
Identification of Key Genes and Pathways in Genotoxic Stress Induced Endothelial Dysfunction: Results of Whole Transcriptome Sequencing

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Whole Transcriptome Sequencing (RNA-Seq)
2.3. Bioinformatical Analysis
2.4. Identification of Differentially Expressed Genes
2.5. Gene Ontology Enrichment Analysis
3. Results
3.1. Identification of DEGs Involved in The Genotoxic Stress Induced Endothelial Disfunction
3.2. Results of GO Enrichment Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Sima, A.V.; Stancu, C.S.; Simionescu, M. Vascular endothelium in atherosclerosis. Cell Tissue Res. 2009, 35, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Bertani, F.; Di Francesco, D.; Corrado, M.D.; Talmon, M.; Fresu, L.G.; Boccafoschi, F. Paracrine Shear-Stress-Dependent Signaling from Endothelial Cells Affects Downstream Endothelial Function and Inflammation. Int. J. Mol. Sci. 2021, 22, 13300. [Google Scholar] [CrossRef]
- Douglas, G.; Channon, K.M. The pathogenesis of atherosclerosis. Medicine 2014, 42, 480–484. [Google Scholar] [CrossRef]
- Mohanan, G.; Das, A.; Rajyaguru, P.I. Genotoxic stress response: What is the role of cytoplasmic mRNA fate? Bioessays 2021, 43, e2000311. [Google Scholar] [CrossRef]
- Sinitsky, M.Y.; Kutikhin, A.G.; Tsepokina, A.V.; Shishkova, D.K.; Asanov, M.A.; Yuzhalin, A.E.; Minina, V.I.; Ponasenko, A.V. Mitomycin C induced genotoxic stress in endothelial cells is associated with differential expression of proinflammatory cytokines. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2020, 858–860, 503252. [Google Scholar] [CrossRef]
- Sinitsky, M.Y.; Tsepokina, A.V.; Kutikhin, A.G.; Shishkova, D.K.; Ponasenko, A.V. The gene expression profile in endothelial cells exposed to mitomycin C. Biochem. Suppl. Ser. B Biomed. Chem. 2021, 15, 255–261. [Google Scholar] [CrossRef]
- Kutikhin, A.G.; Sinitsky, M.Y.; Ponasenko, A.V. The role of mutagenesis in atherosclerosis. Complex Issues Cardiovasc. Dis. 2017, 1, 92–101. [Google Scholar] [CrossRef]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, R.; Harris, M.A.; Huntley, R.; Van Auken, K.; Cherry, J.M. A guide to best practices for Gene Ontology (GO) manual annotation. Database 2013, 2013, bat054. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Shishkova, D.K.; Velikanova, E.A.; Sinitsky, M.Y.; Sinitskaya, A.V.; Markova, V.E. Endothelial Dysfunction in the Context of Blood-Brain Barrier Modeling. J. Evol. Biochem. Physiol. 2022, 58, 781–806. [Google Scholar] [CrossRef] [PubMed]
- Aboyans, V.; Lacroix, P.; Criqui, M.H. Large and small vessels atherosclerosis: Similarities and differences. Prog. Cardiovasc. Dis. 2007, 50, 112–125. [Google Scholar] [CrossRef]
- Dessy, C.; Ferron, O. Pathophysiological roles of nitric oxide: In the heart and the coronary vasculature. Antiinflamm. Antiallergy Agents Med. Chem. 2004, 3, 207–216. [Google Scholar] [CrossRef]
- Schmidt, T.S.; McNeill, E.; Douglas, G.; Crabtree, M.J.; Hale, A.B.; Khoo, L.; O’Neill, C.A.; Cheng, A.; Channon, K.M.; Alp, N.J. Tetrahydrobiopterin supplementation reduces atherosclerosis and vascular inflammation in apolipoprotein E-knockout mice. Clin. Sci. 2010, 119, 131–142. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, M.; Xu, H.; Yu, J. Tetrahydrobiopterin improves endothelial function in cardiovascular disease: A systematic review. Evid. Complement. Alternat. Med. 2014, 2014, 850312. [Google Scholar] [CrossRef]
