1. Introduction
Cardiovascular and cerebrovascular diseases are the leading causes of death worldwide and account for 26% of the total deaths in 2019 [
1]. Thrombosis is a major underlying pathology for these diseases, such as acute coronary syndrome (ACS), stroke, and venous thromboembolism (VTE), with the latter including deep vein thrombosis (DVT) and pulmonary embolism (PE) [
2]. Anticoagulants are the major class of therapeutic agents for thrombosis treatment and prophylaxis. The goal of anticoagulation therapy is to prevent the pathological formation of blood clots during thrombosis, which can result in the occlusion of arteries and veins [
3]. However, a major limitation of anticoagulant therapy is the risk of excessive bleeding [
4]. An ideal anticoagulation therapy should inhibit the formation of unwanted blood clots with minimum perturbation to hemostasis by having the best balance between antithrombotic efficacy and bleeding risk (a high efficacy-to-safety index) [
5].
The activated coagulation factor XI (FXIa) is a potential therapeutic target for anticoagulation with a reduced risk of excessive bleeding [
4]. Contemporary views on blood coagulation hold that the amplification of coagulation occurs mainly through the intrinsic pathway. An initial amount of thrombin is generated through the tissue factor and activated factor VII (FVIIa) complex via the extrinsic pathway. Subsequently, the generated thrombin activates FXI, factor VIII, and factor V to self-amplify the coagulation signals through the intrinsic pathway [
6]. While the inhibition of FXIa shuts down the intrinsic pathway, thrombin generation could still be maintained by the extrinsic pathway to a certain extent. As a result, the inhibition of FXIa tapers but does not completely abolish thrombin generation [
6]. In the treatment of thrombosis, FXIa inhibition can moderate the pathological progression of coagulation and still preserve sufficient potential for hemostasis in response to vascular injury. Therefore, it is likely that FXIa inhibition can provide a favorable overall antithrombotic–hemostasis balance, which would result in minimal bleeding risk [
6].
The hypothesis that FXIa is a valuable target for the development of anticoagulants with minimal risk of bleeding is strongly supported by epidemiological, animal, and clinical data [
6]. One example is the observation that hemophilia C patients rarely have major bleeding complications despite having FXI deficiency [
7]. Patients with severe deficiency in FXI are also found to have a reduced risk of DVT and ischemic stroke [
8,
9]. In addition, FXI knockout mice have reduced thrombosis but do not suffer from excessive bleeding [
10,
11,
12,
13]. Similar observations could be made in the pharmacological inhibition of FXIa in other animal models such as rats, rabbits, and baboons [
14,
15,
16]. A few therapeutic candidates targeting FXI/FXIa have recently completed phase 2 clinical trials [
4]. IONIS-FXI
RX (former name: ISIS-416858), an antisense oligonucleotide that targets FXI mRNA, has shown a statistically significant reduction in the incidence of VTE in patients receiving elective knee arthroplasty as compared to low-molecular-weight heparin (LMWH) enoxaparin [
17]. The incidence of major or clinically relevant bleeding in an IONIS-FXI
RX-treated group is numerically lower than enoxaparin [
17]. In the FOXTROT randomized clinical trial, patients taking pre- or postoperative Osocimab (human IgG1 monoclonal antibody that inhibits FXIa) for knee arthroplasty surgery were similarly reported to have numerically lower major or clinically relevant non-major bleeding as compared to enoxaparin. Patients taking a postoperative dose (0.6 mg/kg, 1.2 mg/kg, and 1.8 mg/kg) of Osocimab were observed to have a similar incidence of VTE with enoxaparin. On the other hand, patients taking a preoperative dose of 1.8 mg/kg were found to have a statistically significant lower incidence of VTE than an enoxaparin-treated group [
18]. The recent AXIOMATIC-TKR clinical trial of Milvexian (an orally available FXIa inhibitor) has also reported that the postoperative inhibition of FXIa by Milevexian could prevent venous thromboembolism in patients undergoing knee arthroplasty without major bleeding [
19]. Thus, FXIa inhibitors can potentially be developed into an anticoagulant drug, with minimal bleeding complications.
