
CDI Exerts Anti-Tumor Effects by Blocking the FoxM1-DNA Interaction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Time-Resolved Fluorescence Energy Transfer-Based FoxM1-DBD Binding Assay
2.3. Cell Culture and Viability Assay
2.4. Cell-Death Analysis
2.5. Real-Time Quantitative PCR Analysis
2.6. Western Blot Analysis
2.7. RNA Isolation and Sequencing
2.8. RNA-Seq Transcriptional Profiling
2.9. Docking and Molecular Dynamics (MD) Simulations
2.10. Statistical Analysis
3. Results
3.1. Inhibitory Effects of CDI on FoxM1-DNA Interaction
3.2. The Effects of CDI on the Proliferation of MDA-MB-231 Breast Cancer Cell Line
3.3. The Effects of CDI on the Expression Levels of FoxM1c and FoxM1 Downstream Target
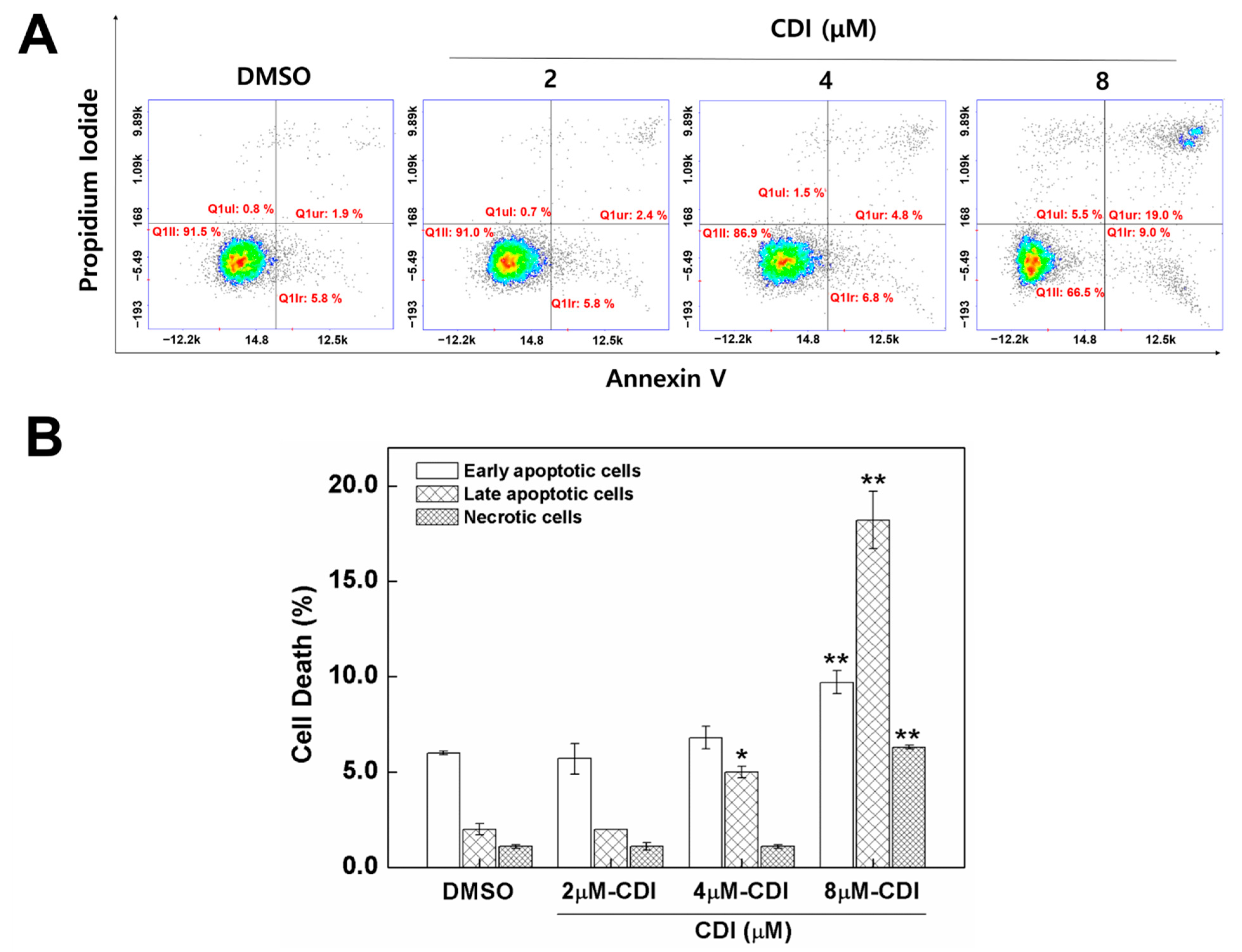
3.4. Apoptosis Induced by CDI in MDA-MB-231 Breast Cancer Cell Line
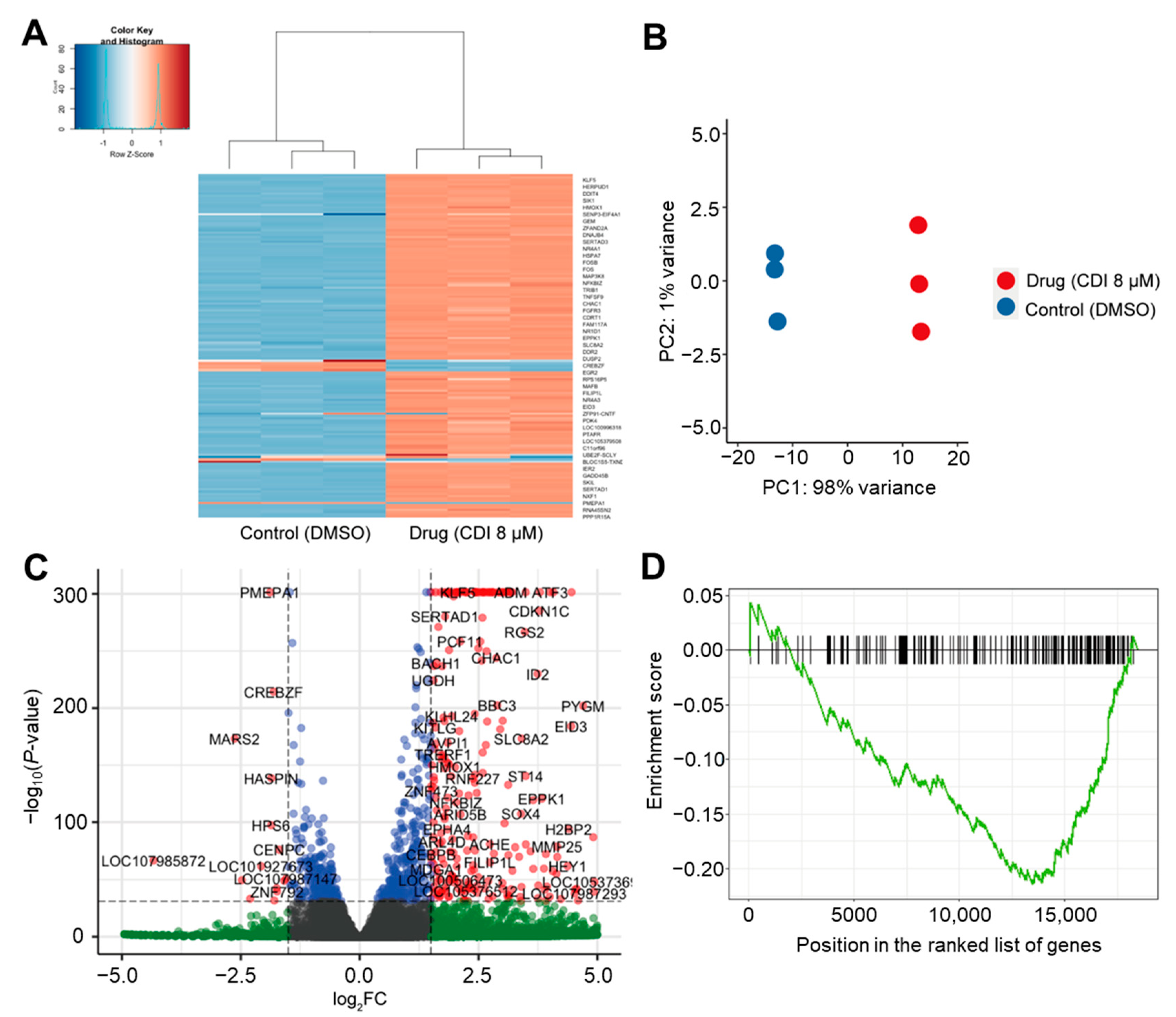
3.5. RNA-Seq Analysis of the Effects of CDI on Global FoxM1 Gene Regulation
3.6. Computational Simulation Results between FoxM1-DBD and CDI
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wierstra, I.; Alves, J. The central domain of transcription factor FOXM1c directly interacts with itself in vivo and switches from an essential to an inhibitory domain depending on the FOXM1c binding site. Biol. Chem. 2007, 388, 805–818. [Google Scholar] [CrossRef]
- Wang, I.C.; Chen, Y.J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, T.; Costa, R.H. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol. Cell Biol. 2005, 25, 10875–10894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Stahl, M.; Medema, R.H. FoxM1: At the crossroads of ageing and cancer. Biochim. Et. Biophys. Acta (BBA)-Rev. Cancer 2007, 1775, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wen, L.; Wen, M.; Liu, T.; Zhao, L.; Wu, B.; Yun, Y.; Liu, W.; Wang, H.; Wang, Y.; et al. FoxM1 promotes epithelial-mesenchymal transition, invasion, and migration of tongue squamous cell carcinoma cells through a c-Met/AKT-dependent positive feedback loop. Anticancer Drugs 2018, 29, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Wierstra, I. FOXM1 (Forkhead box M1) in tumorigenesis: Overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. Adv. Cancer Res. 2013, 119, 191–419. [Google Scholar] [CrossRef]
