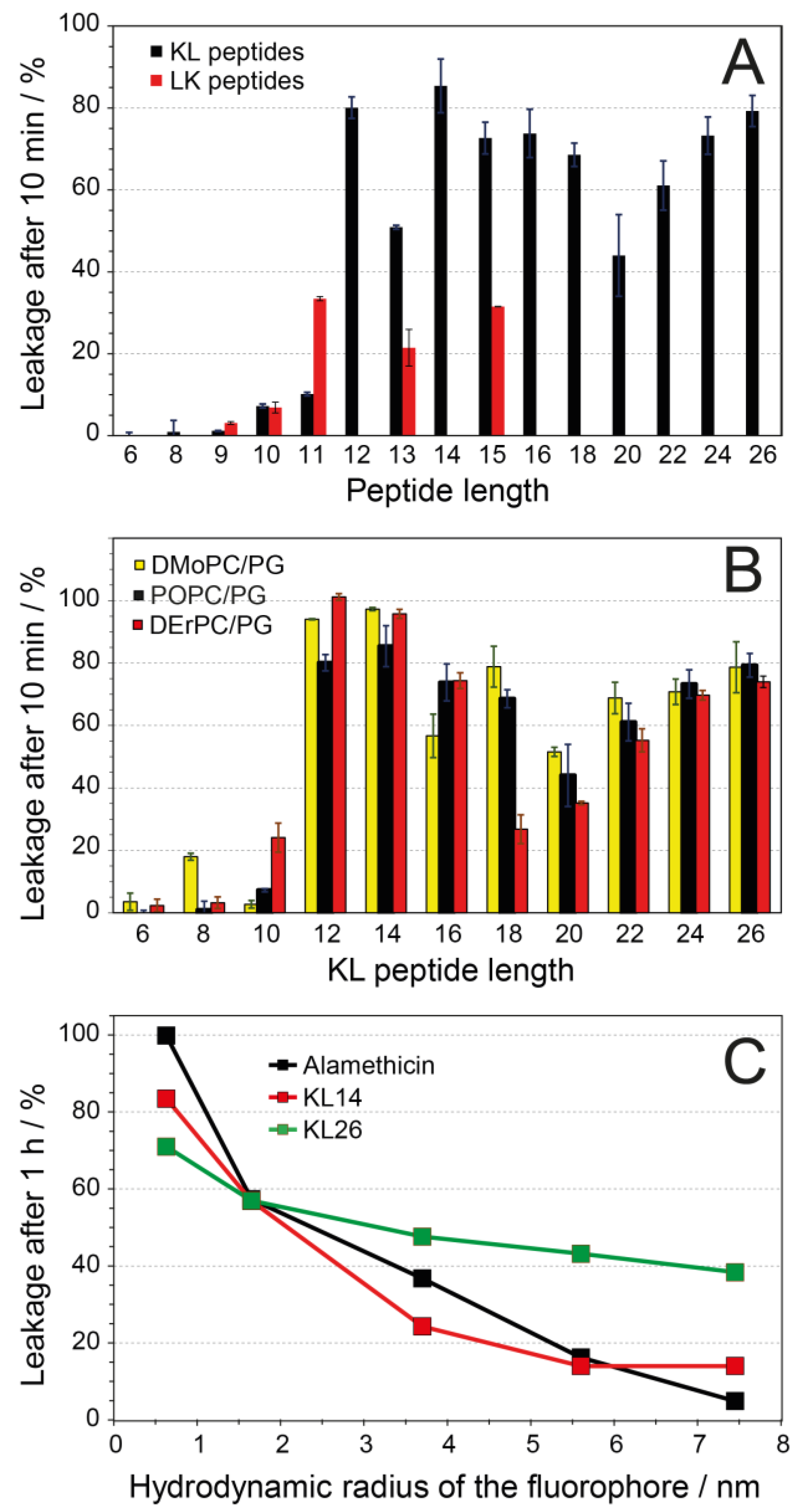
3.6. Vesicle Leakage
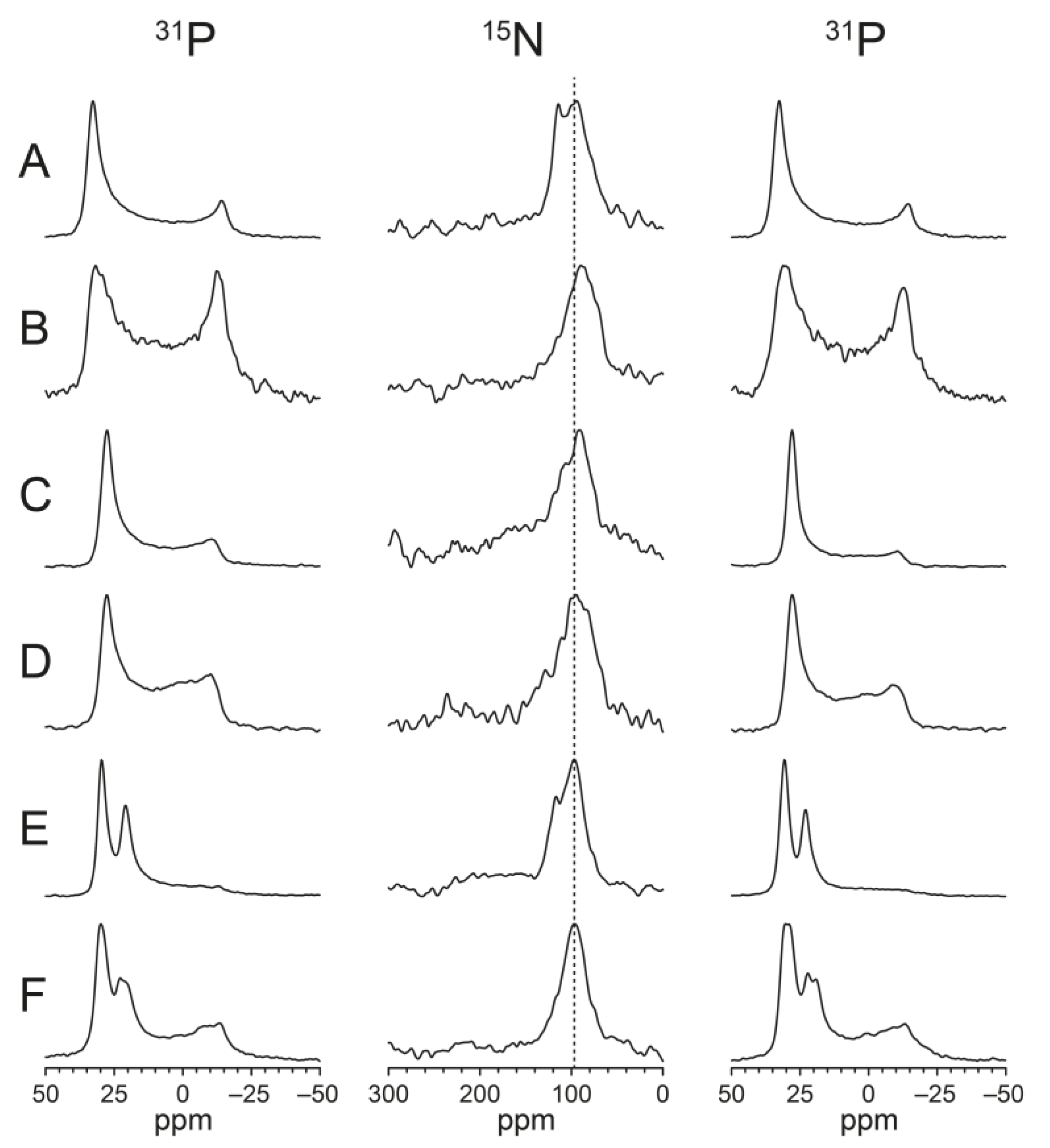
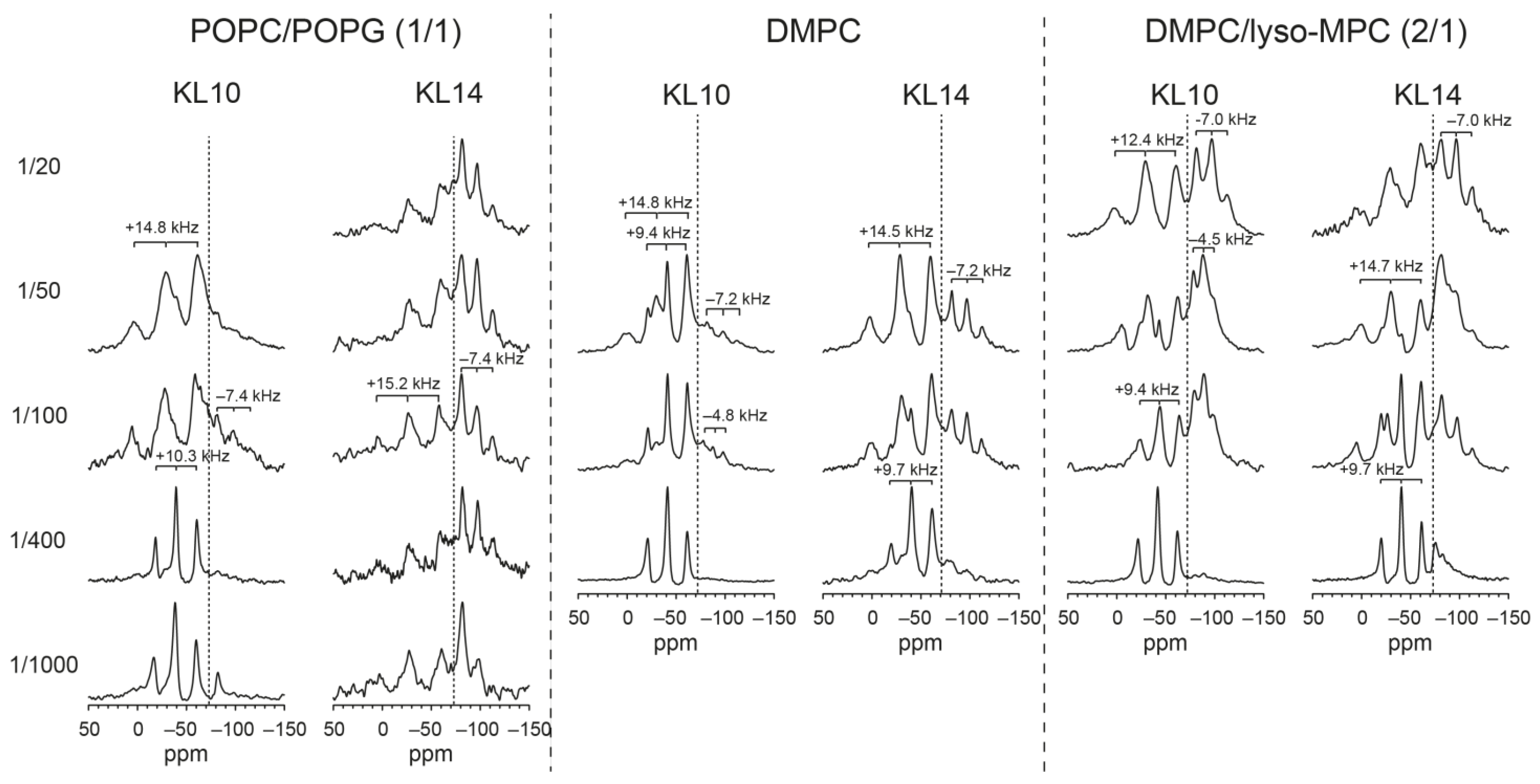
3.7. Solid-State NMR
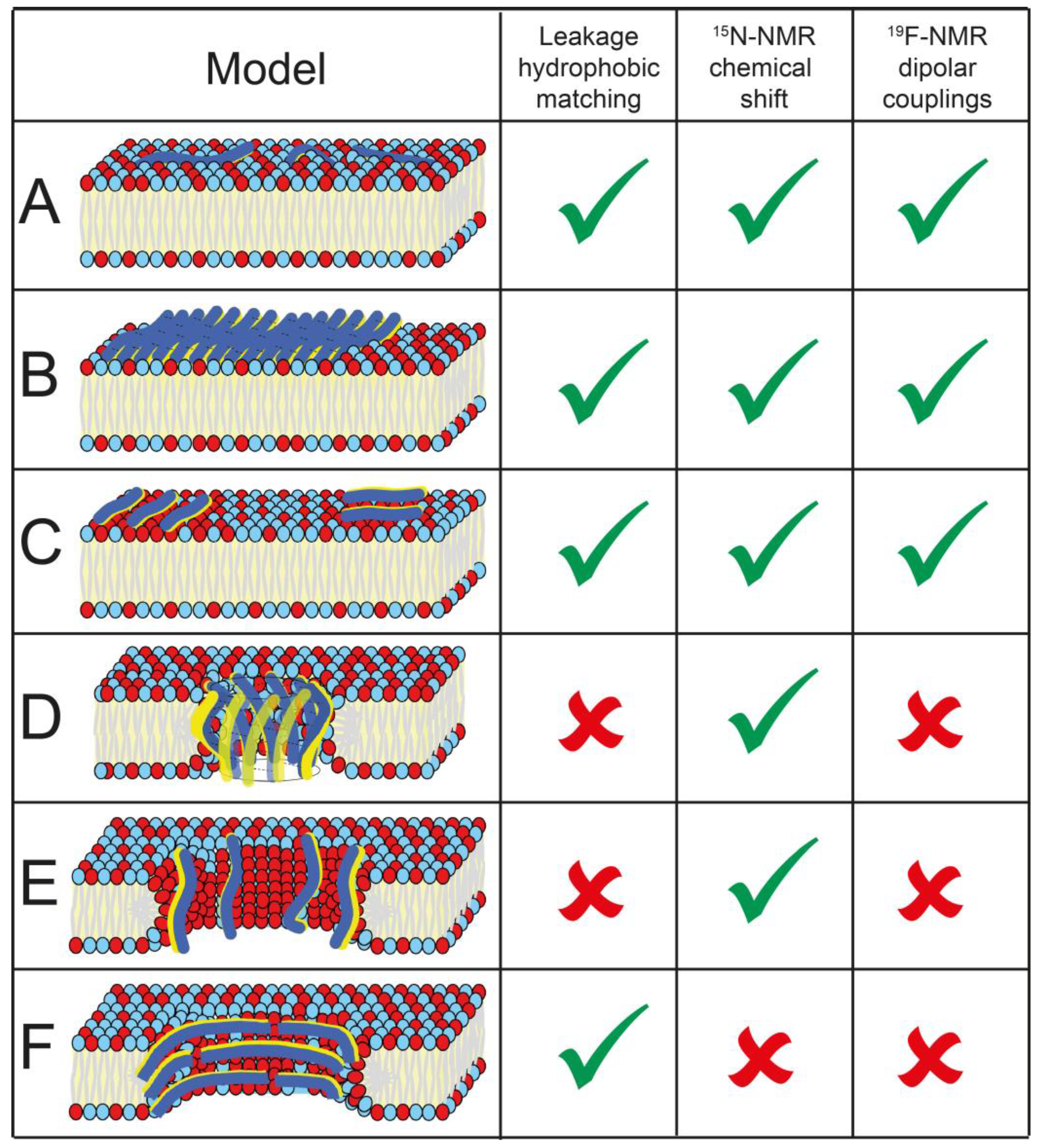
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinheiro da Silva, F.; Machado, M.C. Antimicrobial peptides: Clinical relevance and therapeutic implications. Peptides 2012, 36, 308–314. [Google Scholar] [CrossRef]
- Wimley, W.C.; Hristova, K. Antimicrobial peptides: Successes, challenges and unanswered questions. J. Membr. Biol. 2011, 239, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.A.; Murray, B.E. Antibiotic-resistant bugs in the 21st century—A clinical super-challenge. N. Engl. J. Med. 2009, 360, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Int. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef]
- Bürck, J.; Wadhwani, P.; Fanghänel, S.; Ulrich, A.S. Oriented circular dichroism: A method to characterize membrane-active peptides in oriented lipid bilayers. Acc. Chem. Res. 2016, 49, 184–192. [Google Scholar] [CrossRef]
- Strandberg, E.; Ulrich, A.S. NMR methods for studying membrane-active antimicrobial peptides. Concepts Magn. Reson. A 2004, 23A, 89–120. [Google Scholar] [CrossRef]
- Strandberg, E.; Ulrich, A.S. Solid-state 19F-NMR analysis of peptides in oriented biomembranes. In Modern Magnetic Resonance, 2nd ed.; Webb, G., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–18. [Google Scholar] [CrossRef]
- Strandberg, E.; Ulrich, A.S. Solid-state NMR for studying peptide structures and peptide-lipid interactions in membranes. In Modern Magnetic Resonance, 2nd ed.; Webb, G.A., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–13. [Google Scholar] [CrossRef]
- Wadhwani, P.; Strandberg, E. Structure analysis of membrane-active peptides using 19F-labeled amino acids and solid-state NMR. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; Blackwell Publishing: London, UK, 2009; pp. 463–493. [Google Scholar]
- Strandberg, E.; Ulrich, A.S. Flow charts for the systematic solid-state 19F/2H-NMR structure analysis of membrane-bound peptides. Annu. Rep. NMR Spectrosc. 2020, 99, 80–117. [Google Scholar] [CrossRef]
- Blazyk, J.; Wiegand, R.; Klein, J.; Hammer, J.; Epand, R.M.; Epand, R.F.; Maloy, W.L.; Kari, U.P. A novel linear amphipathic β-sheet cationic antimicrobial peptide with enhanced selectivity for bacterial lipids. J. Biol. Chem. 2001, 276, 27899–27906. [Google Scholar] [CrossRef]