The Kunitz-type protease inhibitor is an important family of serine protease inhibitors that is found in most living organisms [
20]. A single Kunitz domain typically comprises of approximately 60 amino acid residues with alpha/beta folds constrained by three disulfide bonds [
20]. They are functionally diverse, with activities such as the inhibition of ion channels, neutrophil elastase (anti-inflammatory), and blood coagulation factors [
21,
22]. Kunitz-type domains are also found in humans. For example, the tissue factor pathway inhibitor targets the extrinsic tenase complex components, (activated) factor X (FX/FXa) and FVII/FVIIa [
23]. The light chain of the inter-alpha-trypsin inhibitor (bikunin) has been reported to target kallikrein (involved in endothelium permeability and airway inflammation), granzyme (involved in responses of natural killer cells and cytotoxic T-cells), and plasmin (involved in the dissolution of fibrin clots and the activation of matrix metalloproteases) [
24,
25,
26]. The Kunitz domain of protease nexin 2 (PN2KPI) has been reported to be a potent inhibitor of FXIa, which suggests its possible role in the inhibition of FXI-dependent thrombosis in vivo [
27]. The Kunitz-type domain is also a promising miniprotein scaffold being explored for the engineering of non-antibody-based protein therapeutics. Miniproteins are a diverse group of protein scaffolds (1 to 10 kDa) that have good thermal, chemical, and biological stability as well as good tissue penetration capability [
28,
29]. For example, ecallantide is an approved therapeutic for the treatment of hereditary angioedema and is composed of a Kunitz domain [
28,
29].
Previously, we reported the discovery and design of a novel FXIa inhibitor, rFasxiator
N17R,L19E. Fasxiator is a 7 kDa Kunitz-type protease inhibitor that was isolated from the venom of a banded krait snake (
Bungarus fasciatus) [
30]. It inhibits FXIa with moderate potency (IC
50 = 1.5 µM) while also inhibiting plasmin, FXa, and FVIIa. Through the systematic mutations of P1 and P2ʹ residues, we engineered a suitable lead for anticoagulant therapeutic development, with enhanced potency and selectivity. The variant, rFasxiator
N17R,L19E, has >1000-fold improved inhibitory potency towards FXIa. rFasxiator
N17R,L19E is also at least 170-fold more selective towards FXIa as compared to other blood coagulation serine proteases. In addition, the preliminary assessment of in vivo activity by rFasxiator
N17R,L19E has showed some protection against thrombosis in a FeCl
3-induced carotid artery thrombosis (CAT) model. With a fixed amount of inhibitor (0.3 mg/mice) injected through the tail vein, the occlusion time of the carotid artery was extended by approximately 3-fold. In addition to its therapeutic potential, the inhibitor has also been successfully used in a bioassay to quantify FXIa in plasma [
31].
Here, we further characterize rFasxiatorN17R,L19E activity in vivo to understand its potential as a therapeutic candidate. First, serial blood draws were performed on rats injected with rFasxiatorN17R, L19E in order to obtain plasma for coagulation assays that would examine the time course of in vivo anticoagulant effects. Next, we performed dose–response experiments in rodents—namely the FeCl3-induced CAT model in rats and the inferior vena cava (IVC) ligation model in mice—to represent arterial and venous thrombosis, respectively. Unfractionated heparin (UFH) and LMWH enoxaparin were used in head-to-head comparisons of arterial and venous thrombosis through their respective clinically recommended dosages and routes of administration. We were able to demonstrate that rFasxiatorN17R, L19E can achieve a therapeutic antithrombotic level with minimal bleeding, and it is a promising molecule for the development of an FXIa-specific anticoagulant or bioassay reagent.
2. Materials and Methods
2.1. Study Approvals
All animal studies were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at the National University of Singapore (NUS), under protocols R16-0008, R19-1040, and R20-0421.
