
Autophagy: Guardian of Skin Barrier

 ,
,  ,
,  and
and
Abstract
:1. Introduction
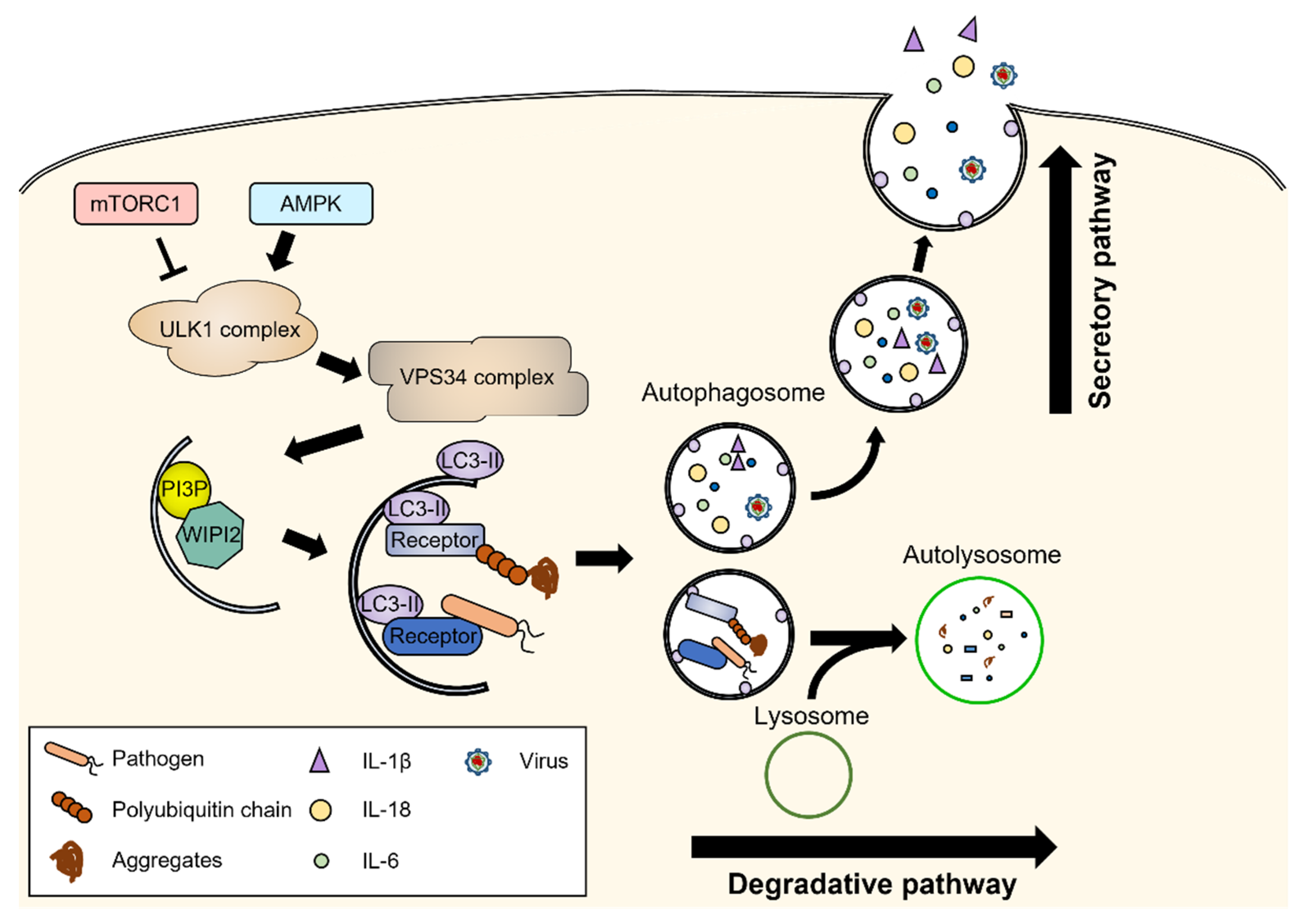
2. Molecular Machinery of Autophagy
3. Autophagy in Keratinocytes
4. Autophagy in Skin Immune Cells
4.1. Langerhans Cells
4.2. Macrophages
4.3. Mast Cells
4.4. Neutrophils
4.5. NK Cells
4.6. T Cells
5. Autophagy in Inflammatory Skin Diseases
5.1. Alopecia Areata
5.2. Psoriasis and Atopic Dermatitis
5.3. Keloids
6. Autophagy in Infectious Skin Diseases
7. Autophagy in Malignant Melanoma
8. Discussion
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Richmond, J.M.; Harris, J.E. Immunology and skin in health and disease. Cold Spring Harb. Perspect. Med. 2014, 4, a015339. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Kang, J.-H.; Lee, S. Autophagy in neurodegenerative diseases: A hunter for aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Zhang, X.-j.; Chen, S.; Huang, K.-x.; Le, W.-d. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Di Rienzo, M.; Romagnoli, A.; Antonioli, M.; Piacentini, M.; Fimia, G.M. TRIM proteins in autophagy: Selective sensors in cell damage and innate immune responses. Cell Death Differ. 2020, 27, 887–902. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12–5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Matoba, K.; Kotani, T.; Tsutsumi, A.; Tsuji, T.; Mori, T.; Noshiro, D.; Sugita, Y.; Nomura, N.; Iwata, S.; Ohsumi, Y. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 2020, 27, 1185–1193. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yu, L. Recent progress in autophagic lysosome reformation. Traffic 2017, 18, 358–361. [Google Scholar] [CrossRef]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Dupont, N.; Castillo, E.F.; Deretic, V. Secretory versus degradative autophagy: Unconventional secretion of inflammatory mediators. J. Innate Immun. 2013, 5, 471–479. [Google Scholar] [CrossRef]
- New, J.; Thomas, S.M. Autophagy-dependent secretion: Mechanism, factors secreted, and disease implications. Autophagy 2019, 15, 1682–1693. [Google Scholar] [CrossRef]
- Jeong, D.; Qomaladewi, N.P.; Lee, J.; Park, S.H.; Cho, J.Y. The role of autophagy in skin fibroblasts, keratinocytes, melanocytes, and epidermal stem cells. J. Investig. Dermatol. 2020, 140, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Akinduro, O.; Sully, K.; Patel, A.; Robinson, D.J.; Chikh, A.; McPhail, G.; Braun, K.M.; Philpott, M.P.; Harwood, C.A.; Byrne, C. Constitutive autophagy and nucleophagy during epidermal differentiation. J. Investig. Dermatol. 2016, 136, 1460–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteleon, C.L.; Agnihotri, T.; Dahal, A.; Liu, M.; Rebecca, V.W.; Beatty, G.L.; Amaravadi, R.K.; Ridky, T.W. Lysosomes support the degradation, signaling, and mitochondrial metabolism necessary for human epidermal differentiation. J. Investig. Dermatol. 2018, 138, 1945–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belleudi, F.; Purpura, V.; Caputo, S.; Torrisi, M.R. FGF7/KGF regulates autophagy in keratinocytes: A novel dual role in the induction of both assembly and turnover of autophagosomes. Autophagy 2014, 10, 803–821. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Dai, H.; Zhuang, B.; Chai, L.; Xie, Y.; Li, Y. Exogenous hydrogen sulfide promotes cell proliferation and differentiation by modulating autophagy in human keratinocytes. Biochem. Biophys. Res. Commun. 2016, 472, 437–443. [Google Scholar] [CrossRef]
