
Suppression of Cardiogenic Edema with Sodium–Glucose Cotransporter-2 Inhibitors in Heart Failure with Reduced Ejection Fraction: Mechanisms and Insights from Pre-Clinical Studies

 , ,
, ,
Abstract
:1. Introduction
2. Contribution of Neurohumoral Activation, Cardiorenal Dysfunction and Cardiac Remodeling to Edema in HFrEF
3. Evidence Supporting Edema Attenuation by SGLT-2i in Pre-Clinical HF Models
3.1. Impact of SGLT-2i on Neurohumoral Activation toward Edema Restraining in HFrEF
3.2. SGLT-2i Positively Affect Natriuretic Peptide System, Diuresis/Natriuresis, HF Signs, and Edema-Associated HF Plasma Biomarkers
3.3. SGLT-2i May Depress Edema by Improving Cardiac Function
3.4. SGLT-2i May Block Edema by Reducing Cardiac Remodeling
3.5. Impact of SGLT-2i on Cardiorenal Function Leading to Edema Suppression
3.6. SGLT-2i Block Activation of Sodium–Hydrogen Exchangers in the Heart and Kidneys That Contribute to the Clinical Progression of HFrEF Associated with Edema
3.7. SGLT-2i May Restrain Edema by Suppressing Chronic Inflammation and ROS
3.8. SGLT-2i May Prevent Vascular Leakage and Edema by Improvement of Endothelial Dysfunction
3.9. Alteration of Cardiac Metabolism and Energy Utilization by SGLT-2i Improves Cardiac Structure and Function, Which May Contribute to Edema Reduction
4. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of Heart Failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of Dilated Cardiomyopathy: Practical Implications for Heart Failure Management. Nat. Rev. Cardiol. 2020, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Coats, A.J.S.; Tsutsui, H.; Abdelhamid, C.M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Bohm, M.; Butler, J.; et al. Universal Definition and Classification of Heart Failure: A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. Eur. J. Heart Fail. 2021, 23, 352–380. [Google Scholar] [CrossRef]
- Dzau, V.J.; Colucci, W.S.; Hollenberg, N.K.; Williams, G.H. Relation of the Renin-Angiotensin-Aldosterone System to Clinical State in Congestive Heart Failure. Circulation 1981, 63, 645–651. [Google Scholar] [CrossRef]
- Clark, A.L.; Cleland, J.G. Causes and Treatment of Oedema in Patients with Heart Failure. Nat. Rev. Cardiol. 2013, 10, 156–170. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 Accf/Aha Guideline for the Management of Heart Failure: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 128, e240–e327. [Google Scholar] [CrossRef]
- Parrinello, G.; Greene, S.J.; Torres, D.; Alderman, M.; Bonventre, J.V.; Di Pasquale, P.; Gargani, L.; Nohria, A.; Fonarow, G.C.; Vaduganathan, M.; et al. Water and Sodium in Heart Failure: A Spotlight on Congestion. Heart Fail. Rev. 2015, 20, 13–24. [Google Scholar] [CrossRef]
- Pellicori, P.; Kaur, K.; Clark, A.L. Fluid Management in Patients with Chronic Heart Failure. Card. Fail. Rev. 2015, 1, 90–95. [Google Scholar] [CrossRef]
- Melenovsky, V.; Andersen, M.J.; Andress, K.; Reddy, Y.N.; Borlaug, B.A. Lung Congestion in Chronic Heart Failure: Haemodynamic, Clinical, and Prognostic Implications. Eur. J. Heart Fail. 2015, 17, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Mentz, R.J.; Stevens, S.R.; DeVore, A.D.; Lala, A.; Vader, J.M.; AbouEzzeddine, O.F.; Khazanie, P.; Redfield, M.M.; Stevenson, L.W.; O’Connor, C.M.; et al. Decongestion Strategies and Renin-Angiotensin-Aldosterone System Activation in Acute Heart Failure. JACC Heart Fail. 2015, 3, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Metra, M.; O’Connor, C.M.; Davison, B.A.; Cleland, J.G.; Ponikowski, P.; Teerlink, J.R.; Voors, A.A.; Givertz, M.M.; Mansoor, G.A.; Bloomfield, D.M.; et al. Early Dyspnoea Relief in Acute Heart Failure: Prevalence, Association with Mortality, and Effect of Rolofylline in the Protect Study. Eur. Heart J. 2011, 32, 1519–1534. [Google Scholar] [CrossRef] [PubMed]
- Aimo, A.; Vergaro, G.; Giannoni, A.; Emdin, M. Wet Is Bad: Residual Congestion Predicts Worse Prognosis in Acute Heart Failure. Int. J. Cardiol. 2018, 258, 201–202. [Google Scholar] [CrossRef]
- Rubio-Gracia, J.; Demissei, B.G.; Ter Maaten, J.M.; Cleland, J.G.; O’Connor, C.M.; Metra, M.; Ponikowski, P.; Teerlink, J.R.; Cotter, G.; Davison, B.A.; et al. Prevalence, Predictors and Clinical Outcome of Residual Congestion in Acute Decompensated Heart Failure. Int. J. Cardiol. 2018, 258, 185–191. [Google Scholar] [CrossRef]
