
Mouse Models for Application in Colorectal Cancer: Understanding the Pathogenesis and Relevance to the Human Condition


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Carcinogen-Induced Mouse Model of Colorectal Cancer
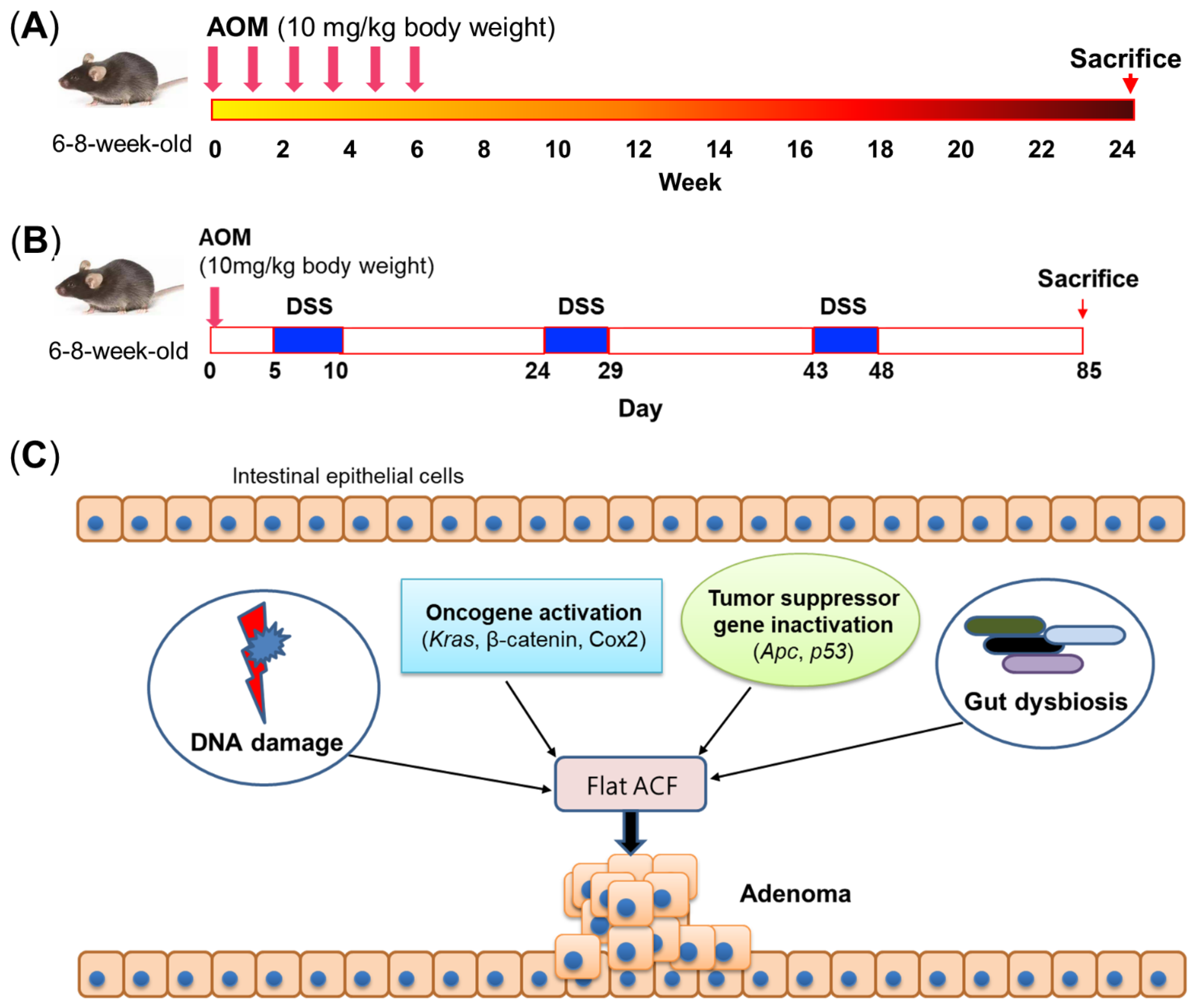
2.1. AOM- or AOM/DSS-Induced Mouse Model of Colorectal Cancer
2.2. Sensitivity to Carcinogen and Tumor Characteristics in Different Mouse Strains
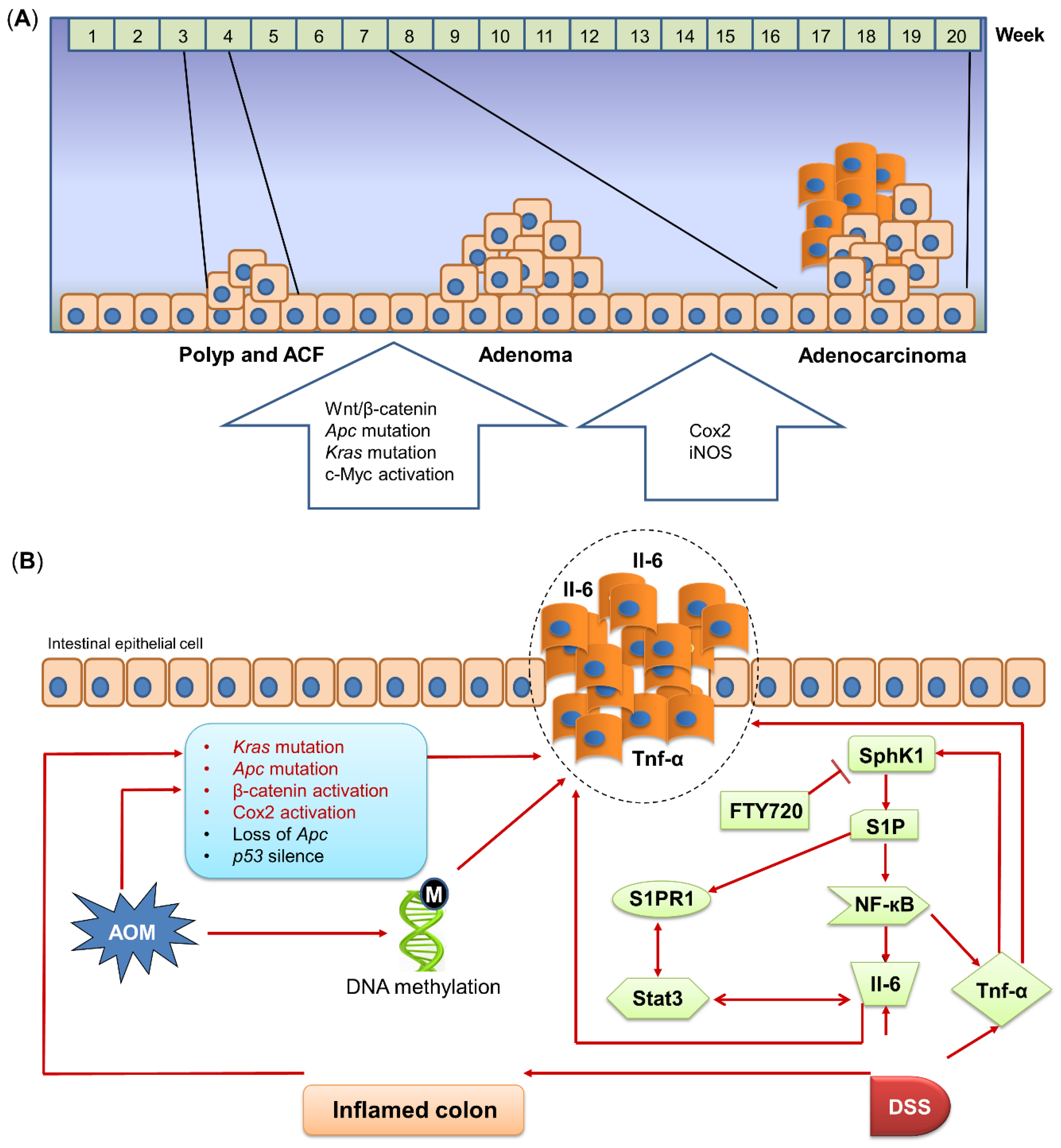
2.3. Modeling Different Stages of Colon Tumorigenesis
2.4. Molecular Mechanisms of Carcinogen-Induced Colon Tumorigenesis
2.5. Gut Microbiota Is Pivotal for Carcinogen-Induced Colon Tumorigenesis
3. Transgenic Mouse Model of Colorectal Cancer
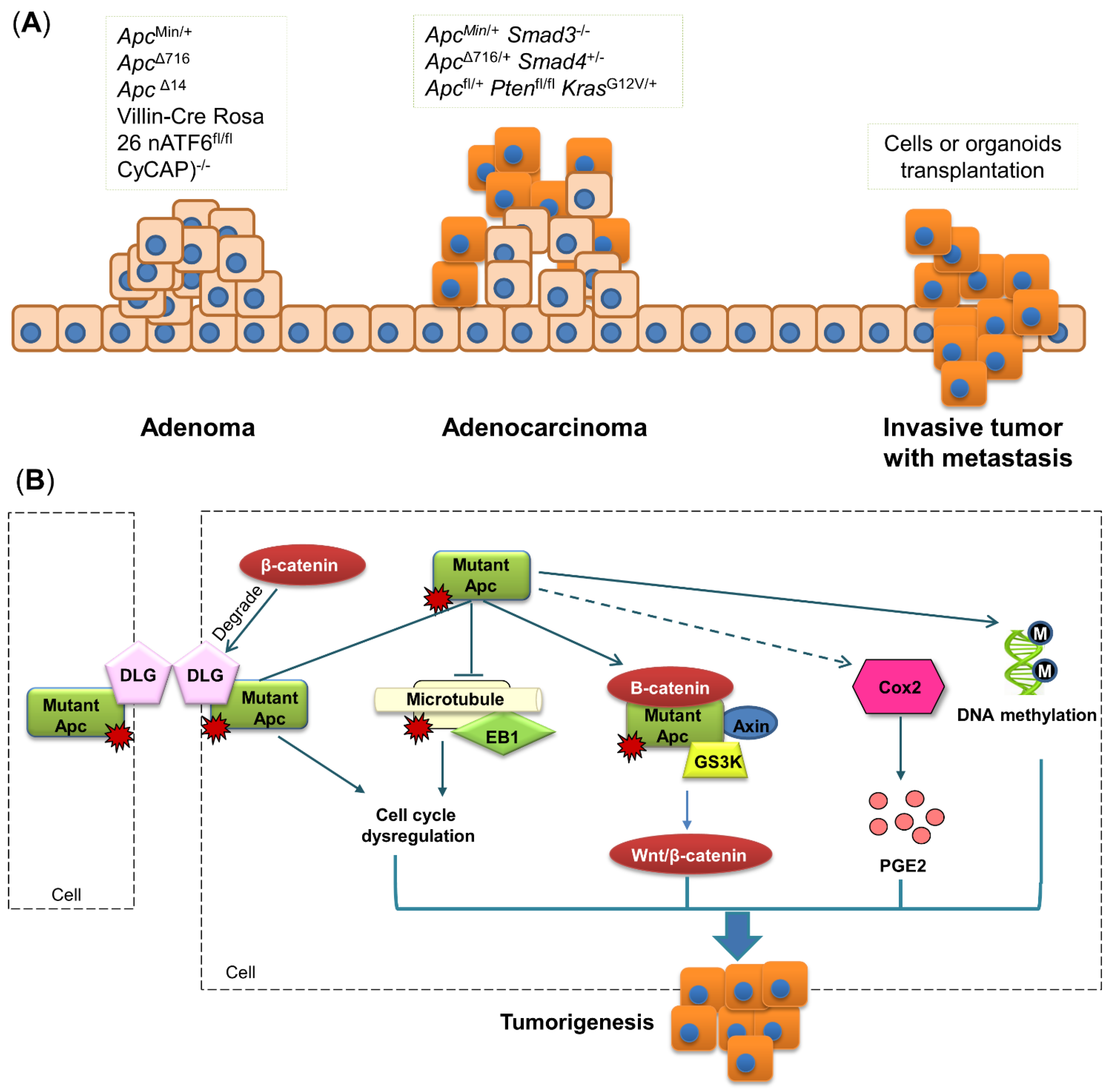
3.1. Apc Mutant Mouse Model of Colorectal Cancer
3.2. Molecular Mechanism of Apc Mutation-Induced Colon Tumorigenesis
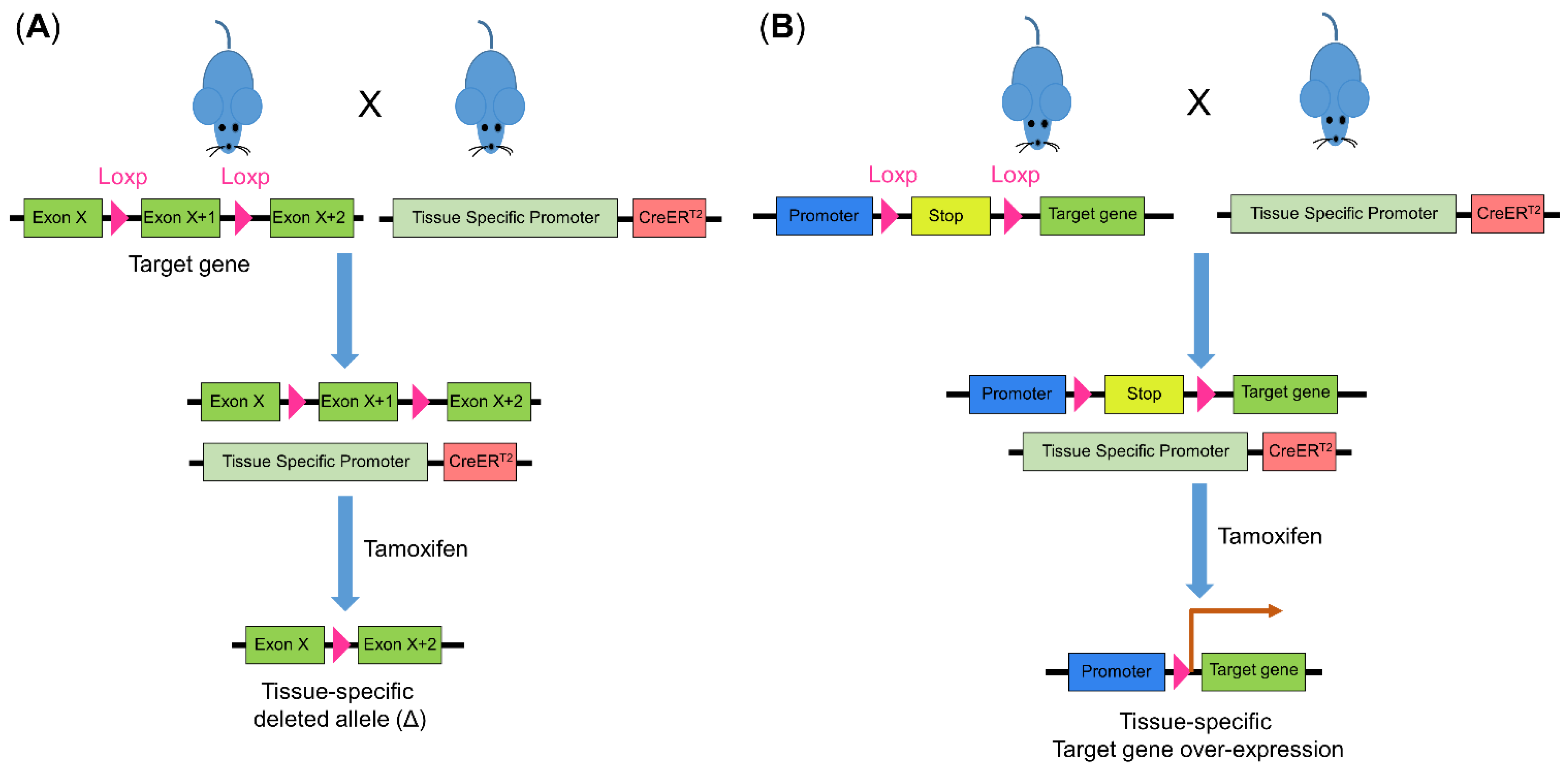
3.3. Cre-Loxp-Based Mouse Models of Colorectal Cancer
4. Colorectal Cancer Metastasis Mouse Models
4.1. Cell and Organoid Xenotransplantation
4.2. Transgenic Mouse Model of Metastasis
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, I.H.; Kim, J.S.; Kim, S.W.; Kim, J.G.; Oh, S.T.; Kang, W.K.; Lee, M.A. Different clinical characteristics in sporadic young-age onset colorectal cancer. Medicine 2016, 95, e4840. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grady, W.M. Genetic testing for high-risk colon cancer patients. Gastroenterology 2003, 124, 1574–1594. [Google Scholar] [CrossRef]
- Aoki, K.; Taketo, M.M. Adenomatous polyposis coli (APC): A multi-functional tumor suppressor gene. J. Cell Sci. 2007, 120, 3327–3335. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, D.W.; Giardina, C.; Tanaka, T. Mouse models for the study of colon carcinogenesis. Carcinogenesis 2009, 30, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Normanno, N.; Tejpar, S.; Morgillo, F.; De Luca, A.; Van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef]
- Chen, J.; Huang, X.F. The signal pathways in azoxymethane-induced colon cancer and preventive implications. Cancer Biol. Ther. 2009, 8, 1313–1317. [Google Scholar] [CrossRef] [Green Version]
- Stastna, M.; Janeckova, L.; Hrckulak, D.; Kriz, V.; Korinek, V. Human Colorectal Cancer from the Perspective of Mouse Models. Genes 2019, 10, 788. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003, 94, 965–973. [Google Scholar] [CrossRef]