- Mijaljica, D.; Devenish, R.J. Nucleophagy at a glance. J. Cell Sci. 2013, 126, 4325–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Narzt, M.S.; Nagelreiter, I.M.; Hohensinner, P.; Terlecki-Zaniewicz, L.; Tschachler, E.; Grillari, J.; Gruber, F. Autophagy deficient keratinocytes display increased DNA damage, senescence and aberrant lipid composition after oxidative stress in vitro and in vivo. Redox Biol. 2017, 11, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Sukseree, S.; Rossiter, H.; Mildner, M.; Pammer, J.; Buchberger, M.; Gruber, F.; Watanapokasin, R.; Tschachler, E.; Eckhart, L. Targeted deletion of Atg5 reveals differential roles of autophagy in keratin K5-expressing epithelia. Biochem. Biophys. Res. Commun. 2013, 430, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, N.; Ueno, T.; Takagi, A.; Trejo, J.A.O.; Haruna, K.; Suga, Y.; Komatsu, M.; Tanaka, K.; Ikeda, S. The significant role of autophagy in the granular layer in normal skin differentiation and hair growth. Arch. Dermatol. Res. 2015, 307, 159–169. [Google Scholar] [CrossRef]
- Noguchi, S.; Honda, S.; Saitoh, T.; Matsumura, H.; Nishimura, E.; Akira, S.; Shimizu, S. Beclin 1 regulates recycling endosome and is required for skin development in mice. Commun. Biol. 2019, 2, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, L.; Yang, S.; Cui, Y.-H.; He, Y.-Y. Keratinocyte autophagy enables the activation of keratinocytes and fibroblastsand facilitates wound healing. Autophagy 2021, 17, 2128–2143. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Liu, W.; Xue, P.; Ma, Y.; Zhang, L.; Zhu, B.; Qi, M.; Li, L.; Zhang, Y.; Wang, Q. Autophagy promotes MSC-mediated vascularization in cutaneous wound healing via regulation of VEGF secretion. Cell Death Dis. 2018, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 2016, 7, 10533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kim, S.K.; Park, H.; Lee, Y.J.; Park, S.H.; Lee, K.J.; Lee, D.G.; Kang, H.; Kim, J.E. Contribution of autophagy-Notch1-mediated NLRP3 inflammasome activation to chronic inflammation and fibrosis in keloid fibroblasts. Int. J. Mol. Sci. 2020, 21, 8050. [Google Scholar] [CrossRef] [PubMed]
- Doebel, T.; Voisin, B.; Nagao, K. Langerhans cells–the macrophage in dendritic cell clothing. Trends Immunol. 2017, 38, 817–828. [Google Scholar] [PubMed]
- Nguyen, A.V.; Soulika, A.M. The dynamics of the skin’s immune system. Int. J. Mol. Sci. 2019, 20, 1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, A.T.; Teles, R.M.; Liu, P.T.; Choi, A.; Legaspi, A.; Sarno, E.N.; Ochoa, M.T.; Parvatiyar, K.; Cheng, G.; Gilliet, M. Autophagy links antimicrobial activity with antigen presentation in Langerhans cells. JCI Insight 2019, 4, e126955. [Google Scholar] [CrossRef]