- Wadhwani, P.; Reichert, J.; Strandberg, E.; Bürck, J.; Misiewicz, J.; Afonin, S.; Heidenreich, N.; Fanghänel, S.; Mykhailiuk, P.K.; Komarov, I.V.; et al. Stereochemical effects on the aggregation and biological properties of the fibril-forming peptide [KIGAKI]3 in membranes. Phys. Chem. Chem. Phys. 2013, 15, 8962–8971. [Google Scholar] [CrossRef]
- Wadhwani, P.; Strandberg, E.; Heidenreich, N.; Bürck, J.; Fanghänel, S.; Ulrich, A.S. Self-assembly of flexible β-strands into immobile amyloid-like β-sheets in membranes as revealed by solid-state 19F NMR. J. Am. Chem. Soc. 2012, 134, 6512–6515. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Schweigardt, F.; Wadhwani, P.; Bürck, J.; Reichert, J.; Cravo, H.L.P.; Burger, L.; Ulrich, A.S. Phosphate-dependent aggregation of [KL]n peptides affects their membranolytic activity. Sci. Rep. 2020, 10, 12300. [Google Scholar] [CrossRef]
- Grage, S.L.; Afonin, S.; Ieronimo, M.; Berditsch, M.; Wadhwani, P.; Ulrich, A.S. Probing and manipulating the lateral pressure profile in lipid bilayers using membrane-active peptides: A solid-state 19F NMR study. Int. J. Mol. Sci. 2022, 23, 4544. [Google Scholar] [CrossRef] [PubMed]
- Silvius, J.R.; Gagné, J. Calcium-induced fusion and lateral phase separations in phosphatidylcholine-phosphatidylserine vesicles–Correlation by calorimetric and fusion measurements. Biochemistry 1984, 23, 3241–3247. [Google Scholar] [CrossRef]
- Serra-Batiste, M.; Ninot-Pedrosa, M.; Bayoumi, M.; Gairi, M.; Maglia, G.; Carulla, N. Aβ42 assembles into specific β-barrel pore-forming oligomers in membrane-mimicking environments. Proc. Natl. Acad. Sci. USA 2016, 113, 10866–10871. [Google Scholar] [CrossRef]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Lal, R.; Nussinov, R. β-Barrel topology of Alzheimer’s β-amyloid ion channels. J. Mol. Biol. 2010, 404, 917–934. [Google Scholar] [CrossRef]
- Kandel, N.; Matos, J.O.; Tatulian, S.A. Structure of amyloid β(25-35) in lipid environment and cholesterol-dependent membrane pore formation. Sci. Rep. 2019, 9, 2689. [Google Scholar] [CrossRef]
- Mani, R.; Cady, S.D.; Tang, M.; Waring, A.J.; Lehrer, R.I.; Hong, M. Membrane-dependent oligomeric structure and pore formation of a β-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA 2006, 103, 16242–16247. [Google Scholar] [CrossRef] [Green Version]
- Ketchem, R.R.; Hu, W.; Cross, T.A. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science 1993, 261, 1457–1460. [Google Scholar] [CrossRef]