- Diergaarde, B.; Braam, H.; Vasen, H.F.; Nagengast, F.M.; van Muijen, G.N.; Kok, F.J.; Kampman, E. Environmental factors and colorectal tumor risk in individuals with hereditary nonpolyposis colorectal cancer. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2007, 5, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, A.; Shank, R.C.; Delker, D.A.; Povey, A.; Cooper, D.P.; Rosenberg, D.W. Initial levels of azoxymethane-induced DNA methyl adducts are not predictive of tumor susceptibility in inbred mice. Toxicol. Appl. Pharmacol. 1998, 150, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Neufert, C.; Becker, C.; Neurath, M.F. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc. 2007, 2, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Guda, K.; Cui, H.; Garg, S.; Dong, M.; Nambiar, P.R.; Achenie, L.E.; Rosenberg, D.W. Multistage gene expression profiling in a differentially susceptible mouse colon cancer model. Cancer Lett. 2003, 191, 17–25. [Google Scholar] [CrossRef]
- Davies, G.R.; Rampton, D.S. Eicosanoids: Role in gastrointestinal inflammation and cancer. Eur. J. Gastroenterol. Hepatol. 1997, 9, 1033–1044. [Google Scholar] [CrossRef]
- Wilson, J.E.; Petrucelli, A.S.; Chen, L.; Koblansky, A.A.; Truax, A.D.; Oyama, Y.; Rogers, A.B.; Brickey, W.J.; Wang, Y.; Schneider, M.; et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat. Med. 2015, 21, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Gupta, J.; del Barco Barrantes, I.; Igea, A.; Sakellariou, S.; Pateras, I.S.; Gorgoulis, V.G.; Nebreda, A.R. Dual function of p38alpha MAPK in colon cancer: Suppression of colitis-associated tumor initiation but requirement for cancer cell survival. Cancer Cell 2014, 25, 484–500. [Google Scholar] [CrossRef] [Green Version]
- Bissahoyo, A.; Pearsall, R.S.; Hanlon, K.; Amann, V.; Hicks, D.; Godfrey, V.L.; Threadgill, D.W. Azoxymethane is a genetic background-dependent colorectal tumor initiator and promoter in mice: Effects of dose, route, and diet. Toxicol. Sci. 2005, 88, 340–345. [Google Scholar] [CrossRef] [Green Version]
- Nambiar, P.R.; Girnun, G.; Lillo, N.A.; Guda, K.; Whiteley, H.E.; Rosenberg, D.W. Preliminary analysis of azoxymethane induced colon tumors in inbred mice commonly used as transgenic/knockout progenitors. Int. J. Oncol. 2003, 22, 145–150. [Google Scholar] [CrossRef]
- Suzuki, R.; Kohno, H.; Sugie, S.; Nakagama, H.; Tanaka, T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis 2006, 27, 162–169. [Google Scholar] [CrossRef]
- Snider, A.J.; Bialkowska, A.B.; Ghaleb, A.M.; Yang, V.W.; Obeid, L.M.; Hannun, Y.A. Murine Model for Colitis-Associated Cancer of the Colon. Methods Mol. Biol. 2016, 1438, 245–254. [Google Scholar] [PubMed] [Green Version]