- Ribeiro, C.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Galle-Treger, L.; Hurrell, B.P.; Lewis, G.; Howard, E.; Jahani, P.S.; Banie, H.; Razani, B.; Soroosh, P.; Akbari, O. Autophagy is critical for group 2 innate lymphoid cell metabolic homeostasis and effector function. J. Allergy Clin. Immunol. 2020, 145, 502–517.e505. [Google Scholar] [CrossRef] [Green Version]
- Sil, P.; Suwanpradid, J.; Muse, G.; Gruzdev, A.; Liu, L.; Corcoran, D.L.; Willson, C.J.; Janardhan, K.; Grimm, S.; Myers, P. Noncanonical autophagy in dermal dendritic cells mediates immunosuppressive effects of UV exposure. J. Allergy Clin. Immunol. 2020, 145, 1389–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.O.; Manzanillo, P.S.; Cox, J.S. Extracellular, M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012, 150, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 2015, 17, 811–819. [Google Scholar]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Shi, C.-S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Lazova, R.; Klump, V.; Pawelek, J. Autophagy in cutaneous malignant melanoma. J. Cutan. Pathol. 2010, 37, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, N.G.; Chaplin, G. Human skin pigmentation as an adaptation to UV radiation. Proc. Natl. Acad. Sci. USA 2010, 107, 8962–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Hong, L.; Kempf, V.R.; Wakamatsu, K.; Ito, S.; Simon, J.D. Ion-exchange and adsorption of Fe (III) by Sepia melanin. Pigment Cell Res. 2004, 17, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Ushio, H.; Ueno, T.; Kojima, Y.; Komatsu, M.; Tanaka, S.; Yamamoto, A.; Ichimura, Y.; Ezaki, J.; Nishida, K.; Komazawa-Sakon, S. Crucial role for autophagy in degranulation of mast cells. J. Allergy Clin. Immunol. 2011, 127, 1267–1276.e1266. [Google Scholar] [CrossRef]
- Wang, B.; Li, X.; Li, M.; Geng, Y.; Wang, N.; Jin, Y.; Zhang, W.; Xu, K.; Wang, J.; Tao, L. Dopamine D3 receptor signaling alleviates mouse rheumatoid arthritis by promoting Toll-like receptor 4 degradation in mast cells. Cell Death Dis. 2022, 13, 240. [Google Scholar] [CrossRef]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Wei, Q.; Shin, J.N.; Fattah, E.A.; Bonilla, D.L.; Xiang, Q.; Eissa, N.T. Autophagy is required for neutrophil-mediated inflammation. Cell Rep. 2015, 12, 1731–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iula, L.; Keitelman, I.A.; Sabbione, F.; Fuentes, F.; Guzman, M.; Galletti, J.G.; Gerber, P.P.; Ostrowski, M.; Geffner, J.R.; Jancic, C.C. Autophagy mediates interleukin-1β secretion in human neutrophils. Front. Immunol. 2018, 9, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yost, C.C.; Cody, M.J.; Harris, E.S.; Thornton, N.L.; McInturff, A.M.; Martinez, M.L.; Chandler, N.B.; Rodesch, C.K.; Albertine, K.H.; Petti, C.A. Impaired neutrophil extracellular trap (NET) formation: A novel innate immune deficiency of human neonates. Blood J. Am. Soc. Hematol. 2009, 113, 6419–6427. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Nishinaka, Y.; Arai, T.; Morita, M.; Mizugishi, K.; Adachi, S.; Takaori-Kondo, A.; Watanabe, T.; Yamashita, K. Uric acid induces NADPH oxidase-independent neutrophil extracellular trap formation. Biochem. Biophys. Res. Commun. 