- Carpino, L.A.; Han, G.Y. 9-Fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 1972, 37, 3404–3409. [Google Scholar] [CrossRef]
- Afonin, S.; Mikhailiuk, P.K.; Komarov, I.V.; Ulrich, A.S. Evaluating the amino acid CF3-bicyclopentylglycine as a new label for solid-state 19F-NMR structure analysis of membrane-bound peptides. J. Pept. Sci. 2007, 13, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Ruden, S.; Hilpert, K.; Berditsch, M.; Wadhwani, P.; Ulrich, A.S. Synergistic interaction between silver nanoparticles and membrane-permeabilizing antimicrobial peptides. Antimicrob. Agents Chemother. 2009, 53, 3538–3540. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Tiltak, D.; Ieronimo, M.; Kanithasen, N.; Wadhwani, P.; Ulrich, A.S. Influence of C-terminal amidation on the antimicrobial and hemolytic activities of cationic α-helical peptides. Pure Appl. Chem. 2007, 79, 717–728. [Google Scholar] [CrossRef]
- Duzgunes, N.; Wilschut, J. Fusion assays monitoring intermixing of aqueous contents. Methods Enzymol. 1993, 220, 3–14. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Prock, S.; Reichert, J.; Wadhwani, P.; Zimpfer, B.; Bürck, J.; Berditsch, M.; Elstner, M.; Ulrich, A.S. Peptide-lipid interactions of the stress-response peptide TisB that induces bacterial persistence. Biophys. J. 2012, 103, 1460–1469. [Google Scholar] [CrossRef]
- Ellens, H.; Bentz, J.; Szoka, F.C. H+- and Ca2+-induced fusion and destabilization of liposomes. Biochemistry 1985, 24, 3099–3106. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; Selsted, M.E.; White, S.H. Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: Pore formation by melittin. Biophys. J. 1997, 72, 1762–1766. [Google Scholar] [CrossRef]
- Stutzin, A. A fluorescence assay for monitoring and analyzing fusion of biological membrane-vesicles in vitro. FEBS Lett. 1986, 197, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Grage, S.L.; Strandberg, E.; Wadhwani, P.; Esteban-Martin, S.; Salgado, J.; Ulrich, A.S. Comparative analysis of the orientation of transmembrane peptides using solid-state 2H- and 15N-NMR: Mobility matters. Eur. Biophys. J. 2012, 41, 475–482. [Google Scholar] [CrossRef]
- Müller, S.D.; De Angelis, A.A.; Walther, T.H.; Grage, S.L.; Lange, C.; Opella, S.J.; Ulrich, A.S. Structural characterization of the pore forming protein TatAd of the twin-arginine translocase in membranes by solid-state 15N-NMR. Biochim. Biophys. Acta 2007, 1768, 3071–3079. [Google Scholar] [CrossRef]