- Palazzuoli, A.; Evangelista, I.; Nuti, R. Congestion Occurrence and Evaluation in Acute Heart Failure Scenario: Time to Reconsider Different Pathways of Volume Overload. Heart Fail. Rev. 2020, 25, 119–131. [Google Scholar] [CrossRef]
- Lombardi, C.M.; Cimino, G.; Pellicori, P.; Bonelli, A.; Inciardi, R.M.; Pagnesi, M.; Tomasoni, D.; Ravera, A.; Adamo, M.; Carubelli, V.; et al. Congestion in Patients with Advanced Heart Failure: Assessment and Treatment. Heart Fail. Clin. 2021, 17, 575–586. [Google Scholar] [CrossRef]
- Greene, S.J.; Fonarow, G.C.; Butler, J. Risk Profiles in Heart Failure: Baseline, Residual, Worsening, and Advanced Heart Failure Risk. Circ. Heart Fail. 2020, 13, e007132. [Google Scholar] [CrossRef]
- Ambrosy, A.P.; Vaduganathan, M.; Huffman, M.D.; Khan, S.; Kwasny, M.J.; Fought, A.J.; Maggioni, A.P.; Swedberg, K.; Konstam, M.A.; Zannad, F.; et al. Clinical Course and Predictive Value of Liver Function Tests in Patients Hospitalized for Worsening Heart Failure with Reduced Ejection Fraction: An Analysis of the Everest Trial. Eur. J. Heart Fail. 2012, 14, 302–311. [Google Scholar] [CrossRef]
- Boorsma, E.M.; Ter Maaten, J.M.; Damman, K.; Dinh, W.; Gustafsson, F.; Goldsmith, S.; Burkhoff, D.; Zannad, F.; Udelson, J.E.; Voors, A.A. Congestion in Heart Failure: A Contemporary Look at Physiology, Diagnosis and Treatment. Nat. Rev. Cardiol. 2020, 17, 641–655. [Google Scholar] [CrossRef]
- Gheorghiade, M.; Gattis, W.A.; O’Connor, C.M.; Adams, K.F., Jr.; Elkayam, U.; Barbagelata, A.; Ghali, J.K.; Benza, R.L.; McGrew, F.A.; Klapholz, M.; et al. Acute Chronic Therapeutic Impact of a Vasopressin Antagonist in Congestive Heart Failure, I. Effects of Tolvaptan, a Vasopressin Antagonist, in Patients Hospitalized with Worsening Heart Failure: A Randomized Controlled Trial. JAMA 2004, 291, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Lala, A.; McNulty, S.E.; Mentz, R.J.; Dunlay, S.M.; Vader, J.M.; AbouEzzeddine, O.F.; DeVore, A.D.; Khazanie, P.; Redfield, M.M.; Goldsmith, S.R.; et al. Relief and Recurrence of Congestion During and after Hospitalization for Acute Heart Failure: Insights from Diuretic Optimization Strategy Evaluation in Acute Decompensated Heart Failure (Dose-Ahf) and Cardiorenal Rescue Study in Acute Decompensated Heart Failure (Caress-Hf). Circ. Heart Fail. 2015, 8, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Claggett, B.; Pozzi, A.; McMurray, J.J.V.; Jhund, P.S.; Packer, M.; Desai, A.S.; Lewis, E.F.; Vaduganathan, M.; Lefkowitz, M.P.; et al. Prognostic Implications of Congestion on Physical Examination among Contemporary Patients with Heart Failure and Reduced Ejection Fraction: Paradigm-Hf. Circulation 2019, 140, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Fluid Volume Overload and Congestion in Heart Failure: Time to Reconsider Pathophysiology and How Volume Is Assessed. Circ. Heart Fail. 2016, 9, e002922. [Google Scholar] [CrossRef]
- Rossignol, P.; Hernandez, A.F.; Solomon, S.D.; Zannad, F. Heart Failure Drug Treatment. Lancet 2019, 393, 1034–1044. [Google Scholar] [CrossRef]
- Sullivan, R.D.; Mehta, R.M.; Tripathi, R.; Reed, G.L.; Gladysheva, I.P. Renin Activity in Heart Failure with Reduced Systolic Function-New Insights. Int. J. Mol. Sci. 2019, 20, 3182. [Google Scholar] [CrossRef]
- Murphy, S.P.; Ibrahim, N.E.; Januzzi, J.L., Jr. Heart Failure with Reduced Ejection Fraction: A Review. JAMA 2020, 324, 488–504. [Google Scholar] [CrossRef]
- Pellicori, P.; Khan, M.J.I.; Graham, F.J.; Cleland, J.G.F. New Perspectives and Future Directions in the Treatment of Heart Failure. Heart Fail. Rev. 2020, 25, 147–159. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 Esc Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Espinoza, C.; Alkhateeb, H.; Siddiqui, T. Updates in Pharmacotherapy of Heart Failure with Reduced Ejection Fraction. Ann. Transl. Med. 2021, 9, 516. [Google Scholar] [CrossRef]
- Patel, N.H.; Bindra, A.S. Transitioning Hfref Patients on Ace Inhibitors to Modern Medical Therapy: What Is the Next Step? JACC Heart Fail. 2021, 9, 319. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Rao, S.; Manek, G.; Fonarow, G.C.; Ghosh, R.K. The Role of Dapagliflozin in the Management of Heart Failure: An Update on the Emerging Evidence. Ther. Clin. Risk Manag. 2021, 17, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Spertus, J.A.; Birmingham, M.C.; Nassif, M.; Damaraju, C.V.; Abbate, A.; Butler, J.; Lanfear, D.E.; Lingvay, I.; Kosiborod, M.N.; Januzzi, J.L. The Sglt2 Inhibitor Canagliflozin in Heart Failure: The Chief-Hf Remote, Patient-Centered Randomized Trial. Nat. Med. 2022, 28, 809–813. [Google Scholar] [CrossRef]