2.2. Expression and Refolding of rFasxiatorN17R, L19E
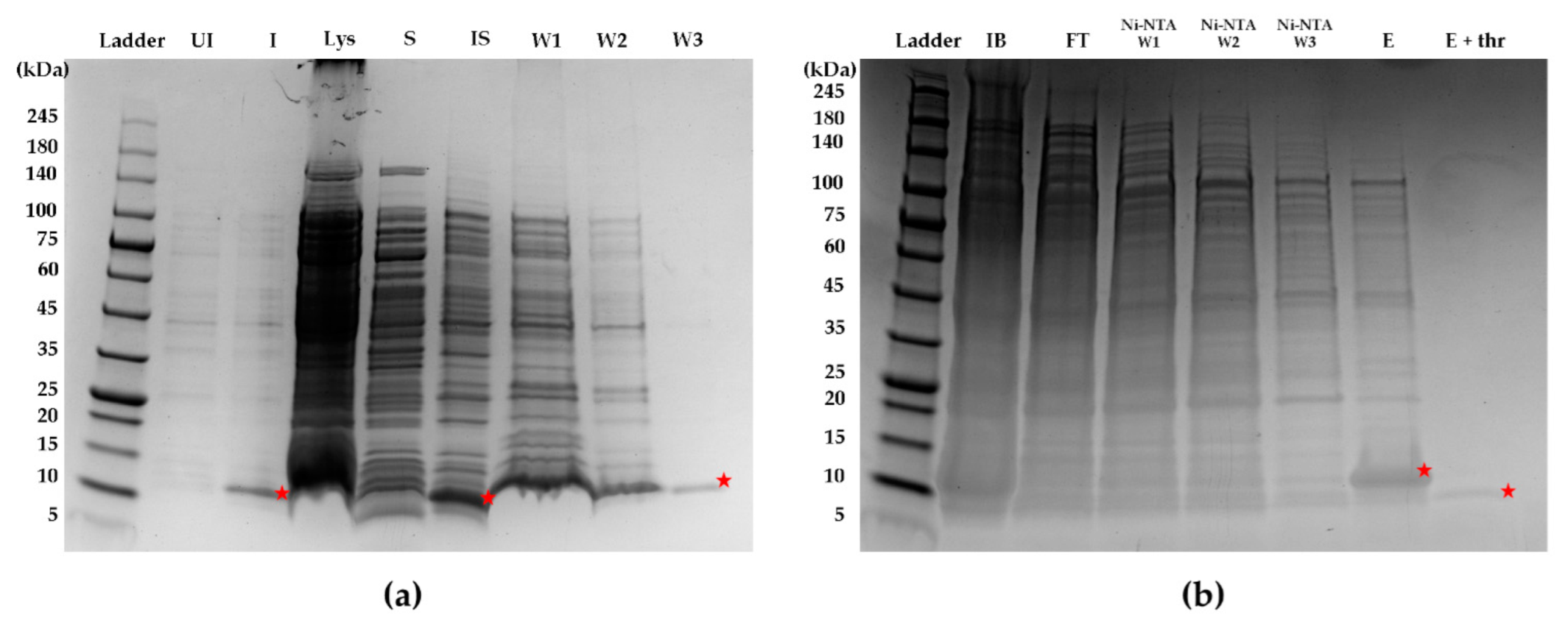
Briefly, a starter culture (containing a final concentration of 50 µg/mL kanamycin) of transformed SHuffle T7 E. coli cells (New England Biolabs, Ipswich, MA, USA) was incubated at 30 °C, with shaking at 250 rpm for 16–18 h overnight. For the expansion of the bacteria culture, 10 mL of starter culture was added to 1 L of LB broth in a 2.8 L Erlenmeyer flask. The E. coli was cultured at 30 °C, with shaking at 250 rpm. Expression of protein was induced using a final concentration of 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) (Bio Basic, Amherst, NY, USA) when the optical density of the E. coli culture at 600 nm reached 0.6. After the addition of IPTG, the culture was incubated at 16 °C, with shaking at 150 rpm. After 18 h, the bacterial cell pellet was collected by centrifugation at 6000 rpm for 30 min. The pellet was resuspended in 10–15 mL of lysis buffer (1× phosphate-buffered saline (PBS), 2% Triton X-100, 5 mM dithiothreitol (DTT)) and incubated on ice, with rocking for 1 h. The resuspended pellet was sonicated for 20 min (5 s on, 5 s off) on ice and subjected to centrifugation at 10,000 rpm for 30 min at 4 °C. Inclusion bodies were obtained by collecting the pellet after the centrifugation of the cell lysate and washed (resuspend pellet with wash buffer, centrifuge at 10,000 rpm for 30 min at 4 °C, and discard supernatant) twice with 12 mL of wash buffer A (1× PBS, 2% Triton X-100, 2 M urea, 400 mM NaCI, 5 mM DTT), followed by another wash with 12 mL of wash buffer B (1× PBS, 200 mM NaCl, 5 mM DTT). For purification through a 6×-His affinity tag, the washed pellet was resuspended in 15 mL of purification buffer comprising 1× PBS, 8 M urea, 5 mM imidazole, 1 mM β-mercaptoethanol, and incubated in a column containing Ni-NTA agarose beads (Qiagen, Hilden, Germany) for 30 min. The washing and on-column refolding of rFasxiatorN17R, L19E were performed by the sequential flowthrough of the above purification buffer without β-mercaptoethanol and the stepwise reduction of urea concentrations from 8 M to 4 M to no urea. rFasxiatorN17R, L19E was subsequently eluted with an elution buffer comprising 1× PBS and 200 mM imidazole. The eluent was diluted 10× using 1× PBS and incubated with thrombin (final concentration of 125 µg/mL) overnight for the cleavage of the 6×-His tag. rFasxiatorN17R, L19E was further purified using reversed-phase high-pressure liquid chromatography (RP-HPLC) on a Jupiter C18 5 µm 300 Å column with a dimension of 250 × 10 mm (Phenomenex, Torrance, CA, USA). An elution gradient of 20–60% over a 10 column volume with buffer B (80% acetonitrile, 0.1% TFA) was used for the elution of rFasxiatorN17R, L19E. The mass of the purified sample was confirmed using electrospray ionization mass spectrometry (ESI-MS).
2.3. Validation of FXIa Inhibition by Purified rFasxiatorN17R, L19E