2014, 443, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Simonson, T.J.; Jondle, C.N.; Mishra, B.B.; Sharma, J. Mincle-mediated neutrophil extracellular trap formation by regulation of autophagy. J. Infect. Dis. 2017, 215, 1040–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germic, N.; Stojkov, D.; Oberson, K.; Yousefi, S.; Simon, H.U. Neither eosinophils nor neutrophils require ATG 5-dependent autophagy for extracellular DNA trap formation. Immunology 2017, 152, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xia, P.; Huang, G.; Zhu, P.; Liu, J.; Ye, B.; Du, Y.; Fan, Z. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat. Commun. 2016, 7, 11023. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.E.; Geary, C.D.; Weizman, O.-E.; Geiger, T.L.; Rapp, M.; Dorn, G.W., II; Overholtzer, M.; Sun, J.C. Atg5 is essential for the development and survival of innate lymphocytes. Cell Rep. 2016, 15, 1910–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, V.; Petty, A.J.; Atwater, A.R.; Wolfe, S.A.; MacLeod, A.S. Skin Viral Infections: Host Antiviral Innate Immunity and Viral Immune Evasion. Front. Immunol. 2020, 11, 593901. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mgrditchian, T.; Arakelian, T.; Paggetti, J.; Noman, M.Z.; Viry, E.; Moussay, E.; Van Moer, K.; Kreis, S.; Guerin, C.; Buart, S. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, E9271–E9279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, R.A. Skin-resident T cells: The ups and downs of on site immunity. J. Investig. Dermatol. 2010, 130, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Texier, L.; Lineburg, K.E.; Cao, B.; McDonald-Hyman, C.; Leveque-El Mouttie, L.; Nicholls, J.; Melino, M.; Nalkurthi, B.C.; Alexander, K.A.; Teal, B. Autophagy-dependent regulatory T cells are critical for the control of graft-versus-host disease. JCI Insight 2016, 1, e86850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alissafi, T.; Banos, A.; Boon, L.; Sparwasser, T.; Ghigo, A.; Wing, K.; Vassilopoulos, D.; Boumpas, D.; Chavakis, T.; Cadwell, K. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J. Clin. Investig. 2017, 127, 2789–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Hu, C.; Wu, X.; Wang, S.; Zhang, A.; Chen, W.; Shen, Y.; Tan, R.; Wu, X.; Sun, Y. Roseotoxin B improves allergic contact dermatitis through a unique anti-inflammatory mechanism involving excessive activation of autophagy in activated T lymphocytes. J. Investig. Dermatol. 2016, 136, 1636–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchonwanit, P.; Kositkuljorn, C.; Pomsoong, C. Alopecia Areata: An Autoimmune Disease of Multiple Players. Immuno Targets Ther. 2021, 10, 299–312. [Google Scholar] [CrossRef]
- Stenn, K.S.; Paus, R. Controls of hair follicle cycling. Physiol. Rev. 2001, 81, 449–494. [Google Scholar] [CrossRef]