- Glaser, R.W.; Sachse, C.; Dürr, U.H.N.; Afonin, S.; Wadhwani, P.; Strandberg, E.; Ulrich, A.S. Concentration-dependent realignment of the antimicrobial peptide PGLa in lipid membranes observed by solid-state 19F-NMR. Biophys. J. 2005, 88, 3392–3397. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Zerweck, J.; Wadhwani, P.; Ulrich, A.S. Synergistic insertion of antimicrobial magainin-family peptides in membranes depends on the lipid spontaneous curvature. Biophys. J. 2013, 104, L9–L11. [Google Scholar] [CrossRef] [PubMed]
- Heinzmann, R.; Grage, S.L.; Schalck, C.; Bürck, J.; Banoczi, Z.; Toke, O.; Ulrich, A.S. A kinked antimicrobial peptide from Bombina maxima. II. Behavior in phospholipid bilayers. Eur. Biophys. J. 2011, 40, 463–470. [Google Scholar] [CrossRef]
- Walther, T.H.; Grage, S.L.; Roth, N.; Ulrich, A.S. Membrane alignment of the pore-forming component TatAd of the twin-arginine translocase from Bacillus subtilis resolved by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2010, 132, 15945–15956. [Google Scholar] [CrossRef] [PubMed]
- Cullis, P.R.; de Kruijff, B. Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim. Biophys. Acta 1979, 559, 399–420. [Google Scholar] [CrossRef]
- Wadhwani, P.; Tremouilhac, P.; Strandberg, E.; Afonin, S.; Grage, S.L.; Ieronimo, M.; Berditsch, M.; Ulrich, A.S. Using fluorinated amino acids for structure analysis of membrane-active peptides by solid-state 19F-NMR. In Current Fluoroorganic Chemistry: New Synthetic Directions, Technologies, Materials, and Biological Applications; Soloshonok, V.A., Mikami, K., Yamazaki, T., Welch, J.T., Honek, J.F., Eds.; ACS Symposium Series 949; American Chemical Society: Washington, DC, USA, 2007; pp. 431–446. [Google Scholar]
- Teng, Q.; Cross, T.A. The in situ determination of the 15N chemical-shift tensor orientation in a polypeptide. J. Magn. Reson. 1989, 85, 439–447. [Google Scholar] [CrossRef]
- Bechinger, B.; Zasloff, M.; Opella, S.J. Structure and orientation of the antibiotic peptide magainin in membranes by solid-state nuclear magnetic resonance spectroscopy. Protein Sci. 1993, 2, 2077–2084. [Google Scholar] [CrossRef]
- Glaser, R.W.; Sachse, C.; Dürr, U.H.N.; Wadhwani, P.; Ulrich, A.S. Orientation of the antimicrobial peptide PGLa in lipid membranes determined from 19F-NMR dipolar couplings of 4-CF3-phenylglycine labels. J. Magn. Reson. 2004, 168, 153–163. [Google Scholar] [CrossRef]
- Dürr, U.H.N.; Grage, S.L.; Witter, R.; Ulrich, A.S. Solid state 19F NMR parameters of fluorine-labeled amino acids. Part I: Aromatic substituents. J. Magn. Reson. 2008, 191, 7–15. [Google Scholar] [CrossRef]