Chromogenic assay was used to determine FXIa inhibition in a buffer containing 50 mM HEPES, 140 mM NaCl, 5 mM CaCl2, and 0.1% BSA at pH 7.4. In a reaction well, 50 µL of human FXIa (final concentration: 0.25 nM; Prolytix, Essex Junction, VT, USA) was preincubated with 50 µL of rFasxiatorN17R, L19E (final concentration: 0.01, 0.03, 0.1, 0.3, 1, 3, 10, or 30 nM) for 45 min at room temperature. After the incubation, 50 µL of the chromogenic substrate S-2366 (final concentration: 1 mM; Werfen, Barcelona, Spain) was added to start the reaction. The rate of product formation was measured according to the increase in absorbance at 405 nm over 15 min. A dose–response curve was plotted using the Prism 6 software, and the IC50 was calculated (GraphPad, San Diego, CA, USA).
2.4. Animals
Rat studies were conducted on male Sprague Dawley rats weighing approximately 280–320 g. Mice studies were conducted on 8 to 9-week-old male C57BL/6NTac mice weighing approximately 25 to 30 g. Since the primary aim of the study was to establish and validate the efficacy and safety of rFasxiator
N17R,L19E in vivo in comparison with reference therapeutics, only animals of a single sex (male) were used to limit the potential variables. Male mice were reported to have a stronger thrombotic response [
32] and were hence chosen as a more stringent challenge for rFasxiator
N17R,L19E. Both rats and mice were obtained from InVivos (Singapore). Rats and mice were kept in microisolator cages within a facility with constant temperature, a 12 h light/dark cycle, and ad libitum access to food and water.
2.5. Time Course Anticoagulant Effect of rFasxiatorN17R, L19E in Rats
Rats were anesthetized with an intraperitoneal (i.p.) injection of ketamine/xylazine (75 mg/10 mg/kg), followed by a maintenance flow of 1% isoflurane in oxygen at 1 L/min. The femoral artery and vein were surgically exposed for blood collection and drug administration, respectively. An i.v. bolus injection (rFasxiatorN17R,L19E 10 mg/kg) was administered via the left femoral vein, while 300 µL blood was drawn from the left femoral artery at various time points (0, 2, 5, 10, 20, 40, 60, 90, and 120 min). The drawn blood was immediately aliquoted into individual microcentrifuge tubes containing 3.2% sodium citrate. Plasma was obtained by centrifugation at 2000× g for 10 min within 30 min of blood collection. The activated partial thromboplastin time (APTT) and prothrombin time (PT) were performed on the Sysmex CA-660 Coagulometer using plasma collected from rats. Actin FSL, APTT reagent (Siemens Healthcare Diagnostics, Malvern, IL, USA) and Innovin, PT reagent (Siemens Healthcare Diagnostics, IL, USA) were used in these analyses. APTT and PT were performed using default machine settings. For APTT, 50 µL of rat plasma was incubated for 1 min at 37 °C. A volume of 50 µL of Actin FSL was added to the reaction mixture and further incubated for 3 min at 37 °C. Finally, 50 µL of CaCl2 was added to start the reaction, and absorbance was measured at 660 nm. For PT, 50 µL of rat plasma was incubated for 3 min at 37 °C. A total of 100 µL of Innovin was added to the reaction, and absorbance was measured at 660 nm.