- Wargovich, M.J.; Brown, V.R.; Morris, J. Aberrant crypt foci: The case for inclusion as a biomarker for colon cancer. Cancers 2010, 2, 1705–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, J.E.; Loberg, E.M.; Olstorn, H.B.; Knutsen, H.; Steffensen, I.L.; Alexander, J. Flat dysplastic aberrant crypt foci are related to tumorigenesis in the colon of azoxymethane-treated rat. Cancer Res. 2005, 65, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Souris, J.S.; Zhang, H.J.; Dougherty, U.; Chen, N.T.; Waller, J.V.; Lo, L.W.; Hart, J.; Chen, C.T.; Bissonnette, M. A novel mouse model of sporadic colon cancer induced by combination of conditional Apc genes and chemical carcinogen in the absence of Cre recombinase. Carcinogenesis 2019, 40, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, M.; Massi, E.; Poeta, M.L.; Carotti, S.; Morini, S.; Cecchetelli, L.; Signori, E.; Fazio, V.M. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog. 2011, 10, 9. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Garrel, S.; Gerber, M.; Maitra, R.; Goel, S. Therapeutic Targets of KRAS in Colorectal Cancer. Cancers 2021, 13, 6233. [Google Scholar] [CrossRef]
- Vaughn, C.P.; Zobell, S.D.; Furtado, L.V.; Baker, C.L.; Samowitz, W.S. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer 2011, 50, 307–312. [Google Scholar] [CrossRef]
- Erdman, S.H.; Wu, H.D.; Hixson, L.J.; Ahnen, D.J.; Gerner, E.W. Assessment of mutations in Ki-ras and p53 in colon cancers from azoxymethane- and dimethylhydrazine-treated rats. Mol. Carcinog. 1997, 19, 137–144. [Google Scholar] [CrossRef]
- Pan, Q.; Lou, X.; Zhang, J.; Zhu, Y.; Li, F.; Shan, Q.; Chen, X.; Xie, Y.; Su, S.; Wei, H.; et al. Genomic variants in mouse model induced by azoxymethane and dextran sodium sulfate improperly mimic human colorectal cancer. Sci. Rep. 2017, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Saraggi, D.; Fassan, M.; Mescoli, C.; Scarpa, M.; Valeri, N.; Michielan, A.; D’Inca, R.; Rugge, M. The molecular landscape of colitis-associated carcinogenesis. Dig. Liver Dis. 2017, 49, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, H.; Watanabe, M.; Ushijima, T.; Toyota, M.; Imai, K.; Weisburger, J.H.; Sugimura, T.; Nagao, M. Specific 5′-GGGA-3′-->5′-GGA-3′ mutation of the Apc gene in rat colon tumors induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Proc. Natl. Acad. Sci. USA 1995, 92, 910–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guda, K.; Upender, M.B.; Belinsky, G.; Flynn, C.; Nakanishi, M.; Marino, J.N.; Ried, T.; Rosenberg, D.W. Carcinogen-induced colon tumors in mice are chromosomally stable and are characterized by low-level microsatellite instability. Oncogene 2004, 23, 3813–3821. [Google Scholar] [CrossRef] [Green Version]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Q.; Wang, M.; Zhao, S.; Ma, J.; Luo, N.; Li, N.; Li, Y.; Xu, G.; Li, J. Interferon-gamma and tumor necrosis factor-alpha disrupt epithelial barrier function by altering lipid composition in membrane microdomains of tight junction. Clin. Immunol. 2008, 126, 67–80. [Google Scholar] [CrossRef]