- Grage, S.L.; Durr, U.H.; Afonin, S.; Mikhailiuk, P.K.; Komarov, I.V.; Ulrich, A.S. Solid state 19F NMR parameters of fluorine-labeled amino acids. Part II: Aliphatic substituents. J. Magn. Reson. 2008, 191, 16–23. [Google Scholar] [CrossRef]
- Miles, A.J.; Wallace, B.A. Circular dichroism spectroscopy of membrane proteins. Chem. Soc. Rev. 2016, 45, 4859–4872. [Google Scholar] [CrossRef] [PubMed]
- Bürck, J.; Roth, S.; Wadhwani, P.; Afonin, S.; Kanithasen, N.; Strandberg, E.; Ulrich, A.S. Conformation and membrane orientation of amphiphilic helical peptides by oriented circular dichroism. Biophys. J. 2008, 95, 3872–3881. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W.; Wu, Y. Lipid-alamethicin interactions influence alamethicin orientation. Biophys. J. 1991, 60, 1079–1087. [Google Scholar] [CrossRef]
- Bazzi, M.D.; Woody, R.W. Interaction of amphipathic polypeptides with phospholipids: Characterization of conformations and the CD of oriented β-sheets. Biopolymers 1987, 26, 1115–1124. [Google Scholar] [CrossRef]
- Woody, R.W. The circular dichroism of oriented β sheets: Theoretical predictions. Tetrahedron Asymmetry 1993, 4, 529–544. [Google Scholar] [CrossRef]
- Strandberg, E.; Bentz, D.; Wadhwani, P.; Ulrich, A.S. Chiral supramolecular architecture of stable transmembrane pores formed by an α-helical antibiotic peptide in the presence of lyso-lipids. Sci. Rep. 2020, 10, 4710. [Google Scholar] [CrossRef]
- Grau-Campistany, A.; Strandberg, E.; Wadhwani, P.; Rabanal, F.; Ulrich, A.S. Extending the hydrophobic mismatch concept to amphiphilic membranolytic peptides. J. Phys. Chem. Lett. 2016, 7, 1116–1120. [Google Scholar] [CrossRef]
- Strandberg, E.; Kanithasen, N.; Bürck, J.; Wadhwani, P.; Tiltak, D.; Zwernemann, O.; Ulrich, A.S. Solid state NMR analysis comparing the designer-made antibiotic MSI-103 with its parent peptide PGLa in lipid bilayers. Biochemistry 2008, 47, 2601–2616. [Google Scholar] [CrossRef]
- Dathe, M.; Wieprecht, T.; Nikolenko, H.; Handel, L.; Maloy, W.L.; MacDonald, D.L.; Beyermann, M.; Bienert, M. Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 1997, 403, 208–212. [Google Scholar] [CrossRef]
- Grau-Campistany, A.; Strandberg, E.; Wadhwani, P.; Reichert, J.; Bürck, J.; Rabanal, F.; Ulrich, A.S. Hydrophobic mismatch demonstrated for membranolytic peptides, and their use as molecular rulers to measure bilayer thickness in native cells. Sci. Rep. 2015, 5, 9388. [Google Scholar] [CrossRef]