- Douglas, G.; Hale, A.B.; Patel, J.; Chuaiphichai, S.; Zen, A.A.H.; Rashbrook, V.S.; Trelfa, L.; Crabtree, M.J.; McNeill, E.; Channon, K.M. Roles for endothelial cell and macrophage Gch1 and tetrahydrobiopterin in atherosclerosis progression. Cardiovasc. Res. 2018, 114, 1385–1399. [Google Scholar] [CrossRef]
- Münzel, T.; Daiber, A. Role of endothelial and macrophage tetrahydrobiopterin in development and progression of atherosclerosis: BH4 puzzle solved? Cardiovasc. Res. 2018, 114, 1310–1312. [Google Scholar] [CrossRef]
- Sims, F.H. A comparison of coronary and internal mammary arteries and implications of the results in the etiology of atherosclerosis. Am. Heart J. 1983, 105, 560–566. [Google Scholar]
- Oliner, J.D.; Saiki, A.Y.; Caenepeel, S. The Role of MDM2 Amplification and Overexpression in Tumorigenesis. Cold Spring Harb. Perspect. Med. 2016, 6, a026336. [Google Scholar] [CrossRef] [PubMed]
- Ihling, C.; Menzel, G.; Wellens, E.; Monting, J.S.; Schaefer, H.E.; Zeiher, A.M. Topographical association between the cyclin-dependent kinases inhibitor P21, p53 accumulation, and cellular proliferation in human atherosclerotic tissue. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Guo, Q.; Gao, H.; Xu, R.; Teng, S.; Wu, Y. Metformin and resveratrol inhibited high glucose-induced metabolic memory of endothelial senescence through SIRT1/p300/p53/p21 pathway. PLoS ONE 2015, 10, e0143814. [Google Scholar]
- Yokoyama, M.; Shimizu, I.; Nagasawa, A.; Yoshida, Y.; Katsuumi, G.; Wakasugi, T.; Hayashi, Y.; Ikegami, R.; Suda, M.; Ota, Y.; et al. p53 plays a crucial role in endothelial dysfunction associated with hyperglycemia and ischemia. J. Mol. Cell. Cardiol. 2019, 129, 105–117. [Google Scholar] [CrossRef]
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Vadivel Gnanasundram, S.; Bonczek, O.; Wang, L.; Chen, S.; Fahraeus, R. p53 mRNA Metabolism Links with the DNA Damage Response. Genes 2021, 12, 1446. [Google Scholar] [CrossRef] [PubMed]
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 2017, 24, 63. [Google Scholar] [CrossRef]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef]
- Lee, J.H.; Kang, Y.; Khare, V.; Jin, Z.Y.; Kang, M.Y.; Yoon, Y.; Hyun, J.W.; Chung, M.H.; Cho, S.I.; Jun, J.Y.; et al. The p53-inducible gene 3 (PIG3) contributes to early cellular response to DNA damage. Oncogene 2010, 29, 1431–1450. [Google Scholar] [CrossRef]
- Li, B.; Shang, Z.F.; Yin, J.J.; Xu, Q.Z.; Liu, X.D.; Wang, Y.; Zhang, S.M.; Guan, H.; Zhou, P.K. PIG3 functions in DNA damage response through regulating DNA-PKcs homeostasis. Int. J. Biol. Sci. 2013, 9, 425–434. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, Y.; Lu, W.; Chen, X. The G protein-coupled receptor 87 is necessary for p53-dependent cell survival in response to genotoxic stress. Cancer Res. 2009, 69, 6049–6056. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Benchimol Ma, W.; Pidd, S. A new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat. Genet. 2000, 26, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Tinel, A.; Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004, 304, 843–846. [Google Scholar] [CrossRef]