- Takahashi, M.; Wakabayashi, K. Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci. 2004, 95, 475–480. [Google Scholar] [CrossRef]
- Ishikawa, T.O.; Herschman, H.R. Tumor formation in a mouse model of colitis-associated colon cancer does not require COX-1 or COX-2 expression. Carcinogenesis 2010, 31, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Burtin, F.; Mullins, C.S.; Linnebacher, M. Mouse models of colorectal cancer: Past, present and future perspectives. World J. Gastroenterol. 2020, 26, 1394–1426. [Google Scholar] [CrossRef]
- Xu, R.; Jin, J.; Hu, W.; Sun, W.; Bielawski, J.; Szulc, Z.; Taha, T.; Obeid, L.M.; Mao, C. Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P. J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 1813–1825. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltzman, T.; Whittington, J.; Driggers, L.; Stephens, J.; Ahnen, D. AOM-induced mouse colon tumors do not express full-length APC protein. Carcinogenesis 1997, 18, 2435–2439. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, Y.; Takata, N.; Jinnai, T.; Morisawa, T.; Shiota, G.; Kawasaki, H.; Hasegawa, J. Sulindac and a cyclooxygenase-2 inhibitor, etodolac, increase APC mRNA in the colon of rats treated with azoxymethane. Gut 2000, 47, 812–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, Y.; Yashima, K.; Morisawa, T.; Ohishi, T.; Marumoto, A.; Sano, A.; Idobe-Fujii, Y.; Miura, N.; Shiota, G.; Murawaki, Y.; et al. Effects of long-term administration of sulindac on APC mRNA and apoptosis in colons of rats treated with azoxymethane. J. Cancer Res. Clin. Oncol. 2002, 128, 589–595. [Google Scholar] [PubMed]
- Mladenova, D.; Daniel, J.J.; Dahlstrom, J.E.; Bean, E.; Gupta, R.; Pickford, R.; Currey, N.; Musgrove, E.A.; Kohonen-Corish, M.R. The NSAID sulindac is chemopreventive in the mouse distal colon but carcinogenic in the proximal colon. Gut 2011, 60, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Nambiar, P.R.; Giardina, C.; Guda, K.; Aizu, W.; Raja, R.; Rosenberg, D.W. Role of the alternating reading frame (P19)-p53 pathway in an in vivo murine colon tumor model. Cancer Res. 2002, 62, 3667–3674. [Google Scholar]
- Entezari Heravi, R.; Hadizadeh, F.; Sankian, M.; Tavakol Afshari, J.; Taghdisi, S.M.; Jafarian, H.; Behravan, J. Novel selective Cox-2 inhibitors induce apoptosis in Caco-2 colorectal carcinoma cell line. Eur. J. Pharm. Sci. 2011, 44, 479–486. [Google Scholar] [CrossRef]