- Subramanya, R.D.; Coda, A.B.; Sinha, A.A. Transcriptional profiling in alopecia areata defines immune and cell cycle control related genes within disease-specific signatures. Genomics 2010, 96, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Parodi, C.; Hardman, J.A.; Allavena, G.; Marotta, R.; Catelani, T.; Bertolini, M.; Paus, R.; Grimaldi, B. Autophagy is essential for maintaining the growth of a human (mini-)organ: Evidence from scalp hair follicle organ culture. PLoS Biol. 2018, 16, e2002864. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Zheng, Y.; Yan, J.; Wang, J.; Liu, X.; Yin, G. BMP2-mediated PTEN enhancement promotes differentiation of hair follicle stem cells by inducing autophagy. Exp. Cell Res. 2019, 385, 111647. [Google Scholar] [CrossRef]
- Betz, R.C.; Petukhova, L.; Ripke, S.; Huang, H.; Menelaou, A.; Redler, S.; Becker, T.; Heilmann, S.; Yamany, T.; Duvic, M.; et al. Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nat. Commun. 2015, 6, 5966. [Google Scholar] [CrossRef] [PubMed]
- Petukhova, L.; Patel, A.V.; Rigo, R.K.; Bian, L.; Verbitsky, M.; Sanna-Cherchi, S.; Erjavec, S.O.; Abdelaziz, A.R.; Cerise, J.E.; Jabbari, A.; et al. Integrative analysis of rare copy number variants and gene expression data in alopecia areata implicates an aetiological role for autophagy. Exp. Dermatol. 2020, 29, 243–253. [Google Scholar] [CrossRef]
- Hardman, J.A.; Nicu, C.; Tai, C.; Harries, M.; Mironov, A.; Purba, T.S.; Paus, R. Does dysfunctional autophagy contribute to immune privilege collapse and alopecia areata pathogenesis? J. Dermatol. Sci. 2020, 100, 75–78. [Google Scholar] [CrossRef]
- Mosca, M.; Hong, J.; Hadeler, E.; Hakimi, M.; Liao, W.; Bhutani, T. The Role of IL-17 Cytokines in Psoriasis. Immuno Targets Ther. 2021, 10, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Bovenschen, H.J.; van de Kerkhof, P.C.; van Erp, P.E.; Woestenenk, R.; Joosten, I.; Koenen, H.J. Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J. Investig. Dermatol. 2011, 131, 1853–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, N.; Peters, A.T. Atopic dermatitis. Allergy Asthma Proc. 2019, 40, 433–436. [Google Scholar] [CrossRef]
- Amer, A.S.; Samaka, R.M.; Moftah, N.H. Beclin1 in psoriasis: An immunohistochemical study. Clin. Exp. Dermatol. 2021, 46, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Karabowicz, P.; Wronski, A.; Ostrowska, H.; Waeg, G.; Zarkovic, N.; Skrzydlewska, E. Reduced Proteasome Activity and Enhanced Autophagy in Blood Cells of Psoriatic Patients. Int. J. Mol. Sci. 2020, 21, 7608. [Google Scholar] [CrossRef] [PubMed]
- Klapan, K.; Frangez, Z.; Markov, N.; Yousefi, S.; Simon, D.; Simon, H.U. Evidence for Lysosomal Dysfunction within the Epidermis in Psoriasis and Atopic Dermatitis. J. Investig. Dermatol. 2021, 141, 2838–2848.e4. [Google Scholar] [CrossRef] [PubMed]
- Hailfinger, S.; Schulze-Osthoff, K. Impaired Autophagy in Psoriasis and Atopic Dermatitis: A New Therapeutic Target? J. Investig. Dermatol. 2021, 141, 2775–2777. [Google Scholar] [CrossRef]