- Janssens, S.; Tinel, A.; Lippens, S.; Tschopp, J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell 2005, 123, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ohki, R. p53-PHLDA3-Akt Network: The Key Regulators of Neuroendocrine Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 4098. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhang, L.; Han, B.; Zhang, Z. PHLDA3 inhibition protects against myocardial ischemia/reperfusion injury by alleviating oxidative stress and inflammatory response via the Akt/Nrf2 axis. Environ. Toxicol. 2021, 36, 2266–2277. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef]
- Carvajal, L.A.; Hamard, P.J.; Tonnessen, C.; Manfredi, J.J. E2F7, a novel target, is up-regulated by p53 and mediates DNA damage-dependent transcriptional repression. Genes Dev. 2012, 26, 1533–1545. [Google Scholar] [CrossRef]
- Weijts, B.G.; Bakker, W.J.; Cornelissen, P.W.; Liang, K.H.; Schaftenaar, F.H.; Westendorp, B.; de Wolf, C.A.; Paciejewska, M.; Scheele, C.L.; Kent, L.; et al. E2F7 and E2F8 promote angiogenesis through transcriptional activation of VEGFA in cooperation with HIF1. EMBO J. 2012, 31, 3871–3884. [Google Scholar] [CrossRef]
- Kotoula, V.; Krikelis, D.; Karavasilis, V.; Koletsa, T.; Eleftheraki, A.G.; Televantou, D.; Christodoulou, C.; Dimoudis, S.; Korantzis, I.; Pectasides, D.; et al. Expression of DNA repair and replication genes in non-small cell lung cancer (NSCLC): A role for thymidylate synthetase (TYMS). BMC Cancer 2012, 12, 342. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Sinha, S.; Kundu, C.N. Nectin cell adhesion molecule-4 (NECTIN-4): A potential target for cancer therapy. Eur. J. Pharmacol. 2021, 911, 174516. [Google Scholar] [CrossRef] [PubMed]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab vedotin antibody–drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Canc. Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Ikeda, W.; Ogita, H.; Rikitake, Y. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu. Rev. Cell Dev. Biol. 2008, 24, 309–342. [Google Scholar] [CrossRef]
- Takai, Y.; Miyoshi, J.; Ikeda, W.; Ogita, H. Nectins and nectin-like molecules: Roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 2008, 9, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Rikitake, Y.; Mandai, K.; Takai, Y. The role of nectins in different types of cell–cell adhesion. J. Cell Sci. 2012, 125, 3713–3722. [Google Scholar] [CrossRef]
- Nishiwada, S.; Sho, M.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Nectin-4 expression contributes to tumor proliferation, angiogenesis and patient prognosis in human pancreatic cancer. J. Exp. Clin. Canc. Res. 2015, 34, 1–9. [Google Scholar] [CrossRef]
- Wang, D.; Day, E.A.; Townsend, L.K.; Djordjevic, D.; Jørgensen, S.B.; Steinberg, G.R. GDF15: Emerging biology and therapeutic applications for obesity and cardiometabolic disease. Nat. Rev. Endocrinol. 2021, 17, 592–607. [Google Scholar] [CrossRef]