- Hernandez, M.; Sullivan, R.D.; McCune, M.E.; Reed, G.L.; Gladysheva, I.P. Sodium-Glucose Cotransporter-2 Inhibitors Improve Heart Failure with Reduced Ejection Fraction Outcomes by Reducing Edema and Congestion. Diagnostics 2022, 12, 989. [Google Scholar] [CrossRef]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of Endothelial Dysfunction in Heart Failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L., Jr. Inflammation in Heart Failure: Jacc State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Guyton, A.C.; Granger, H.J.; Taylor, A.E. Interstitial Fluid Pressure. Physiol. Rev. 1971, 51, 527–563. [Google Scholar] [CrossRef]
- Itkin, M.; Rockson, S.G.; Burkhoff, D. Pathophysiology of the Lymphatic System in Patients with Heart Failure: Jacc State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 278–290. [Google Scholar] [CrossRef]
- Aronson, D. The Interstitial Comartment as a Therapeutic Target in Heart Failure. Front. Cardiovasc. Med. 2022, 9, 933384. [Google Scholar] [CrossRef]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why Do Sglt2 Inhibitors Reduce Heart Failure Hospitalization? A Differential Volume Regulation Hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487. [Google Scholar] [CrossRef]
- Yu, H.; Basu, S.; Hallow, K.M. Cardiac and Renal Function Interactions in Heart Failure with Reduced Ejection Fraction: A Mathematical Modeling Analysis. PLoS Comput. Biol. 2020, 16, e1008074. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, S.; Tanaka, A.; Imai, T.; Shimabukuro, M.; Uehara, H.; Nakamura, I.; Matsunaga, K.; Suzuki, M.; Kashimura, T.; Minamino, T.; et al. Body Fluid Regulation Via Chronic Inhibition of Sodium-Glucose Cotransporter-2 in Patients with Heart Failure: A Post Hoc Analysis of the Candle Trial. Clin. Res. Cardiol. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Tanaka, A.; Shimabukuro, M.; Teragawa, H.; Okada, Y.; Takamura, T.; Taguchi, I.; Toyoda, S.; Tomiyama, H.; Ueda, S.; Higashi, Y.; et al. Reduction of Estimated Fluid Volumes Following Initiation of Empagliflozin in Patients with Type 2 Diabetes and Cardiovascular Disease: A Secondary Analysis of the Placebo-Controlled, Randomized Emblem Trial. Cardiovasc. Diabetol. 2021, 20, 105. [Google Scholar] [CrossRef] [PubMed]
- Rasalam, R.; Atherton, J.J.; Deed, G.; Molloy-Bland, M.; Cohen, N.; Sindone, A. Sodium-Glucose Cotransporter 2 Inhibitor Effects on Heart Failure Hospitalization and Cardiac Function: Systematic Review. ESC Heart Fail. 2021, 8, 4093–4118. [Google Scholar] [CrossRef]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of Renal Glucose Handling Via Sglt1, Sglt2 and Glut2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Yellon, D.M. Sglt2 Inhibitors: Hypotheses on the Mechanism of Cardiovascular Protection. Lancet Diabetes Endocrinol. 2018, 6, 435–437. [Google Scholar] [CrossRef]
- Wojcik, C.; Warden, B.A. Mechanisms and Evidence for Heart Failure Benefits from Sglt2 Inhibitors. Curr. Cardiol. Rep. 2019, 21, 130. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, B. A Review of the Proposed Mechanistic Actions of Sodium Glucose Cotransporter-2 Inhibitors in the Treatment of Heart Failure. Cardiol. Res. 2021, 12, 60–66. [Google Scholar] [CrossRef]
- Fathi, A.; Vickneson, K.; Singh, J.S. Sglt2-Inhibitors. More Than Just Glycosuria and Diuresis. Heart Fail. Rev. 2021, 26, 623–642. [Google Scholar] [CrossRef]
- Sayer, G.; Bhat, G. The Renin-Angiotensin-Aldosterone System and Heart Failure. Cardiol. Clin. 2014, 32, 21–32. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kramer, D.G.; Patel, A.R.; Maron, M.S.; Udelson, J.E. Left Ventricular Remodeling in Heart Failure: Current Concepts in Clinical Significance and Assessment. JACC Cardiovasc. Imaging 2011, 4, 98–108. [Google Scholar] [CrossRef]
- Ibebuogu, U.N.; Gladysheva, I.P.; Houng, A.K.; Reed, G.L. Decompensated Heart Failure Is Associated with Reduced Corin Levels and Decreased Cleavage of Pro-Atrial Natriuretic Peptide. Circ. Heart Fail. 2011, 4, 114–120. [Google Scholar] [CrossRef]