- Hou, T.; Sun, X.; Zhu, J.; Hon, K.L.; Jiang, P.; Chu, I.M.; Tsang, M.S.; Lam, C.W.; Zeng, H.; Wong, C.K. IL-37 Ameliorating Allergic Inflammation in Atopic Dermatitis Through Regulating Microbiota and AMPK-mTOR Signaling Pathway-Modulated Autophagy Mechanism. Front. Immunol. 2020, 11, 752. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.H.; Lim, C.J.; Jung, J.; Kim, H.J.; Park, K.; Shin, J.W.; Huh, C.H.; Park, K.C.; Na, J.I. The effect of autophagy-enhancing peptide in moisturizer on atopic dermatitis: A randomized controlled trial. J. Dermatol. Treat. 2019, 30, 558–564. [Google Scholar] [CrossRef]
- Setta-Kaffetzi, N.; Simpson, M.A.; Navarini, A.A.; Patel, V.M.; Lu, H.-C.; Allen, M.H.; Duckworth, M.; Bachelez, H.; Burden, A.D.; Choon, S.-E. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am. J. Hum. Genet. 2014, 94, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahil, S.K.; Twelves, S.; Farkas, K.; Setta-Kaffetzi, N.; Burden, A.D.; Gach, J.E.; Irvine, A.D.; Képíró, L.; Mockenhaupt, M.; Oon, H.H. AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J. Investig. Dermatol. 2016, 136, 2251–2259. [Google Scholar] [CrossRef]
- Douroudis, K.; Kingo, K.; Traks, T.; Reimann, E.; Raud, K.; Ratsep, R.; Mossner, R.; Silm, H.; Vasar, E.; Koks, S. Polymorphisms in the ATG16L1 gene are associated with psoriasis vulgaris. Acta Derm. Venereol. 2012, 92, 85–87. [Google Scholar] [CrossRef] [Green Version]
- Betarbet, U.; Blalock, T.W. Keloids: A Review of Etiology, Prevention, and Treatment. J. Clin. Aesthetic Dermatol. 2020, 13, 33–43. [Google Scholar]
- Nakashima, M.; Chung, S.; Takahashi, A.; Kamatani, N.; Kawaguchi, T.; Tsunoda, T.; Hosono, N.; Kubo, M.; Nakamura, Y.; Zembutsu, H. A genome-wide association study identifies four susceptibility loci for keloid in the Japanese population. Nat. Genet. 2010, 42, 768–771. [Google Scholar] [CrossRef]
- Jeon, Y.R.; Roh, H.; Jung, J.H.; Ahn, H.M.; Lee, J.H.; Yun, C.O.; Lee, W.J. Antifibrotic Effects of High-Mobility Group Box 1 Protein Inhibitor (Glycyrrhizin) on Keloid Fibroblasts and Keloid Spheroids through Reduction of Autophagy and Induction of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuno, R.; Ito, Y.; Eid, N.; Otsuki, Y.; Kondo, Y.; Ueda, K. Upregulation of autophagy and glycolysis markers in keloid hypoxic-zone fibroblasts: Morphological characteristics and implications. Histol. Histopathol. 2018, 33, 1075–1087. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, P.; Qin, Z.; Yang, X.; Pan, B.; Nie, F.; Bi, H. Altered glucose metabolism and cell function in keloid fibroblasts under hypoxia. Redox Biol. 2021, 38, 101815. [Google Scholar] [CrossRef]
- Ong, C.T.; Khoo, Y.T.; Mukhopadhyay, A.; Do, D.V.; Lim, I.J.; Aalami, O.; Phan, T.T. mTOR as a potential therapeutic target for treatment of keloids and excessive scars. Exp. Dermatol. 2007, 16, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.; Blanchard, S.; MacLeod, A.S. Innate antimicrobial immunity in the skin: A protective barrier against bacteria, viruses, and fungi. PLoS Pathog. 2018, 14, e1007353. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Levine, B. Autophagy, antiviral immunity, and viral countermeasures. Biochim. Biophys. Acta 2009, 1793, 1478–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, J.M.; Mansour, M.K.; Acharya, M.; Sokolovska, A.; Timmons, A.K.; Lacy-Hulbert, A.; Vyas, J.M. The Role of Autophagy-Related Proteins in Candida albicans Infections. Pathogens 2016, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Reyes, A.; Farias, M.A.; Riedel, C.A.; Bueno, S.M.; Kalergis, A.M.; Gonzalez, P.A. Crosstalk between Epithelial Cells, Neurons and Immune Mediators in HSV-1 Skin Infection. Front. Immunol. 2021, 12, 662234. [Google Scholar] [CrossRef]