2.6. Rat Carotid Artery Thrombosis (CAT) Model
Rats were anesthetized with an i.p. injection of ketamine/xylazine (75 mg/10 mg/kg), followed by a maintenance flow of 1% isoflurane in oxygen at 1 L/min. The femoral vein was surgically exposed for drug administration. An incision was made along the midline of the neck, and the right carotid artery was surgically exposed and isolated from the surrounding tissue. A Doppler flow probe (Model: MA1PRB, Transonic System Inc., Ithaca, NY, USA) was attached to the exposed carotid artery, and blood flow was recorded using LabChart 7 Pro (ADInstruments, Colorado Springs, CO, USA). UFH, the parenteral anticoagulant of choice recommended by current clinical guidelines for the treatment of arterial thrombosis such as ACS and PCI, was used as the reference drug [
33]. Saline, rFasxiator
N17R, L19E (0.05, 0.2, 0.5, or 2 mg/kg), or UFH (50, 100, 200, 300, or 432 U/kg) were administered via an i.v. bolus injection through a cannulation at the left femoral vein. Five minutes after the administration of rFasxiator
N17R, L19E, thrombus formation was induced via the application of a FeCl
3-soaked filter paper (2 × 5 mm, 10 µL of 20% FeCl
3 solution) on the surface of the exposed carotid artery in a position distal to the flow probe for 10 min. Blood flow was measured in real time, and time-to-occlusion (TTO) due to thrombus formation was recorded when blood flow decreased to zero. The study was terminated after 60 min. TTO was recorded as 60 min if blood flow persisted throughout.
2.7. Rat Tail Bleeding Model
Rats were anesthetized with an intraperitoneal injection of ketamine/xylazine (75 mg/10 mg/kg), followed by a maintenance flow of 1% isoflurane in oxygen at 1 L/min. The femoral vein was surgically exposed for drug administration. Saline, rFasxiatorN17R, L19E (0.05, 0.2, 0.5, or 2 mg/kg), or UFH (100, 200, 300, or 432U/kg) were administered via an i.v. bolus injection through a cannulation at the left femoral vein. Five minutes after the administration of rFasxiatorN17R, L19E, a spring-loaded blade device (Surgicutt Adult bleeding time device; ITC, Piscataway, NJ, USA) was used to make a longitudinal incision (1 mm depth × 5 mm length) on the ventral side of the rat’s tail (9–9.5 cm from the tip of the tail). Blood from the site of incision was blot dried (from the side of the wound, without touching the wound) with a filter paper every 15 s until the bleeding ceased. Tail bleeding time (TBT) was defined as the time after incision until the cessation of bleeding is observed within eight consecutive blotting periods. The study was terminated 60 min after tail incision. TBT was recorded as 60 min if the bleeding persisted throughout.