- Hsu, L.-A.; Wu, S.; Juang, J.-M.J.; Chiang, F.-T.; Teng, M.-S.; Lin, J.-F.; Huang, H.L.; Ko, Y.L. Growth Differentiation Factor 15 May Predict Mortality of Peripheral and Coronary Artery Diseases and Correlate with Their Risk Factors. Mediat. Inflamm. 2017, 2017, 9398401. [Google Scholar] [CrossRef]
- De Haan, J.J.; Haitjema, S.; den Ruijter, H.M.; Pasterkamp, G.; de Borst, G.J.; Teraa, M.; Verhaar, M.C.; Gremmels, H.; de Jager, S. Growth Differentiation Factor 15 Is Associated with Major Amputation and Mortality in Patients with Peripheral Artery Disease. J. Am. Heart Assoc. 2017, 6, e006225. [Google Scholar] [CrossRef]
- Johnen, H.; Kuffner, T.; Brown, D.A.; Wu, B.J.; Stocker, R.; Breit, S.N. Increased expression of the TGF-b superfamily cytokine MIC-1/GDF15 protects ApoE−/− mice from the development of atherosclerosis. Cardiovasc. Pathol. 2012, 21, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Ha, G.; De Torres, F.; Arouche, N.; Benzoubir, N.; Ferratge, S.; Hatem, E.; Anginot, A.; Uzan, G. GDF15 secreted by senescent endothelial cells improves vascular progenitor cell functions. PLoS ONE 2019, 14, e0216602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar]
- Hasanbasic, I.; Cuerquis, J.; Varnum, B.; Blostein, M.D. Intracellular signaling pathways involved in Gas6-Axl-mediated survival of endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1207-13. [Google Scholar] [CrossRef] [PubMed]
- Hai, T.; Wolfgang, C.D.; Marsee, D.K.; Allen, A.E.; Sivaprasad, U. ATF3 and stress responses. Gene Expr. 1999, 7, 321–335. [Google Scholar] [PubMed]
- Amundson, S.A.; Bittner, M.; Chen, Y.; Trent, J.; Meltzer, P.; Fornace, A.J., Jr. Fluorescent cDNA microarray hybridization reveals complexity and heterogeneity of cellular genotoxic stress responses. Oncogene 1999, 18, 3666–3672. [Google Scholar] [CrossRef]
- Liang, G.; Wolfgang, C.D.; Chen, B.P.C.; Chen, T.H.; Hai, T. ATF3 gene: Genome organization, promoter and regulation. J. Biol. Chem. 1996, 271, 1695–1701. [Google Scholar] [CrossRef]
- Zhang, M.; Zhai, X.; Li, J.; Albers, J.J.; Vuletic, S.; Ren, G. Structural basis of the lipid transfer mechanism of phospholipid transfer protein (PLTP). Biochim. Biophys. Acta Mol. Cell. Biol. Lipids. 2018, 1863, 1082–1094. [Google Scholar] [CrossRef]
- Jiang, X.C.; Yu, Y. The Role of Phospholipid Transfer Protein in the Development of Atherosclerosis. Curr. Atheroscler. Rep. 2021, 23, 9. [Google Scholar] [CrossRef]
- Zhang, K.; Zheng, J.; Chen, Y.; Dong, J.; Li, Z.; Chiang, Y.P.; He, M.; Huang, Q.; Tang, H.; Jiang, X.C. Inducible phospholipid transfer protein deficiency ameliorates atherosclerosis. Atherosclerosis 2021, 324, 9–17. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Barone, G.; Staples, C.J.; Ganesh, A.; Patterson, K.W.; Bryne, D.P.; Myers, K.N.; Patil, A.A.; Eyers, C.E.; Maslen, S.; Skehel, J.M.; et al. Human CDK18 promotes replication stress signaling and genome stability. Nucleic Acids Res. 2016, 44, 8772–8785. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Ng, H.P.; Kim, G.D.; Chan, E.R.; Mahabeleshwar, G.H. BHLHE40 promotes macrophage pro-inflammatory gene expression and functions. FASEB J. 2021, 35, e21940. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.E.; Jarjour, N.N.; Lin, C.C.; Edelson, B.T. Transcription Factor Bhlhe40 in Immunity and Autoimmunity. Trends Immunol. 2020, 41, 1023–1036. [Google Scholar] [CrossRef]