- Wang, I.C.; Meliton, L.; Ren, X.; Zhang, Y.; Balli, D.; Snyder, J.; Whitsett, J.A.; Kalinichenko, V.V.; Kalin, T.V. Deletion of Forkhead Box M1 transcription factor from respiratory epithelial cells inhibits pulmonary tumorigenesis. PLoS ONE 2009, 4, e6609. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Chen, H.; Yu, L.; Shan, L.; Xie, L.; Hu, J.; Chen, T.; Tan, Y. Inhibition of FOXM1 transcription factor suppresses cell proliferation and tumor growth of breast cancer. Cancer Gene Ther. 2013, 20, 117–124. [Google Scholar] [CrossRef]
- Chen, H.; Zou, Y.; Yang, H.; Wang, J.; Pan, H. Downregulation of FoxM1 inhibits proliferation, invasion and angiogenesis of HeLa cells in vitro and in vivo. Int. J. Oncol. 2014, 45, 2355–2364. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, J.; Cui, X.; Yang, Y.; Li, M.; Qu, J.; Li, J.; Wang, J. FoxM1: A novel tumor biomarker of lung cancer. Int J. Clin. Exp. Med. 2015, 8, 3136–3140. [Google Scholar]
- Lu, X.-F.; Zeng, D.; Liang, W.-Q.; Chen, C.-F.; Sun, S.-M.; Lin, H.-Y. FoxM1 is a promising candidate target in the treatment of breast cancer. Oncotarget 2017, 9, 842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.Y.; Jung, A.R.; Kim, G.E.; Yang, J.; Ha, U.S.; Hong, S.-H.; Choi, Y.J.; Moon, M.H.; Kim, S.W.; Lee, J.Y.; et al. High FOXM1 expression is a prognostic marker for poor clinical outcomes in prostate cancer. J. Cancer 2019, 10, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Gormally, M.V.; Dexheimer, T.S.; Marsico, G.; Sanders, D.A.; Lowe, C.; Matak-Vinkovic, D.; Michael, S.; Jadhav, A.; Rai, G.; Maloney, D.J.; et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat. Commun. 2014, 5, 5165. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaei Dakhili, S.A.; Perez, D.J.; Gopal, K.; Tabatabaei Dakhili, S.Y.; Ussher, J.R.; Velazquez-Martinez, C.A. A structure-activity relationship study of Forkhead Domain Inhibitors (FDI): The importance of halogen binding interactions. Bioorg. Chem. 2019, 93, 103269. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Hitchinson, B.; Shah, B.N.; Váraljai, R.; Khan, I.; Benevolenskaya, E.V.; Gaponenko, V.; Arbiser, J.L.; Gartel, A.L. Honokiol is a FOXM1 antagonist. Cell Death Dis. 2018, 9, 84. [Google Scholar] [CrossRef]