- Tripathi, R.; Wang, D.; Sullivan, R.; Fan, T.H.; Gladysheva, I.P.; Reed, G.L. Depressed Corin Levels Indicate Early Systolic Dysfunction before Increases of Atrial Natriuretic Peptide/B-Type Natriuretic Peptide and Heart Failure Development. Hypertension 2016, 67, 362–367. [Google Scholar] [CrossRef]
- Zaidi, S.S.; Ward, R.D.; Ramanathan, K.; Yu, X.; Gladysheva, I.P.; Reed, G.L. Possible Enzymatic Downregulation of the Natriuretic Peptide System in Patients with Reduced Systolic Function and Heart Failure: A Pilot Study. Biomed. Res. Int. 2018, 2018, 7279036. [Google Scholar] [CrossRef]
- Schrier, R.W.; Abraham, W.T. Hormones and Hemodynamics in Heart Failure. N. Engl. J. Med. 1999, 341, 577–585. [Google Scholar] [CrossRef]
- Sullivan, R.D.; Mehta, R.M.; Tripathi, R.; Gladysheva, I.P.; Reed, G.L. Normalizing Plasma Renin Activity in Experimental Dilated Cardiomyopathy: Effects on Edema, Cachexia, and Survival. Int. J. Mol. Sci. 2019, 20, 3886. [Google Scholar] [CrossRef]
- Chiorescu, R.M.; Lazar, R.-D.; Buksa, S.-B.; Mocan, M.; Blendea, D. Biomarkers of Volume Overload and Edema in Heart Failure with Reduced Ejection Fraction. Front. Cardiovasc. Med. 2022, 9, 910100. [Google Scholar] [CrossRef]
- Adams, K.F., Jr. Pathophysiologic Role of the Renin-Angiotensin-Aldosterone and Sympathetic Nervous Systems in Heart Failure. Am. J. Health Syst. Pharm. 2004, 61, S4–S13. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Neurohormonal Activation in Heart Failure with Reduced Ejection Fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef]
- Ansary, T.M.; Nakano, D.; Nishiyama, A. Diuretic Effects of Sodium Glucose Cotransporter 2 Inhibitors and Their Influence on the Renin-Angiotensin System. Int. J. Mol. Sci. 2019, 20, 629. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin Ameliorates Adverse Left Ventricular Remodeling in Nondiabetic Heart Failure by Enhancing Myocardial Energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]
- Borovac, J.A.; D’Amario, D.; Bozic, J.; Glavas, D. Sympathetic Nervous System Activation and Heart Failure: Current State of Evidence and the Pathophysiology in the Light of Novel Biomarkers. World J. Cardiol. 2020, 12, 373–408. [Google Scholar] [CrossRef]
- Shimizu, W.; Kubota, Y.; Hoshika, Y.; Mozawa, K.; Tara, S.; Tokita, Y.; Yodogawa, K.; Iwasaki, Y.K.; Yamamoto, T.; Takano, H.; et al. Effects of Empagliflozin Versus Placebo on Cardiac Sympathetic Activity in Acute Myocardial Infarction Patients with Type 2 Diabetes Mellitus: The Embody Trial. Cardiovasc. Diabetol. 2020, 19, 148. [Google Scholar] [CrossRef]
- Wei, C.M.; Heublein, D.M.; Perrella, M.A.; Lerman, A.; Rodeheffer, R.J.; McGregor, C.G.; Edwards, W.D.; Schaff, H.V.; Burnett, J.C., Jr. Natriuretic Peptide System in Human Heart Failure. Circulation 1993, 88, 1004–1009. [Google Scholar] [CrossRef]
- Gidlof, O. Toward a New Paradigm for Targeted Natriuretic Peptide Enhancement in Heart Failure. Front. Physiol. 2021, 12, 650124. [Google Scholar] [CrossRef]
- Shi, X.; Verma, S.; Yun, J.; Brand-Arzamendi, K.; Singh, K.K.; Liu, X.; Garg, A.; Quan, A.; Wen, X.Y. Effect of Empagliflozin on Cardiac Biomarkers in a Zebrafish Model of Heart Failure: Clues to the Empa-Reg Outcome Trial? Mol. Cell. Biochem. 2017, 433, 97–102. [Google Scholar] [CrossRef]
- Tripathi, R.; Sullivan, R.D.; Fan, T.M.; Houng, A.K.; Mehta, R.M.; Reed, G.L.; Gladysheva, I.P. Cardiac-Specific Overexpression of Catalytically Inactive Corin Reduces Edema, Contractile Dysfunction, and Death in Mice with Dilated Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 203. [Google Scholar] [CrossRef]
- Ma, X.; Tannu, S.; Allocco, J.; Pan, J.; Dipiero, J.; Wong, P. A Mouse Model of Heart Failure Exhibiting Pulmonary Edema and Pleural Effusion: Useful for Testing New Drugs. J. Pharmacol. Toxicol. Methods 2019, 96, 78–86. [Google Scholar] [CrossRef]
- Tripathi, R.; Sullivan, R.D.; Fan, T.M.; Mehta, R.M.; Gladysheva, I.P.; Reed, G.L. A Low-Sodium Diet Boosts Ang (1-7) Production and No-Cgmp Bioavailability to Reduce Edema and Enhance Survival in Experimental Heart Failure. Int. J. Mol. Sci. 2021, 22, 4035. [Google Scholar] [CrossRef]
- Lee, H.C.; Shiou, Y.L.; Jhuo, S.J.; Chang, C.Y.; Liu, P.L.; Jhuang, W.J.; Dai, Z.K.; Chen, W.Y.; Chen, Y.F.; Lee, A.S. The Sodium-Glucose Co-Transporter 2 Inhibitor Empagliflozin Attenuates Cardiac Fibrosis and Improves Ventricular Hemodynamics in Hypertensive Heart Failure Rats. Cardiovasc. Diabetol. 2019, 18, 45. [Google Scholar] [CrossRef]