- Huynh, J.P.; Lin, C.C.; Kimmey, J.M.; Jarjour, N.N.; Schwarzkopf, E.A.; Bradstreet, T.R.; Shchukina, I.; Shpynov, O.; Weaver, C.T.; Taneja, R.; et al. Bhlhe40 is an essential repressor of IL-10 during Mycobacterium tuberculosis infection. J. Exp. Med. 2018, 215, 1823–1838. [Google Scholar] [CrossRef]
- Teng, Y.S.; Zhao, Y.L.; Li, M.S.; Liu, Y.G.; Cheng, P.; Lv, Y.P.; Mao, F.Y.; Chen, W.; Yang, S.M.; Hao, C.J.; et al. Upexpression of BHLHE40 in gastric epithelial cells increases CXCL12 production through interaction with p-STAT3 in Helicobacter pylori-associated gastritis. FASEB J. 2020, 34, 1169–1181. [Google Scholar] [CrossRef]
- Lukosz, M.; Mlynek, A.; Czypiorski, P.; Altschmied, J.; Haendeler, J. The transcription factor Grainyhead like 3 (GRHL3) affects endothelial cell apoptosis and migration in a NO-dependent manner. Biochem. Biophys. Res. Commun. 2011, 412, 648–653. [Google Scholar] [CrossRef]
- Zhou, X.; Michal, J.J.; Zhang, L.; Ding, B.; Lunney, J.K.; Liu, B.; Jiang, Z. Interferon induced IFIT family genes in host antiviral defense. Int. J. Biol. Sci. 2013, 9, 200–208. [Google Scholar] [CrossRef]
- Imaizumi, T.; Hashimoto, S.; Sato, R.; Umetsu, H.; Aizawa, T.; Watanabe, S.; Kawaguchi, S.; Matsumiya, T.; Seya, K.; Ding, J.; et al. IFIT Proteins Are Involved in CXCL10 Expression in Human Glomerular Endothelial Cells Treated with a Toll-Like Receptor 3 Agonist. Kidney Blood Press. Res. 2021, 46, 74–83. [Google Scholar] [CrossRef]
- Yang, Z.; Liang, H.; Zhou, Q.; Li, Y.; Chen, H.; Ye, W.; Chen, D.; Fleming, J.; Shu, H.; Liu, Y. Crystal structure of ISG54 reveals a novel RNA binding structure and potential functional mechanisms. Cell Res. 2012, 22, 1328–1338. [Google Scholar] [CrossRef]
- Imamichi, Y.; Mizutani, T.; Ju, Y.; Matsumura, T.; Kawabe, S.; Kanno, M.; Yazawa, T.; Miyamoto, K. Transcriptional regulation of human ferredoxin reductase through an intronic enhancer in steroidogenic cells. Biochim. Biophys. Acta. 2014, 1839, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Chen, X. The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene 2002, 21, 7195–7204. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.; Kinzler, K.W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothkamm, K.; Beinke, C.; Romm, H.; Badie, C.; Balagurunathan, Y.; Barnard, S.; Bernard, N.; Boulay-Greene, H.; Brengues, M.; De Amicis, A.; et al. Comparison of established and emerging biodosimetry assays. Radiat. Res. 2013, 180, 111–119. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, G.; Cruz-Garcia, L.; Majewski, M.; Grepl, J.; Abend, M.; Port, M.; Tichý, A.; Sirak, I.; Malkova, A.; Donovan, E.; et al. FDXR is a biomarker of radiation exposure in vivo. Sci. Rep. 2018, 8, 684. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium-Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Eng, E.T.; Zhu, J.; Lu, C.; Walz, T.; Springer, T.A. Sequence and structure relationships within von Willebrand factor. Blood 2012, 120, 449–458. [Google Scholar] [CrossRef]