- Shao, J.; Fujiwara, T.; Kadowaki, Y.; Fukazawa, T.; Waku, T.; Itoshima, T.; Yamatsuji, T.; Nishizaki, M.; Roth, J.A.; Tanaka, N. Overexpression of the wild-type p53 gene inhibits NF-kappaB activity and synergizes with aspirin to induce apoptosis in human colon cancer cells. Oncogene 2000, 19, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Li, H.; Tian, G.; Li, S. Dynamic microbe and molecule networks in a mouse model of colitis-associated colorectal cancer. Sci. Rep. 2014, 4, 4985. [Google Scholar] [CrossRef] [Green Version]
- Uronis, J.M.; Muhlbauer, M.; Herfarth, H.H.; Rubinas, T.C.; Jones, G.S.; Jobin, C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS ONE 2009, 4, e6026. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wei, H.; Zhou, Y.; Szeto, C.H.; Li, C.; Lin, Y.; Coker, O.O.; Lau, H.C.H.; Chan, A.W.H.; Sung, J.J.Y.; et al. High-Fat Diet Promotes Colorectal Tumorigenesis Through Modulating Gut Microbiota and Metabolites. Gastroenterology 2022, 162, 135–149.e2. [Google Scholar] [CrossRef] [PubMed]
- Coleman, O.I.; Lobner, E.M.; Bierwirth, S.; Sorbie, A.; Waldschmitt, N.; Rath, E.; Berger, E.; Lagkouvardos, I.; Clavel, T.; McCoy, K.D.; et al. Activated ATF6 Induces Intestinal Dysbiosis and Innate Immune Response to Promote Colorectal Tumorigenesis. Gastroenterology 2018, 155, 1539–1552.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhang, X.; Xu, H.; Li, S.; Lau, H.C.; Chen, Q.; Zhang, B.; Zhao, L.; Chen, H.; Sung, J.J.; et al. Microbial Community Heterogeneity Within Colorectal Neoplasia and its Correlation With Colorectal Carcinogenesis. Gastroenterology 2021, 160, 2395–2408. [Google Scholar] [CrossRef] [PubMed]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Jackstadt, R.; Sansom, O.J. Mouse models of intestinal cancer. J. Pathol. 2016, 238, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Moser, A.R.; Luongo, C.; Gould, K.A.; McNeley, M.K.; Shoemaker, A.R.; Dove, W.F. ApcMin: A mouse model for intestinal and mammary tumorigenesis. Eur. J. Cancer 1995, 31A, 1061–1064. [Google Scholar] [CrossRef]
- Kwong, L.N.; Dove, W.F. APC and its modifiers in colon cancer. Adv. Exp. Med. Biol. 2009, 656, 85–106. [Google Scholar]
- Zhou, X.; Geng, L.; Wang, D.; Yi, H.; Talmon, G.; Wang, J. R-Spondin1/LGR5 Activates TGFbeta Signaling and Suppresses Colon Cancer Metastasis. Cancer Res. 2017, 77, 6589–6602. [Google Scholar] [CrossRef] [Green Version]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998, 92, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Sodir, N.M.; Chen, X.; Park, R.; Nickel, A.E.; Conti, P.S.; Moats, R.; Bading, J.R.; Shibata, D.; Laird, P.W. Smad3 deficiency promotes tumorigenesis in the distal colon of ApcMin/+ mice. Cancer Res. 2006, 66, 8430–8438. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef]