- Abdurrachim, D.; Teo, X.Q.; Woo, C.C.; Chan, W.X.; Lalic, J.; Lam, C.S.P.; Lee, P.T.H. Empagliflozin Reduces Myocardial Ketone Utilization While Preserving Glucose Utilization in Diabetic Hypertensive Heart Disease: A Hyperpolarized (13) C Magnetic Resonance Spectroscopy Study. Diabetes Obes. Metab. 2019, 21, 357–365. [Google Scholar] [CrossRef]
- Yurista, S.R.; Sillje, H.H.W.; Oberdorf-Maass, S.U.; Schouten, E.M.; Pavez Giani, M.G.; Hillebrands, J.L.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium-Glucose Co-Transporter 2 Inhibition with Empagliflozin Improves Cardiac Function in Non-Diabetic Rats with Left Ventricular Dysfunction after Myocardial Infarction. Eur. J. Heart Fail. 2019, 21, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Byrne, N.J.; Matsumura, N.; Maayah, Z.H.; Ferdaoussi, M.; Takahara, S.; Darwesh, A.M.; Levasseur, J.L.; Jahng, J.W.S.; Vos, D.; Parajuli, N.; et al. Empagliflozin Blunts Worsening Cardiac Dysfunction Associated with Reduced Nlrp3 (Nucleotide-Binding Domain-Like Receptor Protein 3) Inflammasome Activation in Heart Failure. Circ. Heart Fail. 2020, 13, e006277. [Google Scholar] [CrossRef] [PubMed]
- Tanajak, P.; Sa-Nguanmoo, P.; Sivasinprasasn, S.; Thummasorn, S.; Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Cardioprotection of Dapagliflozin and Vildagliptin in Rats with Cardiac Ischemia-Reperfusion Injury. J Endocrinol 2018, 236, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.A.; Zhang, Y.; Desjardins, J.F.; Nghiem, L.; Visram, A.; Batchu, S.N.; Yerra, V.G.; Kabir, G.; Thai, K.; Advani, A.; et al. Load-Independent Effects of Empagliflozin Contribute to Improved Cardiac Function in Experimental Heart Failure with Reduced Ejection Fraction. Cardiovasc. Diabetol. 2020, 19, 13. [Google Scholar] [CrossRef]
- Sayour, A.A.; Korkmaz-Icoz, S.; Loganathan, S.; Ruppert, M.; Sayour, V.N.; Olah, A.; Benke, K.; Brune, M.; Benko, R.; Horvath, E.M.; et al. Acute Canagliflozin Treatment Protects against in Vivo Myocardial Ischemia-Reperfusion Injury in Non-Diabetic Male Rats and Enhances Endothelium-Dependent Vasorelaxation. J. Transl. Med. 2019, 17, 127. [Google Scholar] [CrossRef]
- Wang, K.; Li, Z.; Sun, Y.; Liu, X.; Ma, W.; Ding, Y.; Hong, J.; Qian, L.; Xu, D. Dapagliflozin Improves Cardiac Function, Remodeling, Myocardial Apoptosis, and Inflammatory Cytokines in Mice with Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021; (online ahead of print). [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Garcia-Ropero, A.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Vargas-Delgado, A.P.; Flores-Umanzor, E.J.; Sanz, J.; et al. Empagliflozin Ameliorates Diastolic Dysfunction and Left Ventricular Fibrosis/Stiffness in Nondiabetic Heart Failure: A Multimodality Study. JACC Cardiovasc. Imaging 2021, 14, 393–407. [Google Scholar] [CrossRef]
- Arow, M.; Waldman, M.; Yadin, D.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Freimark, D.; Kornowski, R.; Aravot, D.; Hochhauser, E.; et al. Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Attenuates Diabetic Cardiomyopathy. Cardiovasc. Diabetol. 2020, 19, 7. [Google Scholar] [CrossRef]
- Kraker, K.; Herse, F.; Golic, M.; Reichhart, N.; Crespo-Garcia, S.; Strauss, O.; Grune, J.; Kintscher, U.; Ebrahim, M.; Bader, M.; et al. Effects of Empagliflozin and Target-Organ Damage in a Novel Rodent Model of Heart Failure Induced by Combined Hypertension and Diabetes. Sci. Rep. 2020, 10, 14061. [Google Scholar] [CrossRef]
- Sabatino, J.; De Rosa, S.; Tamme, L.; Iaconetti, C.; Sorrentino, S.; Polimeni, A.; Mignogna, C.; Amorosi, A.; Spaccarotella, C.; Yasuda, M.; et al. Empagliflozin Prevents Doxorubicin-Induced Myocardial Dysfunction. Cardiovasc. Diabetol. 2020, 19, 66. [Google Scholar] [CrossRef]
- Li, X.; Lu, Q.; Qiu, Y.; do Carmo, J.M.; Wang, Z.; da Silva, A.A.; Mouton, A.; Omoto, A.C.M.; Hall, M.E.; Li, J.; et al. Direct Cardiac Actions of the Sodium Glucose Co-Transporter 2 Inhibitor Empagliflozin Improve Myocardial Oxidative Phosphorylation and Attenuate Pressure-Overload Heart Failure. J. Am. Heart Assoc. 2021, 10, e018298. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, M.; Xu, J.; Xu, B.; Kang, L. Empagliflozin Prevents from Early Cardiac Injury Post Myocardial Infarction in Non-Diabetic Mice. Eur. J. Pharm. Sci. 2021, 161, 105788. [Google Scholar] [CrossRef]