- Epand, R.F.; Savage, P.B.; Epand, R.M. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim. Biophys. Acta 2007, 1768, 2500–2509. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D. Energetics of hydrophobic matching in lipid-protein interactions. Biophys. J. 2008, 94, 3996–4013. [Google Scholar] [CrossRef] [PubMed]
- Kucerka, N.; Tristram-Nagle, S.; Nagle, J.F. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 2006, 208, 193–202. [Google Scholar] [CrossRef]
- He, K.; Ludtke, S.J.; Worcester, D.L.; Huang, H.W. Neutron scattering in the plane of membranes: Structure of alamethicin pores. Biophys. J. 1996, 70, 2659–2666. [Google Scholar] [CrossRef]
- Qian, S.; Wang, W.C.; Yang, L.; Huang, H.W. Structure of the alamethicin pore reconstructed by x-ray diffraction analysis. Biophys. J. 2008, 94, 3512–3522. [Google Scholar] [CrossRef]
- Bohrer, M.P.; Deen, W.M.; Robertson, C.R.; Troy, J.L.; Brenner, B.M. Influence of molecular configuration on the passage of macromolecules across the glomerular capillary wall. J. Gen. Physiol. 1979, 74, 583–593. [Google Scholar] [CrossRef]
- Chen, Y.X.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef]
- Zamora-Carreras, H.; Strandberg, E.; Mühlhäuser, P.; Bürck, J.; Wadhwani, P.; Jiménez, M.Á.; Bruix, M.; Ulrich, A.S. Alanine scan and 2H NMR analysis of the membrane-active peptide BP100 point to a distinct carpet mechanism of action. Biochim. Biophys. Acta 2016, 1858, 1328–1338. [Google Scholar] [CrossRef]
- Strandberg, E.; Ulrich, A.S. AMPs and OMPs: Is the folding and bilayer insertion of β-stranded outer membrane proteins governed by the same biophysical principles as for α-helical antimicrobial peptides? Biochim. Biophys. Acta 2015, 1848, 1944–1954. [Google Scholar] [CrossRef]
- Strandberg, E.; Tiltak, D.; Ehni, S.; Wadhwani, P.; Ulrich, A.S. Lipid shape is a key factor for membrane interactions of amphipathic helical peptides. Biochim. Biophys. Acta 2012, 1818, 1764–1776. [Google Scholar] [CrossRef]
- Leitgeb, B.; Szekeres, A.; Manczinger, L.; Vagvolgyi, C.; Kredics, L. The history of alamethicin: A review of the most extensively studied peptaibol. Chem. Biodivers. 2007, 4, 1027–1051. [Google Scholar] [CrossRef]
- Matsuzaki, K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta 1998, 1376, 391–400. [Google Scholar] [CrossRef]
- Yang, L.; Weiss, T.M.; Lehrer, R.I.; Huang, H.W. Crystallization of antimicrobial pores in membranes: Magainin and protegrin. Biophys. J. 2000, 79, 2002–2009. [Google Scholar] [CrossRef]