- Sansom, O.J.; Meniel, V.; Wilkins, J.A.; Cole, A.M.; Oien, K.A.; Marsh, V.; Jamieson, T.J.; Guerra, C.; Ashton, G.H.; Barbacid, M.; et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 14122–14127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemer, B.; Warren, R.D.; Barrett, M.P.; Cisek, K.; Das, A.; Jeffery, I.B.; Hurley, E.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. The oral microbiota in colorectal cancer is distinctive and predictive. Gut 2018, 67, 1454–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomkovich, S.; Yang, Y.; Winglee, K.; Gauthier, J.; Muhlbauer, M.; Sun, X.; Mohamadzadeh, M.; Liu, X.; Martin, P.; Wang, G.P.; et al. Locoregional Effects of Microbiota in a Preclinical Model of Colon Carcinogenesis. Cancer Res. 2017, 77, 2620–2632. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Sarnaik, A.A.; Bonney, T.M.; Keirsey, J.; Combs, K.A.; Steigerwald, K.; Acharya, S.; Behbehani, G.K.; Barton, M.C.; Lowy, A.M.; et al. The APC tumor suppressor inhibits DNA replication by directly binding to DNA via its carboxyl terminus. Gastroenterology 2008, 135, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Kimelman, D.; Xu, W. beta-catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Nakamura, M.; Zhou, X.Z.; Lu, K.P. Critical role for the EB1 and APC interaction in the regulation of microtubule polymerization. Curr. Biol. 2001, 11, 1062–1067. [Google Scholar] [CrossRef] [Green Version]
- Stypula-Cyrus, Y.; Mutyal, N.N.; Dela Cruz, M.; Kunte, D.P.; Radosevich, A.J.; Wali, R.; Roy, H.K.; Backman, V. End-binding protein 1 (EB1) up-regulation is an early event in colorectal carcinogenesis. FEBS Lett. 2014, 588, 829–835. [Google Scholar] [CrossRef] [Green Version]
- Ishidate, T.; Matsumine, A.; Toyoshima, K.; Akiyama, T. The APC-hDLG complex negatively regulates cell cycle progression from the G0/G1 to S phase. Oncogene 2000, 19, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbaiah, V.K.; Narayan, N.; Massimi, P.; Banks, L. Regulation of the DLG tumor suppressor by beta-catenin. Int. J. Cancer 2012, 131, 2223–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Lee, J.H.; Shu, L.; Huang, Y.; Li, W.; Zhang, C.; Yang, A.Y.; Boyanapalli, S.S.; Perekatt, A.; Hart, R.P.; et al. Association of aberrant DNA methylation in Apc(min/+) mice with the epithelial-mesenchymal transition and Wnt/beta-catenin pathways: Genome-wide analysis using MeDIP-seq. Cell Biosci. 2015, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, K.; Sarkar, S.; Broadbent, T.J.; Voas, M.; Grossmann, K.F.; Nadauld, L.D.; Dehghanizadeh, S.; Hagos, F.T.; Li, Y.; Toth, R.K.; et al. DNA demethylase activity maintains intestinal cells in an undifferentiated state following loss of APC. Cell 2010, 142, 930–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, G.; Schafer, A.; Marhold, J.; Stach, D.; Swaminathan, S.K.; Handa, V.; Doderlein, G.; Maltry, N.; Wu, W.; Lyko, F.; et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 2007, 445, 671–675. [Google Scholar] [CrossRef]