2.8. Mouse Inferior Vena Cava (IVC) Ligation Model
Mice were anesthetized with an intraperitoneal injection of ketamine/xylazine (75 mg/10 mg/kg), followed by a maintenance flow of 1% isoflurane in oxygen at 1 L/min. An incision was made with surgical scissors along the abdominal midline, 1.5 cm down from the sternum. The abdomen was then accessed through the linea alba, and the intestines were exteriorized to the mouse’s left with the use of sterile cotton buds moistened with sterile warm (37 °C) saline. The intestines were then covered with sterile gauze soaked in warm (37 °C) saline to prevent them from drying out. The IVC was then identified, and with forceps, a careful diverging movement was applied to create a hole between the aorta and the IVC. A 7-0 inert Prolene suture was carefully threaded through the hole with forceps, and a loose knot was tied around the IVC, just below the left renal vein. The loose knot was then tightened to ligate the IVC, and another knot was tied to secure the ligation. All visible side branches connected to the IVC were also ligated. After ligation, the intestines were carefully pushed back into the peritoneal cavity and distributed equally. The incision was then sutured in 2 layers. The peritoneum was closed with 6-0 vicryl sutures using continuous loops, with both ends secured with knots. The skin was closed with 6-0 monofilament sutures through interrupted loops. Iodine was applied over the stitches to minimize the risk of infection. Anesthetic reversal (atipamezole, 0.1 mL/10 g) was administered through i.p. injection to speed up recovery. LMWH enoxaparin was chosen as the reference drug because the parenteral anticoagulant is recommended as the initial or lead-in treatment for acute DVT and PE by current clinical guidelines [
34,
35]. Saline, rFasxiator
N17R,L19E (5 mg/kg or 10 mg/kg), or enoxaparin (18.5 mg/kg), together with 0.5 mL of warm (37 °C) saline (for hydration) and buprenophrine (0.1 mg/kg, for analgesic treatment), were injected subcutaneously (s.c.) before placing the mice (with heat pad) back into their cage to recover. Twenty-four hours after the surgery, the mice were euthanized after the tail bleeding experiment (see
Section 2.9), and their IVCs were excised. The full length of the thrombi formed in the IVCs were extracted, dried for 30 min, and weighed.
2.9. Mouse Tail Bleeding Model
Mice that had undergone IVC ligation and had been injected s.c. with saline, rFasxiatorN17R, L19E (5 mg/kg or 10 mg/kg), or enoxaparin (18.5 mg/kg) 24 h prior were subjected to tail bleeding experiments before euthanization. The mice were anesthetized with an i.p. injection of ketamine/xylazine (75 mg/10 mg/kg), followed by a maintenance flow of 1% isoflurane in oxygen at 1 L/min. The mice were placed in prone position on a 37 °C heating plate, with their tails hanging off the edge of the heating plate. Their tails were then soaked in warm (37 °C) saline for 5 min. After soaking, the mice tails were first dried with paper towels before slotting their tails through a snipped-off 10 uL pipette tip. A scalpel blade was used to transect the tail that had passed through the pipette tip. The transacted tail was then immediately placed into a new warm (37 °C) saline for a maximum period of 5 min. TBT was observed. The collected blood was lysed using the freeze–thawing method, and its hemoglobin content was analyzed according to the protocol of the Hemoglobin Assay Kit (Sigma-Aldrich, St. Louis, MO, USA).
2.10. Statistical Analysis
All statistical analyses and curve-fitting by non-linear regression were performed using Prism 6 (GraphPad, CA, USA). The dose-response curve to determine the IC50 of rFasxiatorN17R, L19E was used and ascertained with goodness-of-fit analyses, as implemented by Prism 6. The statistical difference between clotting times for saline and rFasxiatorN17R,L19E for the analysis of time course anticoagulant effect in rats, was analyzed by two-way analysis of variance (ANOVA), followed by Fisher’s Least Significant Difference (LSD) test for significance at each of the time points. Statistical difference among the saline and treatment groups in CAT model was analyzed by one-way ANOVA, followed by Tukey’s multiple comparison test. All data are expressed as mean standard error of mean (SEM), unless stated otherwise.
4. Discussion
The assessment of in vivo efficacy in disease-relevant animal models of molecules of interest in comparison to appropriate reference treatments is a crucial step in the development of any therapeutic. For anticoagulants, bleeding tendency can also be studied in animals as a safety indicator. In the present study, we evaluated rFasxiator
N17R,L19E for these purposes. We expressed and refolded rFasxiator
N17R, L19E to its functional conformation, with an IC
50 similar to a previously reported value [
30]. We observed that rFasxiator
N17R, L19E inhibits the intrinsic pathway (prolongation of APTT) in rats for at least 60 min, while the extrinsic pathway remains unmodified (no considerable change in PT over time). In our CAT model, for the assessment of arterial thrombosis, we showed that 2 mg/kg of rFasxiator
N17R, L19E i.v. achieved a similar antithrombotic efficacy (60 min occlusion time) to that of UFH at the clinically recommended dose for PCI, with 3.5× lower bleeding time. In our IVC ligation model, for the assessment of venous thrombosis, we showed that 10 mg/kg of rFasxiator
N17R, L19E s.c. achieved a similar antithrombotic efficacy to that of the therapeutic dose of enoxaparin without increasing baseline bleeding time at 24 h. Overall, we have shown that our FXIa-inhibiting miniprotein rFasxiator
N17R, L19E can achieve a similar efficacy to that of reference therapeutics in both arterial and venous thrombosis models.