- Nie, L.; Guo, X.; Esmailzadeh, L.; Zhang, J.; Asadi, A.; Collinge, M.; Li, X.; Kim, J.D.; Woolls, M.; Jin, S.W.; et al. Transmembrane protein ESDN promotes endothelial VEGF signaling and regulates angiogenesis. J. Clin. Investig. 2013, 123, 5082–5097. [Google Scholar] [CrossRef]
- Li, B.; Lin, Z.; Liang, Q.; Hu, Y.; Xu, W.F. PAQR6 Expression Enhancement Suggests a Worse Prognosis in Prostate Cancer Patients. Open Life Sci. 2018, 13, 511–517. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal. Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Naderi, A. Molecular functions of brain expressed X-linked 2 (BEX2) in malignancies. Exp. Cell Res. 2019, 376, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, X.; Li, J.; Lu, Y.; Zhao, S.; Tang, X.; Chen, X.; Li, J.; Zheng, Y.; Li, S.; et al. APOBEC3B interaction with PRC2 modulates microenvironment to promote HCC progression. Gut 2019, 68, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- McMahan, R.S.; Birkland, T.P.; Smigiel, K.S.; Vandivort, T.C.; Rohani, M.G.; Manicone, A.M.; McGuire, J.K.; Gharib, S.A.; Parks, W.C. Stromelysin-2 (MMP10) Moderates Inflammation by Controlling Macrophage Activation. J. Immunol. 2016, 197, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.W.; Shi, H.Y.; Liu, S.; Feng, S.X.; Feng, S.Q.; Gong, B.Q. miR-496/MMP10 Is Involved in the Proliferation of IL-1β-Induced Fibroblast-Like Synoviocytes via Mediating the NF-κB Signaling Pathway. Inflammation 2021, 44, 1359–1369. [Google Scholar] [CrossRef]
- Ma, Q.; Lu, Y.; Gu, Y. ENKUR Is Involved in the Regulation of Cellular Biology in Colorectal Cancer Cells via PI3K/Akt Signaling Pathway. Technol. Cancer Res. Treat. 2019, 18, 1533033819841433. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, J.; Du, J.; Jiang, X.; Xu, X.; Liu, Y.; He, Q.; Liang, H.; Fang, P.; Zhan, H.; et al. HMGCS1 drives drug-resistance in acute myeloid leukemia through endoplasmic reticulum-UPR-mitochondria axis. Biomed. Pharmacother. 2021, 137, 111378. [Google Scholar] [CrossRef]
- Chen, B.; Li, P.; Li, J.; Chen, J. Putative genes and pathways involved in the acne treatment of isotretinoin via microarray data analyses. Biomed. Res. Int. 2020, 2020, 5842795. [Google Scholar] [CrossRef]
- Wang, I.H.; Huang, T.T.; Chen, J.L.; Chu, L.W.; Ping, Y.H.; Hsu, K.W.; Huang, K.H.; Fang, W.L.; Lee, H.C.; Chen, C.F.; et al. Mevalonate pathway enzyme HMGCS1 contributes to gastric cancer progression. Cancers 2020, 12, 1088. [Google Scholar] [CrossRef]
- Ying, X.; Zhu, Y.; Jin, X.; Chang, X. Umbilical cord plasma-derived exosomes from preeclamptic women induce vascular dysfunction by targeting HMGCS1 in endothelial cells. Placenta 2021, 103, 86–93. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer. 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Scherer, D.; Deutelmoser, H.; Balavarca, Y.; Toth, R.; Habermann, N.; Buck, K.; Kap, E.J.; Botma, A.; Seibold, P.; Jansen, L.; et al. Polymorphisms in the Angiogenesis-Related Genes EFNB2, MMP2 and JAG1 Are Associated with Survival of Colorectal Cancer Patients. Int. J. Mol. Sci. 2020, 21, 5395. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; An, P.; Zhang, R.; He, X.; Yin, G.; Min, W. Etk/Bmx as a tumor necrosis factor receptor type 2-specific kinase: Role in endothelial cell migration and angiogenesis. Mol. Cell. Biol. 2002, 22, 7512–7523. [Google Scholar] [CrossRef] [PubMed]