- Woo, V.; Eshleman, E.M.; Hashimoto-Hill, S.; Whitt, J.; Wu, S.E.; Engleman, L.; Rice, T.; Karns, R.; Qualls, J.E.; Haslam, D.B.; et al. Commensal segmented filamentous bacteria-derived retinoic acid primes host defense to intestinal infection. Cell Host Microbe 2021, 29, 1744–1756.e5. [Google Scholar] [CrossRef]
- Son, J.S.; Khair, S.; Pettet, D.W., 3rd; Ouyang, N.; Tian, X.; Zhang, Y.; Zhu, W.; Mackenzie, G.G.; Robertson, C.E.; Ir, D.; et al. Altered Interactions between the Gut Microbiome and Colonic Mucosa Precede Polyposis in APCMin/+ Mice. PLoS ONE 2015, 10, e0127985. [Google Scholar] [CrossRef]
- Jager, R.; Maurer, J.; Jacob, A.; Schorle, H. Cell type-specific conditional regulation of the c-myc proto-oncogene by combining Cre/loxP recombination and tamoxifen-mediated activation. Genesis 2004, 38, 145–150. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Hanaoka, M.; Ishikawa, T.; Ishiguro, M.; Tokura, M.; Yamauchi, S.; Kikuchi, A.; Uetake, H.; Yasuno, M.; Kawano, T. Expression of ATF6 as a marker of pre-cancerous atypical change in ulcerative colitis-associated colorectal cancer: A potential role in the management of dysplasia. J. Gastroenterol. 2018, 53, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Trahey, M.; Weissman, I.L. Cyclophilin C-associated protein: A normal secreted glycoprotein that down-modulates endotoxin and proinflammatory responses in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 3006–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torlakovic, E.E.; Keeler, V.; Wang, C.; Lim, H.J.; Lining, L.A.; Laferte, S. Cyclophilin C-associated protein (CyCAP) knock-out mice spontaneously develop colonic mucosal hyperplasia and exaggerated tumorigenesis after treatment with carcinogen azoxymethane. BMC Cancer 2009, 9, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Davis, C.; Ryan, J.; Janney, C.; Pena, M.M. Development and characterization of a reliable mouse model of colorectal cancer metastasis to the liver. Clin. Exp. Metastasis 2013, 30, 903–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Zhang, Y.; Wong, C.C.; Zhang, J.; Dong, Y.; Li, X.; Kang, W.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. RNF6 Promotes Colorectal Cancer by Activating the Wnt/beta-Catenin Pathway via Ubiquitination of TLE3. Cancer Res. 2018, 78, 1958–1971. [Google Scholar] [CrossRef] [Green Version]