There is considerable interest in developing the Kunitz domain as miniprotein therapeutics [
28,
29]. Several exogenous inhibitors of FXIa with the Kunitz domain were identified from venomous animals or the saliva of hematophagous animals. In addition to rFasxiator
N17R, L19E, other examples include Ir-CPI from the castor bean tick (
Ixodes ricinus), Desmolaris from the vampire bat (
Desmodus rotundus), WPK5 (and its mutant WPK5-Mut) from the leech (
Whitmania pigra), and DAKS1 from the sharp-nosed viper (
Deinagkistrodon acutus) [
38,
39,
40,
41]. Each of these inhibitors have their own unique characteristics. All molecules generally showed good antithrombotic efficacy with low bleeding risk in animal models [
38,
39,
40,
41]. However, the selectivity profiles of these inhibitors against different coagulation and fibrinolytic serine proteases vary. Among all coagulation and fibrinolytic enzymes, rFasxiator
N17R, L19E is most selective towards FXIa (K
i = 0.86 nM), while the K
i(s) against other coagulation enzymes is at least 170-fold higher (next low K
i is against plasmin at 146 nM) [
30]. In contrast, Ir-CPI binds to FXIa, FXIIa, and plasmin with similar affinities at the nanomolar level, and to kallikrein at the micromolar level [
40]. Similarly, Desmolaris binds to FXIa and FXa with similar K
i(s) of 12 and 15 nM, while inhibiting kallikrein with a slightly weaker activity [
39]. WPK5-Mut and DAKS1 also appeared to be FXIa-selective by at least 15- and 24-fold against other coagulation and fibrinolytic enzymes (86% inhibition when the WPK5-Mut:FXIa ratio was at 50:1, 28% inhibition when the WPK5-Mut:FXa ratio was at 775:1; 70% inhibition when the DAKS1:FXIa ratio was at 125:1, 62% inhibition when the DAKS1:Kallikrein ratio was at 3000:1) [
38,
41]. Although we did not perform head-to-head comparisons of rFasxiator
N17R, L19E and the other exogenous FXIa inhibitors, the in vivo activity of rFasxiator
N17R, L19E was consistent with these molecules. For example, in the CAT model, 2 mg/kg rFasxiator
N17R, L19E (i.v.) showed maximum antithrombotic efficacy, similar to the therapeutic dose of UFH. In comparison, it was reported that 3 mg/kg of WPK5-Mut (i.v.) or 2.6 mg/kg of DAKS1 showed similar antithrombotic efficacy compared to UFH as positive control [
38,
41]. Other than rFasxiator
N17R,L19E, Ir-CPI was the only other molecule tested in a venous thrombosis model. Although Ir-CPI at 1 mg/kg appeared to reduced thrombus formation in the IVC ligation model, major differences in the experiments made the comparison with rFasxiator
N17R, L19E challenging. For example, rFasxiator
N17R, L19E was injected s.c. to allow for direct comparison with the reference therapeutic enoxaparin. In contrast, Ir-CPI was injected i.v., and the experiments did not include a reference therapeutic such as enoxaparin. Nevertheless, the discovery of different exogenous inhibitors continues to enrich the development pipeline of molecules targeting FXIa, an important antithrombotic drug target.