- O’Connell, D.; Liang, C. Autophagy interaction with herpes simplex virus type-1 infection. Autophagy 2016, 12, 451–459. [Google Scholar] [CrossRef]
- Kennedy, P.G.E.; Mogensen, T.H.; Cohrs, R.J. Recent Issues in Varicella-Zoster Virus Latency. Viruses 2021, 13, 2018. [Google Scholar] [CrossRef]
- Clebak, K.T.; Malone, M.A. Skin infections. Prim. Care Clin. Off. Pract. 2018, 45, 433–454. [Google Scholar] [CrossRef]
- Hansen, N.S.; Leth, S.; Nielsen, L.T. Toksisk shock-syndrom. Ugeskr. Laeger 2020, 182, V11190673. [Google Scholar]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Na-kata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef]
- Lin, C.Y.; Nozawa, T.; Minowa-Nozawa, A.; Toh, H.; Hikichi, M.; Iibushi, J.; Nakagawa, I. Autophagy Receptor Tollip Facilitates Bacterial Autophagy by Recruiting Galectin-7 in Response to Group A Streptococcus Infection. Front. Cell. Infect. Microbiol. 2020, 10, 583137. [Google Scholar] [CrossRef]
- Lu, S.L.; Kuo, C.F.; Chen, H.W.; Yang, Y.S.; Liu, C.C.; Anderson, R.; Wu, J.J.; Lin, Y.S. Insufficient Acidification of Autophagosomes Facilitates Group A Streptococcus Survival and Growth in Endothelial Cells. mBio 2015, 6, e01435-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, T.C.; Liebl, D.; Seymour, L.M.; Gillen, C.M.; Lim, J.Y.; Larock, C.N.; Davies, M.R.; Schulz, B.L.; Nizet, V.; Teasdale, R.D.; et al. The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe 2013, 14, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surewaard, B.G.; Deniset, J.F.; Zemp, F.J.; Amrein, M.; Otto, M.; Conly, J.; Omri, A.; Yates, R.M.; Kubes, P. Identification and treatment of the Staphylococcus aureus reservoir in vivo. J. Exp. Med. 2016, 213, 1141–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Fan, Z.; Han, H. Autophagy in Staphylococcus aureus Infection. Front. Cell. Infect. Microbiol. 2021, 11, 750222. [Google Scholar] [CrossRef]
- Franco-Paredes, C.; Marcos, L.A.; Henao-Martínez, A.F.; Rodríguez-Morales, A.J.; Villamil-Gómez, W.E.; Gotuzzo, E.; Bonifaz, A. Cutaneous mycobacterial infections. Clin. Microbiol. Rev. 2018, 32, e00069-18. [Google Scholar] [CrossRef] [Green Version]
- Griffith, D.E.; Aksamit, T.; Brown-Elliott, B.A.; Catanzaro, A.; Daley, C.; Gordin, F.; Holland, S.M.; Horsburgh, R.; Huitt, G.; Iademarco, M.F.; et al. An official ATS/IDSA statement: Diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med. 2007, 175, 367–416. [Google Scholar] [CrossRef] [Green Version]
- Silwal, P.; Kim, I.S.; Jo, E.K. Autophagy and Host Defense in Nontuberculous Mycobacterial Infection. Front. Immunol. 2021, 12, 728742. [Google Scholar] [CrossRef]