- Ludtke, S.J.; He, K.; Heller, W.T.; Harroun, T.A.; Yang, L.; Huang, H.W. Membrane pores induced by magainin. Biochemistry 1996, 35, 13723–13728. [Google Scholar] [CrossRef]
- Strandberg, E.; Bentz, D.; Wadhwani, P.; Bürck, J.; Ulrich, A.S. Terminal charges modulate the pore forming activity of cationic amphipathic helices. BBA-Biomembranes 2020, 1862, 183243. [Google Scholar] [CrossRef]
- Gagnon, M.C.; Strandberg, E.; Grau-Campistany, A.; Wadhwani, P.; Reichert, J.; Bürck, J.; Rabanal, F.; Auger, M.; Paquin, J.F.; Ulrich, A.S. Influence of the length and charge on the activity of α-helical amphipathic antimicrobial peptides. Biochemistry 2017, 56, 1680–1695. [Google Scholar] [CrossRef]
- Pan, J.; Heberle, F.A.; Tristram-Nagle, S.; Szymanski, M.; Koepfinger, M.; Katsaras, J.; Kucerka, N. Molecular structures of fluid phase phosphatidylglycerol bilayers as determined by small angle neutron and X-ray scattering. Biochim. Biophys. Acta 2012, 1818, 2135–2148. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Gierasch, L.M.; Montal, M.; Zasloff, M.; Opella, S.J. Orientations of helical peptides in membrane bilayers by solid state NMR spectroscopy. Solid State Nucl. Magn. Reson. 1996, 7, 185–191. [Google Scholar] [CrossRef]
- Rance, M.; Byrd, R.A. Obtaining high-fidelity spin-1/2 powder spectra in anisotropic media–Phase-cycled Hahn echo spectroscopy. J. Magn. Reson. 1983, 52, 221–240. [Google Scholar] [CrossRef]
- Levitt, M.H.; Suter, D.; Ernst, R.R. Spin dynamics and thermodynamics in solid-state NMR cross polarization. J. Chem. Phys. 1986, 84, 4243–4255. [Google Scholar] [CrossRef]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wu, X.L.; Mehring, M. Elimination of ringing effects in multiple-pulse sequences. Chem. Phys. Lett. 1990, 173, 481–484. [Google Scholar] [CrossRef]
- Bennett, A.E.; Rienstra, C.M.; Auger, M.; Lakshmi, K.V.; Griffin, R.G. Heteronuclear decoupling in rotating solids. J. Chem. Phys. 1995, 103, 6951–6958. [Google Scholar] [CrossRef]
- Reichert, J.; Grasnick, D.; Afonin, S.; Bürck, J.; Wadhwani, P.; Ulrich, A.S. A critical evaluation of the conformational requirements of fusogenic peptides in membranes. Eur. Biophys. J. 2007, 36, 405–413. [Google Scholar] [CrossRef]
- Sigma-Aldrich. Fluorescein Isothiocyanate-Dextran. Available online: https://www.sigmaaldrich.com/technical-documents/protocols/biology/fluorescein-isothiocyanate-dextran.html#ref (accessed on 14 July 2022).