- Asensio Lopez, M.D.C.; Lax, A.; Hernandez Vicente, A.; Saura Guillen, E.; Hernandez-Martinez, A.; Fernandez Del Palacio, M.J.; Bayes-Genis, A.; Pascual Figal, D.A. Empagliflozin Improves Post-Infarction Cardiac Remodeling through Gtp Enzyme Cyclohydrolase 1 and Irrespective of Diabetes Status. Sci. Rep. 2020, 10, 13553. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin Normalizes the Size and Number of Mitochondria and Prevents Reduction in Mitochondrial Size after Myocardial Infarction in Diabetic Hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef] [PubMed]
- Koyani, C.N.; Plastira, I.; Sourij, H.; Hallstrom, S.; Schmidt, A.; Rainer, P.P.; Bugger, H.; Frank, S.; Malle, E.; von Lewinski, D. Empagliflozin Protects Heart from Inflammation and Energy Depletion Via Ampk Activation. Pharmacol. Res. 2020, 158, 104870. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Nakamura, K.; Miyoshi, T.; Yoshida, M.; Akazawa, K.; Saito, Y.; Akagi, S.; Ohno, Y.; Kondo, M.; Miura, D.; et al. Inhibitory Effects of Tofogliflozin on Cardiac Hypertrophy in Dahl Salt-Sensitive and Salt-Resistant Rats Fed a High-Fat Diet. Int. Heart J. 2019, 60, 728–735. [Google Scholar] [CrossRef]
- Kuriyama, S. A Potential Mechanism of Cardio-Renal Protection with Sodium-Glucose Cotransporter 2 Inhibitors: Amelioration of Renal Congestion. Kidney Blood Press. Res. 2019, 44, 449–456. [Google Scholar] [CrossRef]
- Yang, C.C.; Chen, Y.T.; Wallace, C.G.; Chen, K.H.; Cheng, B.C.; Sung, P.H.; Li, Y.C.; Ko, S.F.; Chang, H.W.; Yip, H.K. Early Administration of Empagliflozin Preserved Heart Function in Cardiorenal Syndrome in Rat. Biomed. Pharmacother. 2019, 109, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Activation and Inhibition of Sodium-Hydrogen Exchanger Is a Mechanism That Links the Pathophysiology and Treatment of Diabetes Mellitus with That of Heart Failure. Circulation 2017, 136, 1548–1559. [Google Scholar] [CrossRef]
- Philippaert, K.; Kalyaanamoorthy, S.; Fatehi, M.; Long, W.; Soni, S.; Byrne, N.J.; Barr, A.; Singh, J.; Wong, J.; Palechuk, T.; et al. Cardiac Late Sodium Channel Current Is a Molecular Target for the Sodium/Glucose Cotransporter 2 Inhibitor Empagliflozin. Circulation 2021, 143, 2188–2204. [Google Scholar] [CrossRef]
- Morris, K. Targeting the Myocardial Sodium-Hydrogen Exchange for Treatment of Heart Failure. Expert. Opin. Ther. Targets 2002, 6, 291–298. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.T.; Coronel, R. Sglt2 Inhibitor Empagliflozin Inhibits the Cardiac Na+/H+ Exchanger 1: Persistent Inhibition under Various Experimental Conditions. Cardiovasc. Res. 2021, 117, 2699–2701. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Hardziyenka, M.; Schumacher, C.A.; Belterman, C.N.; van Borren, M.M.; Verkerk, A.O.; Coronel, R.; Fiolet, J.W. Chronic Inhibition of the Na+/H+—Exchanger Causes Regression of Hypertrophy, Heart Failure, and Ionic and Electrophysiological Remodelling. Br. J. Pharmacol. 2008, 154, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Iborra-Egea, O.; Santiago-Vacas, E.; Yurista, S.R.; Lupon, J.; Packer, M.; Heymans, S.; Zannad, F.; Butler, J.; Pascual-Figal, D.; Lax, A.; et al. Unraveling the Molecular Mechanism of Action of Empagliflozin in Heart Failure with Reduced Ejection Fraction with or without Diabetes. JACC Basic Transl. Sci. 2019, 4, 831–840. [Google Scholar] [CrossRef]
- Chung, Y.J.; Park, K.C.; Tokar, S.; Eykyn, T.R.; Fuller, W.; Pavlovic, D.; Swietach, P.; Shattock, M.J. Off-Target Effects of Sglt2 Blockers: Empagliflozin Does Not Inhibit Na+/H+ Exchanger-1 or Lower [Na+]i in the Heart. Cardiovasc. Res. 2020, 117, 2794–2806. [Google Scholar] [CrossRef]
- Chung, Y.J.; Park, K.C.; Tokar, S.; Eykyn, T.R.; Fuller, W.; Pavlovic, D.; Swietach, P.; Shattock, M.J. Sglt2 Inhibitors and the Cardiac Na+/H+ Exchanger-1: The Plot Thickens. Cardiovasc. Res. 2021, 117, 2702–2704. [Google Scholar] [CrossRef] [PubMed]
- Wiig, H. Pathophysiology of Tissue Fluid Accumulation in Inflammation. J Physiol 2011, 589, 2945–2953. [Google Scholar] [CrossRef]
- Van Linthout, S.; Tschope, C. Inflammation—Cause or Consequence of Heart Failure or Both? Curr. Heart Fail. Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef]