- Gridley, T. Notch signaling in the vasculature. Curr. Top. Dev. Biol. 2010, 92, 277–309. [Google Scholar] [PubMed] [Green Version]
- Blanco, R.; Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Rho, S.S.; Choi, D.H.; Kwon, Y.G. LDB2 regulates the expression of DLL4 through the formation of oligomeric complexes in endothelial cells. BMB Rep. 2018, 51, 21–26. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| HCAEC | HITAEC |
|---|---|
| Upregulated genes | |
| NECTIN4 (1175.6-fold), ENSG00000288684 (999.7-fold), SIGLEC14 (728.6-fold), ENSG00000285188 (697.5-fold), ENSG00000285245 (683.8-fold), ENSG00000144785 (654.5-fold), RBM14-RBM4 (647.1-fold), MUC19 (564.9-fold), ENSG00000287542 (542.6-fold), ATP5MF-PTCD1 (493.5-fold), ENSG00000261732 (467.4-fold), COL7A1 (431.6-fold), CD70 (384.7-fold), PAQR6 (361.5-fold), ENSG00000279686 (357-fold), ENSG00000258461 (353.4-fold), ST20-MTHFS (316.3-fold), GPR87 (307.7-fold), DENND2C (279-fold), GRHL3 (269.2-fold), UNC13A (215-fold), ENKUR (147.6-fold), IFIT2 (145.5-fold), GRIP2 (114-fold), B3GNT7 (95.6-fold), CDK18 (93.5-fold), TEDC2 (82.9-fold), BEX2 (61.2-fold), APOBEC3B (59.3-fold), CCL5 (18.8-fold), EPS8L2 (5.8-fold), PLTP (5.7-fold), SLC7A2 (5.1-fold), ATF3 (3.9-fold), CXCL8 (3.5-fold), VWCE (3.5-fold), GDF15 (3.3-fold), KLHL21 (3-fold), PIDD1 (2.9-fold), TYMS (2.8-fold), CX3CL1 (2.7-fold), E2F7 (2.7-fold), FDXR (2.7-fold), MDM2 (2.4-fold), PHLDA3 (2.4-fold), MMP10 (2.4-fold), SOD2 (2.3-fold), SELE (2.3-fold), BHLHE40 (2.3-fold), RUSC2 (2.2-fold), CDKN1A (2.2-fold), DCBLD2 (2.1-fold), AXL (2.1-fold), ATP13A3 (2.1-fold), MT2A (2.1-fold), TP53I3 (2-fold) | MDM2 (2.3-fold) |
| Downregulated genes | |
| IQCJ-SCHIP1 (3-fold), LDB2 (2.2-fold), GIMAP8 (2.1-fold), BMX (2.1-fold), EFNB2 (2-fold), HMGCS1 (2-fold) | None detected |
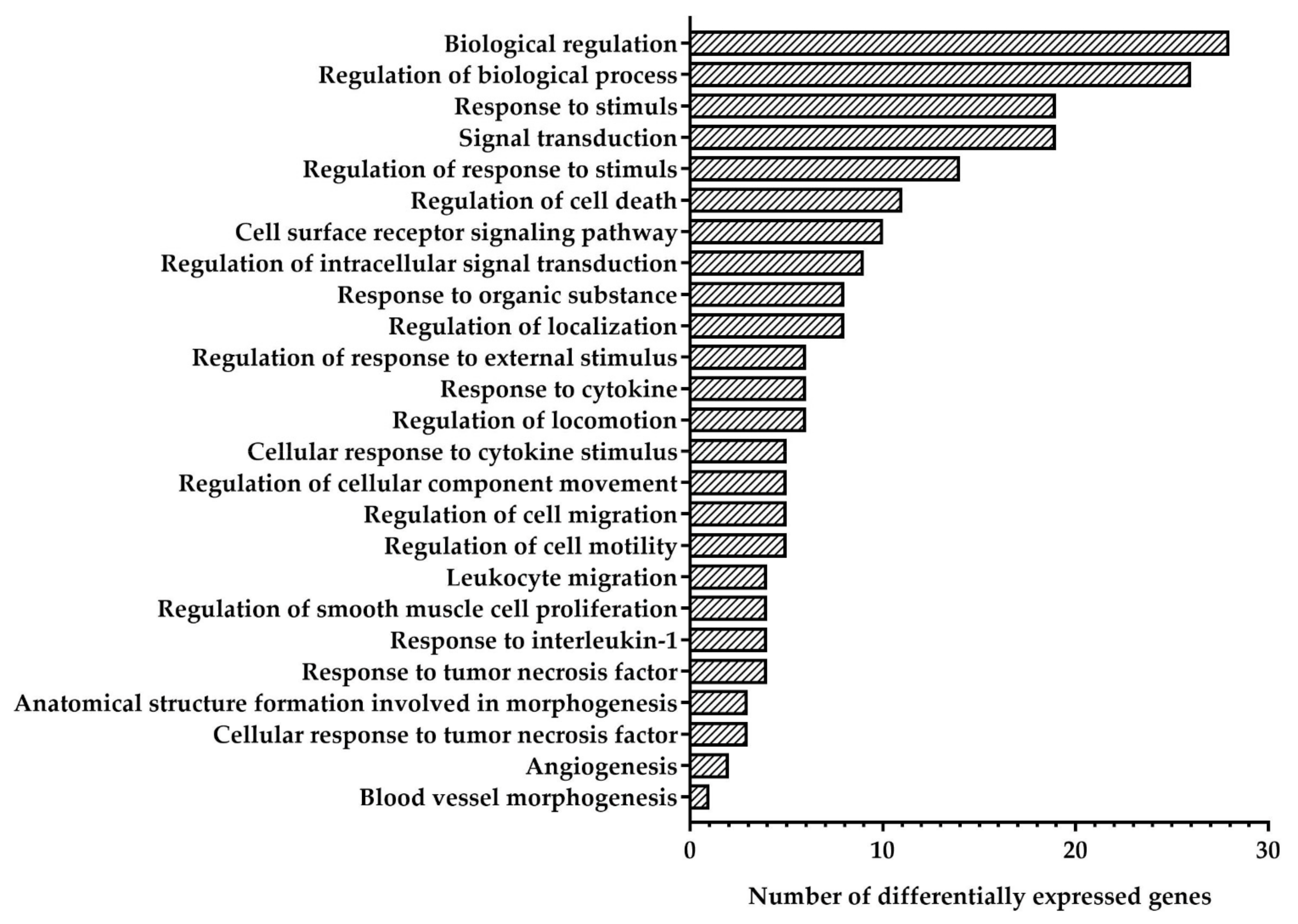
| Functional Group (GO Term) | Genes |
|---|---|
| Upregulated genes | |
| Biological regulation (GO:0065007) | MDM2, GDF15, CDKN1A, SOD2, PLTP, TP53I3, SELE, DCBLD2, CX3CL1, PHLDA3, MT2A, TYMS, ATP13A3, CXCL8, FDXR, AXL, E2F7, EPS8L2, KLHL21, BHLHE40, PIDD1, ATF3, CCL5 |
| Regulation of biological processes (GO:0050789) | MDM2, GDF15, CDKN1A, SOD2, PLTP, TP53I3, SELE, DCBLD2, CX3CL1, PHLDA3, MT2A, TYMS, CXCL8, AXL, E2F7, EPS8L2, KLHL21, BHLHE40, PIDD1, ATF3, CCL5 |
| Response to stimulus (GO:0050896) | MDM2, CDKN1A, SOD2, SELE, DCBLD2, CX3CL1, PHLDA3, MT2A, TYMS, VWCE, CXCL8, AXL, E2F7, EPS8L2, BHLHE40, PIDD1, ATF3, CCL5 |
| Signal transduction (GO:0007165) | MDM2, GDF15, CDKN1A, SOD2, SELE, DCBLD2, CX3CL1, PHLDA3, MT2A, CXCL8, AXL, E2F7, EPS8L2, BHLHE40, PIDD1, ATF3, CCL5 |
| Regulation of response to stimulus (GO:0048583) | MDM2, GDF15, CDKN1A, SOD2, SELE, CX3CL1, PHLDA3, CXCL8, AXL, PIDD1, ATF3, CCL5 |
| Regulation of cell death (GO:0010941) | MDM2, GDF15, SOD2, TP53I3, CX3CL1, AXL, BHLHE40, PIDD1, ATF3, CCL5 |
| Cell surface receptor signaling pathway (GO:0007166) | GDF15, CDKN1A, SOD2, CX3CL1, MT2A, CXCL8, AXL, BHLHE40, CCL5 |
| Regulation of intracellular signal transduction (GO:1902531) | MDM2, GDF15, SOD2, CX3CL1, PHLDA3, AXL, EPS8L2, PIDD1, CCL5 |