Despite the availability of several anticoagulant therapeutics, rFasxiator
N17R,L19E as a miniprotein-based, potent, and specific FXIa inhibitor has a niche as a parenteral, short-term (a course that lasts between an hour and a few days) anticoagulant for acute arterial and venous arterial thrombosis. For prophylaxis and treatment against acute arterial thrombosis such as ACS and PCI, parenteral injectable anticoagulants with a fast onset and a short duration of action are preferable since the antithrombotic effect can be easily adjustable on demand [
42]. For example, during PCI, UFH is given as a bolus i.v. injection of 70–100 U/kg (with top-ups, when necessary) [
33] and has a duration of action of up to 4 h [
43]. Bivalirudin, an FDA-approved thrombin inhibitor indicated for PCI, is usually given as a bolus i.v. injection of 0.75 mg/kg, followed by a 1.75 mg/kg/h continuous i.v. infusion, with a short half-life of 25 min [
44]. Our study suggests that rFasxiator
N17R, L19E is likely to have a slightly longer duration of action (between 60 min and 90 min) when administered i.v., which is consistent with the short half-life of other low-molecular-weight proteins and peptides [
45]. However, rFasxiator
N17R, L19E may be advantageous for indications such as PCI, whereby a single i.v. bolus will generate enough antithrombotic efficacy for peri-procedural protection against stent thrombosis, with a reduced risk of post-procedural bleeding due to fast clearance (typical PCI procedure lasts for 30–60 min). Furthermore, rFasxiator
N17R, L19E as an FXIa-targeting inhibitor is likely to have a low inherent bleeding risk. In contrast, the use of UFH comes with many challenges and health risks. Apart from the risk of bleeding, the unpredictable and variable pharmacokinetic profile of UFH makes it challenging to provide an optimal dose for treatment [
46]. It is reported that the failure to achieve the appropriate therapeutic concentration of UFH during the early treatment of an acute thrombotic event is associated with a several-fold increase in the recurrence of DVT and myocardial infarction (MI) in patients [
46]. UFH can also cause serious drawbacks such as heparin-induced thrombocytopenia, osteoporosis, and hypersensitivity reaction [
4]. On the other hand, bivalirudin is expensive and may not offer obvious clinical advantages over UFH in the management of patients undergoing PCI [
47]. Moreover, a meta-analysis study has reported that the use of bivalirudin has a higher risk of MI and stent thrombosis as compared to UFH [
48]. Parenteral FXIa inhibitors under clinical development may also not be suitable for the treatment of acute thrombosis. IONIS-FXI
RX has slow onset and offset of action [
3]. The maximum reduction of FXIa using IONIS-FXI
RX would take 3–4 weeks of treatment, and the restoration of FXIa back to its baseline level would take several weeks after stopping treatment [
3]. Although Osocimab has a fast onset of activity after its infusion [
49], it has a long half-life of 30 to 40 days, which would take several weeks to be completely eliminated from the body after treatment [
18]. The orally available Milvexian only reached steady-state plasma concentration within 3 to 6 days [
50]. Therefore, rFasxiator
N17R, L19E remained the good therapeutic candidate for the development of a parenteral anticoagulant against acute thrombosis diseases.
Similarly, for the inpatient treatment of acute VTE, parenteral, fast-onset anticoagulants remain crucial. Short-term (days), repeated s.c. dosing of LMWH is usually used at the initiation of anticoagulant treatments for DVT and PE [
34,
35]. Therefore, we tested rFasxiator
N17R, L19E in comparison with LMWH enoxaparin in the IVC ligation model in mice. Despite the apparent short duration of action for rFasxiator
N17R, L19E when injected i.v. (
Figure 3), the s.c. injection of rFasxiator
N17R, L19E, albeit at the higher dose of 10 mg/kg, allowed for the effective prevention of clot formation in the ligated IVC, which was sustained throughout the 24 h experimental period. This shows that rFasxiator
N17R, L19E may also be a viable option as a parenteral anticoagulant for the treatment of acute venous thromboembolism.
Our study is not without limitations. First, the bacterial expression of rFasxiator
N17R, L19E resulted in an insoluble protein and a low overall yield, posing as scale-up and manufacturing challenge for therapeutic development. An optimized recombinant expression process in mammalian or yeast cells would be needed in the future. Second, due to the fast clearance of rFasxiator
N17R,L19E, we had to use a high concentration of rFasxiator
N17R,L19E in addition to the s.c. injection in the IVC ligation model to demonstrate its sustainable efficacy over a 24 h period. However, appropriate modifications of the molecule such as conjugation with polyethylene glycol, fatty acid chain, and fusion with IgG Fc may drastically prolong the half-life of the molecule [
51]. Finally, animal in vivo data may not be directly applicable to humans. Further studies should be performed on human samples to validate the effect of rFasxiator
N17R, L19E for human use.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