- Moret, R.; Hellmers, L.; Zhang, X.; Gills, J.; Hite, N.; Klinger, A.; Maresh, G.A.; Canter, D.; Bardot, S.; Margolin, D.A.; et al. Patient-derived Orthotopic Xenograft Models for Human Urothelial Cell Carcinoma and Colorectal Cancer Tumor Growth and Spontaneous Metastasis. J. Vis. Exp. 2019, 147, e5922. [Google Scholar] [CrossRef]
- Zhu, G.; Zhao, M.; Han, Q.; Tan, Y.; Sun, Y.; Bouvet, M.; Clary, B.; Singh, S.R.; Ye, J.; Hoffman, R.M. Temozolomide and Pazopanib Combined with FOLFOX Regressed a Primary Colorectal Cancer in a Patient-derived Orthotopic Xenograft Mouse Model. Transl. Oncol. 2020, 13, 100739. [Google Scholar] [CrossRef]
- Kwon, J.; Oh, S.; Park, M.; Kong, J.S.; Lee, S.; Lee, H.; Kim, Y.; Kang, K.T.; Shin, U.S.; Jung, J. Advanced Xenograft Model with Cotransplantation of Patient-Derived Organoids and Endothelial Colony-Forming Cells for Precision Medicine. J. Oncol. 2021, 2021, 9994535. [Google Scholar] [CrossRef]
- Kang, K.T.; Lin, R.Z.; Kuppermann, D.; Melero-Martin, J.M.; Bischoff, J. Endothelial colony forming cells and mesenchymal progenitor cells form blood vessels and increase blood flow in ischemic muscle. Sci. Rep. 2017, 7, 770. [Google Scholar] [CrossRef]
- Whisner, C.M.; Athena Aktipis, C. The Role of the Microbiome in Cancer Initiation and Progression: How Microbes and Cancer Cells Utilize Excess Energy and Promote One Another’s Growth. Curr. Nutr. Rep. 2019, 8, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Rad, R.; Cadinanos, J.; Rad, L.; Varela, I.; Strong, A.; Kriegl, L.; Constantino-Casas, F.; Eser, S.; Hieber, M.; Seidler, B.; et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell 2013, 24, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Davies, E.J.; Marsh Durban, V.; Meniel, V.; Williams, G.T.; Clarke, A.R. PTEN loss and KRAS activation leads to the formation of serrated adenomas and metastatic carcinoma in the mouse intestine. J. Pathol. 2014, 233, 27–38. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Lau, H.C.-H.; Zhang, X.; Yu, J. Mouse Models for Application in Colorectal Cancer: Understanding the Pathogenesis and Relevance to the Human Condition. Biomedicines 2022, 10, 1710. https://doi.org/10.3390/biomedicines10071710
Li C, Lau HC-H, Zhang X, Yu J. Mouse Models for Application in Colorectal Cancer: Understanding the Pathogenesis and Relevance to the Human Condition. Biomedicines. 2022; 10(7):1710. https://doi.org/10.3390/biomedicines10071710
Chicago/Turabian StyleLi, Chuangen, Harry Cheuk-Hay Lau, Xiang Zhang, and Jun Yu. 2022. "Mouse Models for Application in Colorectal Cancer: Understanding the Pathogenesis and Relevance to the Human Condition" Biomedicines 10, no. 7: 1710. https://doi.org/10.3390/biomedicines10071710