- Aimo, A.; Castiglione, V.; Borrelli, C.; Saccaro, L.F.; Franzini, M.; Masi, S.; Emdin, M.; Giannoni, A. Oxidative Stress and Inflammation in the Evolution of Heart Failure: From Pathophysiology to Therapeutic Strategies. Eur. J. Prev. Cardiol. 2020, 27, 494–510. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Emdin, M.; Aimo, A.; Castiglione, V.; Vergaro, G.; Georgiopoulos, G.; Saccaro, L.F.; Lombardi, C.M.; Passino, C.; Cerbai, E.; Metra, M.; et al. Targeting Cyclic Guanosine Monophosphate to Treat Heart Failure: Jacc Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 76, 1795–1807. [Google Scholar] [CrossRef]
- Fischer, D.; Rossa, S.; Landmesser, U.; Spiekermann, S.; Engberding, N.; Hornig, B.; Drexler, H. Endothelial Dysfunction in Patients with Chronic Heart Failure Is Independently Associated with Increased Incidence of Hospitalization, Cardiac Transplantation, or Death. Eur. Heart J. 2005, 26, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Kerem, A.; Yin, J.; Kaestle, S.M.; Hoffmann, J.; Schoene, A.M.; Singh, B.; Kuppe, H.; Borst, M.M.; Kuebler, W.M. Lung Endothelial Dysfunction in Congestive Heart Failure: Role of Impaired Ca2+ Signaling and Cytoskeletal Reorganization. Circ. Res. 2010, 106, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Giannitsi, S.; Bougiakli, M.; Bechlioulis, A.; Naka, K. Endothelial Dysfunction and Heart Failure: A Review of the Existing Bibliography with Emphasis on Flow Mediated Dilation. JRSM Cardiovasc. Dis. 2019, 8, 2048004019843047. [Google Scholar] [CrossRef] [PubMed]
- Alshnbari, A.S.; Millar, S.A.; O’Sullivan, S.E.; Idris, I. Effect of Sodium-Glucose Cotransporter-2 Inhibitors on Endothelial Function: A Systematic Review of Preclinical Studies. Diabetes Ther. 2020, 11, 1947–1963. [Google Scholar] [CrossRef]
- Ugusman, A.; Kumar, J.; Aminuddin, A. Endothelial Function and Dysfunction: Impact of Sodium-Glucose Cotransporter 2 Inhibitors. Pharmacol. Ther. 2021, 224, 107832. [Google Scholar] [CrossRef]
- Dou, L.; Burtey, S. Reversing Endothelial Dysfunction with Empagliflozin to Improve Cardiomyocyte Function in Cardiorenal Syndrome. Kidney Int. 2021, 99, 1062–1064. [Google Scholar] [CrossRef]
- Juni, R.P.; Al-Shama, R.; Kuster, D.W.D.; van der Velden, J.; Hamer, H.M.; Vervloet, M.G.; Eringa, E.C.; Koolwijk, P.; van Hinsbergh, V.W.M. Empagliflozin Restores Chronic Kidney Disease-Induced Impairment of Endothelial Regulation of Cardiomyocyte Relaxation and Contraction. Kidney Int. 2021, 99, 1088–1101. [Google Scholar] [CrossRef]
- Han, Y.; Cho, Y.E.; Ayon, R.; Guo, R.; Youssef, K.D.; Pan, M.; Dai, A.; Yuan, J.X.; Makino, A. Sglt Inhibitors Attenuate No-Dependent Vascular Relaxation in the Pulmonary Artery but Not in the Coronary Artery. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1027–L1036. [Google Scholar] [CrossRef]
- Sala, V.; Gatti, S.; Gallo, S.; Medico, E.; Cantarella, D.; Cimino, J.; Ponzetto, A.; Crepaldi, T. A New Transgenic Mouse Model of Heart Failure and Cardiac Cachexia Raised by Sustained Activation of Met Tyrosine Kinase in the Heart. Biomed. Res. Int. 2016, 2016, 9549036. [Google Scholar] [CrossRef]
- Packer, M. Cardioprotective Effects of Sirtuin-1 and Its Downstream Effectors: Potential Role in Mediating the Heart Failure Benefits of Sglt2 (Sodium-Glucose Cotransporter 2) Inhibitors. Circ. Heart Fail. 2020, 13, e007197. [Google Scholar] [CrossRef]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the Empa-Reg Outcome Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.; Luptak, I.; Chambers, J.M.; Hobai, I.; Panagia, M.; Pimentel, D.R.; Siwik, D.A.; Qin, F.; Colucci, W.S. Effects of Sodium-Glucose Linked Transporter 2 Inhibition with Ertugliflozin on Mitochondrial Function, Energetics, and Metabolic Gene Expression in the Presence and Absence of Diabetes Mellitus in Mice. J. Am. Heart Assoc. 2021, 10, e019995. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Ramirez, W.; Mills, A.M.; Huckestein, B.R.; Anderson, M.; Pangburn, M.M.; Lang, E.Y.; Mullet, S.J.; Chuan, B.W.; Guo, L.; et al. Empagliflozin Restores Cardiac Metabolic Flexibility in Diet-Induced Obese C57bl6/J Mice. Curr. Res. Physiol. 2022, 5, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.D.; Houng, A.K.; Gladysheva, I.P.; Fan, T.M.; Tripathi, R.; Reed, G.L.; Wang, D. Corin Overexpression Reduces Myocardial Infarct Size and Modulates Cardiomyocyte Apoptotic Cell Death. Int. J. Mol. Sci. 2020, 21, 3456. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.L.; Gladysheva, I.P.; Sullivan, R.D.; Mehta, R.M. Method of Personalized Treatment for Cardiomyopathy and Heart Failure and Associated Diseases by Measuring Edema and Cachexia/Sarcopenia. United States Patent Application Serial Number 17/313,904, 20210263120, 26 August 2021. [Google Scholar]