| Response to organic substance (GO:0010033) | MDM2, SELE, CX3CL1, MT2A, TYMS, CXCL8, AXL, CCL5 |
| Regulation of localization (GO:0032879) | SOD2, PLTP, SELE, CX3CL1, CXCL8, AXL, CCL5 |
| Regulation of response to external stimulus (GO:0032101) | CDKN1A, SELE, CX3CL1, CXCL8, CCL5 |
| Response to cytokine (GO:0034097) | SELE, CX3CL1, MT2A, CXCL8, AXL, CCL5 |
| Regulation of locomotion (GO:0040012) | SOD2, CX3CL1, CXCL8, CCL5 |
| Cellular response to cytokine stimulus (GO:0071345) | CX3CL1, MT2A, CXCL8, AXL, CCL5 |
| Regulation of cellular component movement (GO:0051270) | SOD2, CX3CL1, CXCL8, CCL5 |
| Regulation of cell migration (GO:0030334) | SOD2, CX3CL1, CXCL8, CCL5 |
| Regulation of cell motility (GO:2000145) | SOD2, CX3CL1, CXCL8, CCL5 |
| Leucocyte migration (GO:0050900) | SELE, CX3CL1, CXCL8, CCL5 |
| Regulation of smooth muscle cell proliferation (GO:0048660) | CDKN1A, SOD2, CX3CL1, CCL5 |
| Response to interleukin-1 (GO:0070555) | SELE, CX3CL1, CXCL8, CCL5 |
| Response to tumor necrosis factor (GO:0034612) | SELE, CX3CL1, CXCL8, CCL5 |
| Anatomical structure formation involved in morphogenesis (GO:0048446) | CXCL8, E2F7, BHLHE40 |
| Cellular response to tumor necrosis factor (GO:0071356) | CX3CL1, CXCL8, CCL5 |
| Angiogenesis (GO:0001525) | CXCL8, E2F7 |
| Downregulated genes | |
| Biological regulation (GO:0065007) | BMX, EFNB2, HMGSC1, LDB2, IQCJ-SCHIP1 |
| Regulation of biological processes (GO:0050789) | BMX, EFNB2, HMGSC1, LDB2, IQCJ-SCHIP1 |
| Response to stimulus (GO:0050896) | BMX |
| Signal transduction (GO:0007165) | BMX, EFNB2 |
| Regulation of response to stimulus (GO:0048583) | BMX, EFNB2 |
| Regulation of cell death (GO:0010941) | EFNB2 |
| Cell surface receptor signaling pathway (GO:0007166) | BMX |
| Regulation of localization (GO:0032879) | LDB2 |
| Regulation of response to external stimulus (GO:0032101) | EFNB2 |
| Regulation of locomotion (GO:0040012) | EFNB2, LDB2 |
| Regulation of cellular component movement (GO:0051270) | LDB2 |
| Regulation of cell migration (GO:0030334) | LDB2 |
| Regulation of cell motility (GO:2000145) | LDB2 |
| Blood vessel morphogenesis (GO:0048514) | EFNB2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinitsky, M.; Sinitskaya, A.; Shishkova, D.; Tupikin, A.; Asanov, M.; Khutornaya, M.; Kabilov, M.; Ponasenko, A. Identification of Key Genes and Pathways in Genotoxic Stress Induced Endothelial Dysfunction: Results of Whole Transcriptome Sequencing. Biomedicines 2022, 10, 2067. https://doi.org/10.3390/biomedicines10092067
Sinitsky M, Sinitskaya A, Shishkova D, Tupikin A, Asanov M, Khutornaya M, Kabilov M, Ponasenko A. Identification of Key Genes and Pathways in Genotoxic Stress Induced Endothelial Dysfunction: Results of Whole Transcriptome Sequencing. Biomedicines. 2022; 10(9):2067. https://doi.org/10.3390/biomedicines10092067
Chicago/Turabian StyleSinitsky, Maxim, Anna Sinitskaya, Daria Shishkova, Alexey Tupikin, Maxim Asanov, Maria Khutornaya, Marsel Kabilov, and Anastasia Ponasenko. 2022. "Identification of Key Genes and Pathways in Genotoxic Stress Induced Endothelial Dysfunction: Results of Whole Transcriptome Sequencing" Biomedicines 10, no. 9: 2067. https://doi.org/10.3390/biomedicines10092067