- Kwok, J.M.; Myatt, S.S.; Marson, C.M.; Coombes, R.C.; Constantinidou, D.; Lam, E.W. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol. Cancer Ther. 2008, 7, 2022–2032. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.K.; Bhat, U.G.; Hughes, D.E.; Wang, I.C.; Costa, R.H.; Gartel, A.L. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 2006, 66, 9731–9735. [Google Scholar] [CrossRef] [Green Version]
- Gartel, A.L. Thiostrepton, proteasome inhibitors and FOXM1. Cell Cycle 2011, 10, 4341–4342. [Google Scholar] [CrossRef] [PubMed]
- Klinhom-On, N.; Seubwai, W.; Sawanyawisuth, K.; Obchoei, S.; Mahalapbutr, P.; Wongkham, S. FOXM1 inhibitor, Siomycin A, synergizes and restores 5-FU cytotoxicity in human cholangiocarcinoma cell lines via targeting thymidylate synthase. Life Sci. 2021, 286, 120072. [Google Scholar] [CrossRef]
- Avtanski, D.; Poretsky, L. Phyto-polyphenols as potential inhibitors of breast cancer metastasis. Mol. Med. 2018, 24, 29. [Google Scholar] [CrossRef]
- Ziegler, Y.; Laws, M.J.; Sanabria Guillen, V.; Kim, S.H.; Dey, P.; Smith, B.P.; Gong, P.; Bindman, N.; Zhao, Y.; Carlson, K.; et al. Suppression of FOXM1 activities and breast cancer growth in vitro and in vivo by a new class of compounds. NPJ Breast Cancer 2019, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tretyakova, E.V.; Smirnova, I.E.; Kazakova, O.B.; Tolstikov, G.A.; Yavorskaya, N.P.; Golubeva, I.S.; Pugacheva, R.B.; Apryshko, G.N.; Poroikov, V.V. Synthesis and anticancer activity of quinopimaric and maleopimaric acids’ derivatives. Bioorg. Med. Chem. 2014, 22, 6481–6489. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ruben, E.A.; Rajadas, J.; Teng, N.N.H. In silico investigation of FOXM1 binding and novel inhibitors in epithelial ovarian cancer. Bioorg. Med. Chem. 2015, 23, 4576–4582. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Littler, D.R.; Alvarez-Fernandez, M.; Stein, A.; Hibbert, R.G.; Heidebrecht, T.; Aloy, P.; Medema, R.H.; Perrakis, A. Structure of the FoxM1 DNA-recognition domain bound to a promoter sequence. Nucleic Acids Res. 2010, 38, 4527–4538. [Google Scholar] [CrossRef] [Green Version]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.-y.; Ye, M.-y.; Huang, R.-z.; Li, Y.-j.; Zhu, Y.-t.; Pan, Y.-m.; Liao, Z.-X.; Wang, H.-s. Synthesis and antitumor activity evaluation of maleopimaric acid N-aryl imide atropisomers. Bioorg. Med. Chem. Lett. 2013, 23, 6755–6758. [Google Scholar] [CrossRef]
- Lee, M.Y.; Haam, C.E.; Mun, J.; Lim, G.; Lee, B.H.; Oh, K.-S. Development of a FOXM1-DBD Binding Assay for High-Throughput Screening Using TR-FRET Assay. Biol. Pharm. Bull. 2021, 44, 1484–1491. [Google Scholar] [CrossRef]
- Wang, Y.; Ung, M.H.; Xia, T.; Cheng, W.; Cheng, C. Cancer cell line specific co-factors modulate the FOXM1 cistrome. Oncotarget 2017, 8, 76498–76515. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.Y.; Lee, M.Y.; Lee, J.H.; Lee, B.H.; Oh, K.S. DeepHIT: A deep learning framework for prediction of hERG-induced cardiotoxicity. Bioinformatics 2020, 36, 3049–3055. [Google Scholar] [CrossRef] [PubMed]
- Shaker, B.; Yu, M.S.; Song, J.S.; Ahn, S.; Ryu, J.Y.; Oh, K.S.; Na, D. LightBBB: Computational prediction model of blood-brain-barrier penetration based on LightGBM. Bioinformatics 2021, 37, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Yu, M.S.; Kazmi, S.R.; Oh, S.Y.; Rhee, K.H.; Bae, M.A.; Lee, B.H.; Shin, D.S.; Oh, K.S.; Ceong, H.; et al. Computational determination of hERG-related cardiotoxicity of drug candidates. BMC Bioinform. 2019, 20, 250. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.Y.; Lee, J.H.; Lee, B.H.; Song, J.S.; Ahn, S.; Oh, K.S. PredMS: A random Forest model for predicting metabolic stability of drug candidates in human liver microsomes. Bioinformatics 2021, btab547. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yu, M.-S.; Na, D. DILI-Stk: An Ensemble Model for the Prediction of Drug-induced Liver Injury of Drug Candidates. Curr. Bioinform. 2022, 17, 296–303. [Google Scholar] [CrossRef]