- DiGiacinto, D.; Bagley, J.; Goldsbury, A.M. The Value of Sonography in the Assessment of Skin Cancers and Their Metastases. J. Diagn. Med. Sonogr. 2016, 32, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Jung, T.; Haist, M.; Kuske, M.; Grabbe, S.; Bros, M. Immunomodulatory Properties of BRAF and MEK Inhibitors Used for Melanoma Therapy-Paradoxical ERK Activation and Beyond. Int. J. Mol. Sci. 2021, 22, 9890. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeldt, M.T.; Ryan, K.M. The multiple roles of autophagy in cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.H.; Piao, S.; Wang, D.; McAfee, Q.W.; Nathanson, K.L.; Lum, J.J.; Li, L.Z.; Amaravadi, R.K. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin. Cancer Res. 2011, 17, 3478–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmati, M.; Ebrahim, S.; Hashemi, S.; Motamedi, M.; Moosavi, M.A. New insights on the role of autophagy in the pathogenesis and treatment of melanoma. Mol. Biol. Rep. 2020, 47, 9021–9032. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, L.; Bodemeyer, V.; De Zio, D. The complex role of autophagy in melanoma evolution: New perspectives from mouse models. Front. Oncol. 2020, 9, 1506. [Google Scholar] [CrossRef] [Green Version]
- Foth, M.; McMahon, M. Autophagy Inhibition in BRAF-Driven Cancers. Cancers 2021, 13, 3498. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 2019, 10, 1693. [Google Scholar] [CrossRef] [Green Version]
- Mehnert, J.M.; Mitchell, T.C.; Huang, A.C.; Aleman, T.S.; Kim, B.J.; Schuchter, L.M.; Linette, G.P.; Karakousis, G.C.; Mitnick, S.; Giles, L.; et al. BAMM (BRAF Autophagy and MEK Inhibition in Melanoma): A Phase I/II Trial of Dabrafenib, Trametinib, and Hydroxychloroquine in Advanced BRAFV600-mutant Melanoma. Clin. Cancer Res. 2022, 28, 1098–1106. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Skin Diseases | Relevant Roles of Autophagy |
|---|---|
| Alopecia areata (AA) | Maintenance of the stage of growth (anagen) during the hair cycle and differentiation of hair follicle stem cells |
| Psoriasis (PS) and atopic dermatitis (AD) | Keratinocyte differentiation, skin barrier function, and inflammation prevention |
| Keloids | Notch1-NLRP3 inflammasome signaling activation and myofibroblast differentiation |
| Infectious disease | Pathogen elimination (anti-viral pathway) or viral replication and exit from the host (pro-viral pathway) |
| Melanoma | Tumor suppression by reducing harmful materials and limiting proliferation during initial stages and tumor promotion by supplying nutrients during advanced stages of tumorigenesis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.J.; Park, J.; Kim, S.K.; Park, H.; Kim, J.E.; Lee, S. Autophagy: Guardian of Skin Barrier. Biomedicines 2022, 10, 1817. https://doi.org/10.3390/biomedicines10081817
Kim HJ, Park J, Kim SK, Park H, Kim JE, Lee S. Autophagy: Guardian of Skin Barrier. Biomedicines. 2022; 10(8):1817. https://doi.org/10.3390/biomedicines10081817
Chicago/Turabian StyleKim, Hyun Jee, Jisoo Park, Sun Kyeon Kim, Hyungsun Park, Jung Eun Kim, and Seongju Lee. 2022. "Autophagy: Guardian of Skin Barrier" Biomedicines 10, no. 8: 1817. https://doi.org/10.3390/biomedicines10081817