- Wooten, M.K.C.; Koganti, V.R.; Zhou, S.S.; Rankin, S.E.; Knutson, B.L. Synthesis and nanofiltration membrane performance of oriented mesoporous silica thin films on macroporous supports. ACS Appl. Mater. Interfaces 2016, 8, 21806–21815. [Google Scholar] [CrossRef]
- Mayer, L.D.; Hope, M.J.; Cullis, P.R. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta 1986, 858, 161–168. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Net Charge | Length a/Å |
|---|---|---|---|
| KL6 | KLKLKL-NH2 | +4 | 22.2 |
| KL8 | KLKLKLKL-NH2 | +5 | 29.6 |
| KL9 | KLKLKLKLK-NH2 | +6 | 33.3 |
| LK9 | LKLKLKLKL-NH2 | +5 | 33.3 |
| KL10 | KLKLKLKLKL-NH2 | +6 | 37.0 |
| LK10 | LKLKLKLKLK-NH2 | +6 | 37.0 |
| KL11 | KLKLKLKLKLK-NH2 | +7 | 40.7 |
| LK11 | LKLKLKLKLKL-NH2 | +6 | 40.7 |
| KL12 | KLKLKLKLKLKL-NH2 | +7 | 44.4 |
| KL13 | KLKLKLKLKLKLK-NH2 | +8 | 48.1 |
| LK13 | LKLKLKLKLKLKL-NH2 | +7 | 48.1 |
| KL14 | KLKLKLKLKLKLKL-NH2 | +8 | 51.8 |
| KL15 | KLKLKLKLKLKLKLK-NH2 | +9 | 55.5 |
| LK15 | LKLKLKLKLKLKLKL-NH2 | +8 | 55.5 |
| KL16 | KLKLKLKLKLKLKLKL-NH2 | +9 | 59.2 |
| KL18 | KLKLKLKLKLKLKLKLKL-NH2 | +10 | 66.6 |
| KL20 | KLKLKLKLKLKLKLKLKLKL-NH2 | +11 | 74.0 |
| KL22 | KLKLKLKLKLKLKLKLKLKLKL-NH2 | +12 | 81.4 |
| KL24 | KLKLKLKLKLKLKLKLKLKLKLKL-NH2 | +13 | 88.8 |
| KL26 | KLKLKLKLKLKLKLKLKLKLKLKLKL-NH2 | +14 | 96.2 |
| KL10-15N | KLKLK-(15N-Leu)-KLKL-NH2 | +6 | 37.0 |
| KL14-15N | KLKLK-(15N-Leu)-KLKLKLKL-NH2 | +8 | 51.8 |
| KL10-19F | KLKLK-(CF3-Bpg)-KLKL-NH2 | +6 | 37.0 |
| KL14-19F | KLKLK-(CF3-Bpg)-KLKLKLKL-NH2 | +8 | 51.8 |
| Peptide | Gram-Negative | Gram-Positive | Hemolysis a | ||
|---|---|---|---|---|---|
| E. coli | E. helveticus | B. subtilis | S. xylosus | HC50 | |
| KL6 | >256 | 128 | >256 | 256 | >256 |
| KL8 | 64 | 16 | 16 | 8 | >256 |
| KL9 | 32 | 8 | 16 | 2 | >256 |
| LK9 | 16 | 4 | 4 | 2 | 145 |
| KL10 b | 4 | 2 | 2 | 2 | 47 |
| LK10 b | 8 | 2 | 4 | 1 | 256 |
| KL11 b | 4 | 2 | 4 | 1 | >256 |
| LK11 | 2 | 1 | 2 | 1 | 3 |
| KL12 | 2 | 2 | 2 | 2 | 8 |
| KL13 | 2 | 1 | 2 | 1 | 9 |
| LK13 | 8 | 4 | 8 | 4 | 2 |
| KL14 | 8 | 4 | 4 | 4 | 1 |
| KL15 | 8 | 2 | 4 | 2 | 2 |
| LK15 | 64 | 16 | 16 | 16 | <1 |
| KL16 | 32 | 8 | 16 | 8 | 2 |
| KL18 | 32 | 16 | 32 | 8 | 1 |
| KL20 | 32 | 32 | 32 | 16 | <1 |
| KL22 | 32 | 32 | 32 | 16 | <1 |
| KL24 | 32 | 16 | 32 | 8 | <1 |
| KL26 | 64 | 16 | 32 | 16 | <1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schweigardt, F.; Strandberg, E.; Wadhwani, P.; Reichert, J.; Bürck, J.; Cravo, H.L.P.; Burger, L.; Ulrich, A.S. Membranolytic Mechanism of Amphiphilic Antimicrobial β-Stranded [KL]n Peptides. Biomedicines 2022, 10, 2071. https://doi.org/10.3390/biomedicines10092071
Schweigardt F, Strandberg E, Wadhwani P, Reichert J, Bürck J, Cravo HLP, Burger L, Ulrich AS. Membranolytic Mechanism of Amphiphilic Antimicrobial β-Stranded [KL]n Peptides. Biomedicines. 2022; 10(9):2071. https://doi.org/10.3390/biomedicines10092071
Chicago/Turabian StyleSchweigardt, Fabian, Erik Strandberg, Parvesh Wadhwani, Johannes Reichert, Jochen Bürck, Haroldo L. P. Cravo, Luisa Burger, and Anne S. Ulrich. 2022. "Membranolytic Mechanism of Amphiphilic Antimicrobial β-Stranded [KL]n Peptides" Biomedicines 10, no. 9: 2071. https://doi.org/10.3390/biomedicines10092071