- Weech, A.A.; Goettsch, E.; Reeves, E.B. Nutritional Edema in the Dog: I. Development of Hypoproteinemia on a Diet Deficient in Protein. J. Exp. Med. 1935, 61, 299–317. [Google Scholar] [CrossRef]
- Kizhakke Puliyakote, A.S.; Holverda, S.; Sa, R.C.; Arai, T.J.; Theilmann, R.J.; Botros, L.; Bogaard, H.J.; Prisk, G.K.; Hopkins, S.R. Prone Positioning Redistributes Gravitational Stress in the Lung in Normal Conditions and in Simulations of Oedema. Exp. Physiol. 2022, 107, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, R.J. Evolution of Cardiovascular Baroreceptor Control. Biol. Rev. Camb. Philos. Soc. 1985, 60, 121–162. [Google Scholar] [CrossRef]
- Nakos, G.; Batistatou, A.; Galiatsou, E.; Konstanti, E.; Koulouras, V.; Kanavaros, P.; Doulis, A.; Kitsakos, A.; Karachaliou, A.; Lekka, M.E.; et al. Lung and ‘End Organ’ Injury Due to Mechanical Ventilation in Animals: Comparison between the Prone and Supine Positions. Crit. Care 2006, 10, R38. [Google Scholar] [CrossRef]
- Di Fusco, S.A.; Gronda, E.; Mocini, E.; Luca, F.; Bisceglia, I.; De Luca, L.; Caldarola, P.; Cipriani, M.; Corda, M.; De Nardo, A.; et al. Anmco Statement on the Use of Sodium-Glucose Cotransporter 2 Inhibitors in Patients with Heart Failure: A Practical Guide for a Streamlined Implementation. Eur. Heart J. Suppl. 2022, 24, C272–C277. [Google Scholar] [CrossRef]
- Pittampalli, S.; Upadyayula, S.; Mekala, H.M.; Lippmann, S. Risks Vs Benefits for Sglt2 Inhibitor Medications. Fed. Pract. 2018, 35, 45–48. [Google Scholar]
- Butler, J.; Usman, M.S.; Khan, M.S.; Greene, S.J.; Friede, T.; Vaduganathan, M.; Filippatos, G.; Coats, A.J.S.; Anker, S.D. Efficacy and Safety of Sglt2 Inhibitors in Heart Failure: Systematic Review and Meta-Analysis. ESC Heart Fail. 2020, 7, 3298–3309. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, R.; Ng, K.; Alkabbani, W.; Labib, Y.; Mourad, N.; Gamble, J.M. Adverse Events Associated with Sodium Glucose Co-Transporter 2 Inhibitors: An Overview of Quantitative Systematic Reviews. Ther. Adv. Drug Saf. 2021, 12, 2042098621989134. [Google Scholar] [CrossRef] [PubMed]


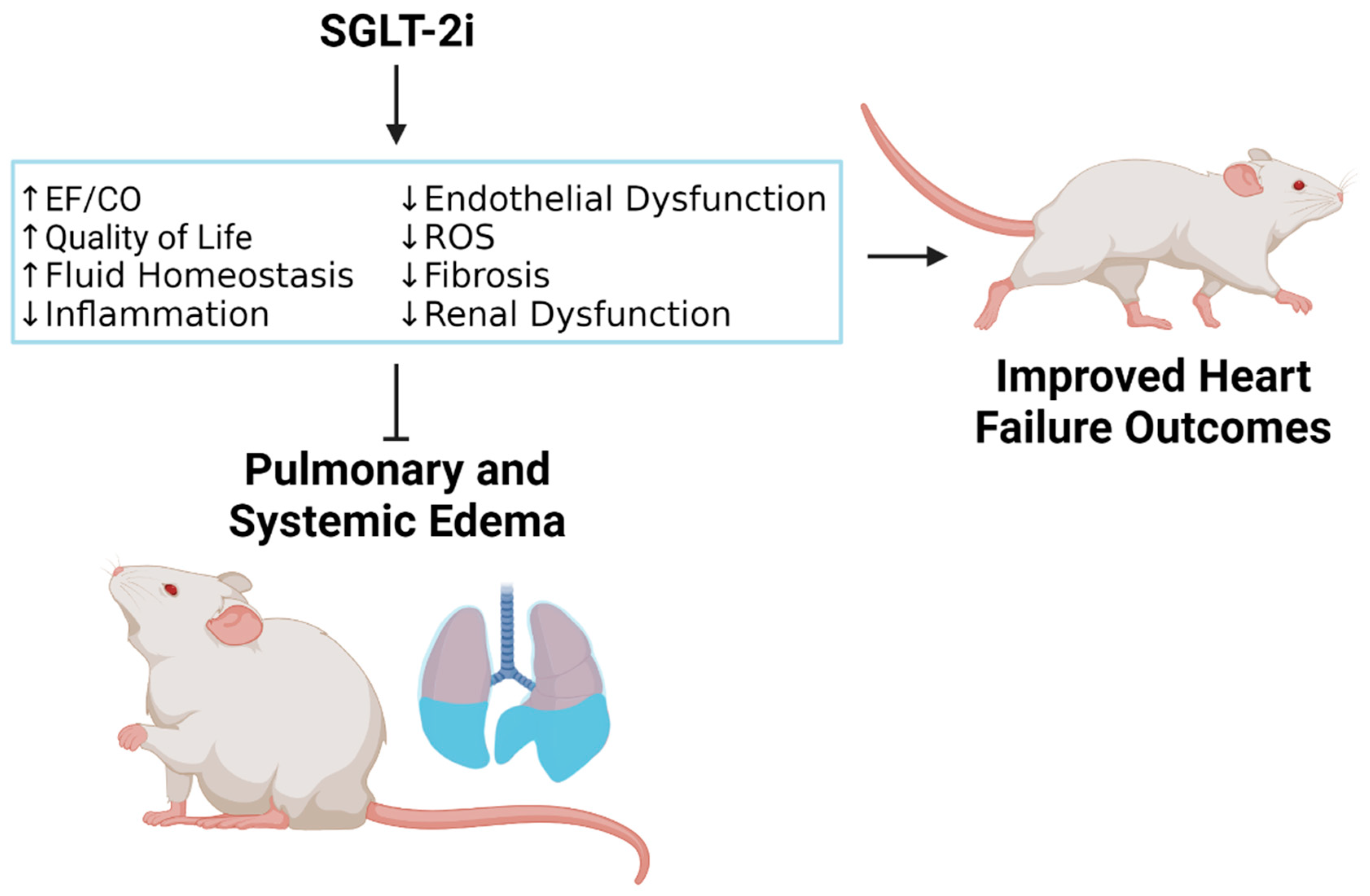{kind=link}
| Preclinical Models (species) | Drug Dosage Duration | Cardiac Function | Cardiac Remodeling | Inflammation, ROS, ED |
|---|---|---|---|---|
| Hypertensive HF model (rat) | Empagliflozin 20 mg/kg/day 12 weeks [70] |
|
| |
| Genetic HFrEF model (rat) | Empagliflozin 10 mg/kg/day 4 weeks |
|
| |
| Post-MI HFrEF model (rat; mouse, pig) | Empagliflozin 10mg orally 2 months [61] |
|
| |
| 20 mg/kg/day 6 weeks [75] |
|
| ||
| 30 mg/kg/day 2 weeks [72] |
|
| ||
| 10 mg/day 2 months [78] |
|
| ||
| 10 mg/kg/day 2 weeks [81] |
| |||
| Dapagliflozin 1 mg/kg/day 28 days [72, 74] | ||||
| 1.5 mg/kg/day 4 weeks [77] |
|
|
| |
| Canagliflozin 3 µg/kg 5 mins [76, 82] | ||||
| Ang II-induced cardiomyopathy (mouse) | Dapagliflozin 1.5 mg/kg/day 30 days [79] |
|
| |
| DOX-induced cardiomyopathy (mouse) | Empagliflozin (not provided) [81] |
|
| |
| LPS-induced cardiomyopathy (mouse) | Empagliflozin 5 mg/kg [86] |
|
| |
| TAC-induced HFrEF (mouse) | Empagliflozin 10 mg/kg/day 2 weeks post-surgery [73, 82] |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sullivan, R.D.; McCune, M.E.; Hernandez, M.; Reed, G.L.; Gladysheva, I.P. Suppression of Cardiogenic Edema with Sodium–Glucose Cotransporter-2 Inhibitors in Heart Failure with Reduced Ejection Fraction: Mechanisms and Insights from Pre-Clinical Studies. Biomedicines 2022, 10, 2016. https://doi.org/10.3390/biomedicines10082016
Sullivan RD, McCune ME, Hernandez M, Reed GL, Gladysheva IP. Suppression of Cardiogenic Edema with Sodium–Glucose Cotransporter-2 Inhibitors in Heart Failure with Reduced Ejection Fraction: Mechanisms and Insights from Pre-Clinical Studies. Biomedicines. 2022; 10(8):2016. https://doi.org/10.3390/biomedicines10082016
Chicago/Turabian StyleSullivan, Ryan D., Mariana E. McCune, Michelle Hernandez, Guy L. Reed, and Inna P. Gladysheva. 2022. "Suppression of Cardiogenic Edema with Sodium–Glucose Cotransporter-2 Inhibitors in Heart Failure with Reduced Ejection Fraction: Mechanisms and Insights from Pre-Clinical Studies" Biomedicines 10, no. 8: 2016. https://doi.org/10.3390/biomedicines10082016