- Yu, M.S.; Lee, J.; Lee, Y.; Na, D. 2-D chemical structure image-based in silico model to predict agonist activity for androgen receptor. BMC Bioinform. 2020, 21, 245. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Gartel, A.L. A novel mode of FoxM1 regulation: Positive auto-regulatory loop. Cell Cycle 2009, 8, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Langlet, F.; Haeusler, R.A.; Linden, D.; Ericson, E.; Norris, T.; Johansson, A.; Cook, J.R.; Aizawa, K.; Wang, L.; Buettner, C.; et al. Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell 2017, 171, 824–835.e818. [Google Scholar] [CrossRef] [PubMed]
- Chesnokov, M.S.; Halasi, M.; Borhani, S.; Arbieva, Z.; Shah, B.N.; Oerlemans, R.; Khan, I.; Camacho, C.J.; Gartel, A.L. Novel FOXM1 inhibitor identified via gene network analysis induces autophagic FOXM1 degradation to overcome chemoresistance of human cancer cells. Cell Death Dis. 2021, 12, 704. [Google Scholar] [CrossRef]
- Wonsey, D.R.; Follettie, M.T. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005, 65, 5181–5189. [Google Scholar] [CrossRef] [Green Version]
- Laoukili, J.; Kooistra, M.R.H.; Brás, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Costa, R.H. FoxM1 dances with mitosis. Nat. Cell Biol. 2005, 7, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Hegde, N.S.; Sanders, D.A.; Rodriguez, R.; Balasubramanian, S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat. Chem. 2011, 3, 725–731. [Google Scholar] [CrossRef]
- Tabatabaei-Dakhili, S.A.; Aguayo-Ortiz, R.; Dominguez, L.; Velazquez-Martinez, C.A. Untying the knot of transcription factor druggability: Molecular modeling study of FOXM1 inhibitors. J. Mol. Graph. Model. 2018, 80, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Kongsema, M.; Wongkhieo, S.; Khongkow, M.; Lam, E.W.; Boonnoy, P.; Vongsangnak, W.; Wong-Ekkabut, J. Molecular mechanism of Forkhead box M1 inhibition by thiostrepton in breast cancer cells. Oncol. Rep. 2019, 42, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Ulhaka, K.; Kanokwiroon, K.; Khongkow, M.; Bissanum, R.; Khunpitak, T.; Khongkow, P. The Anticancer Effects of FDI-6, a FOXM1 Inhibitor, on Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 6685. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, W.D.; Lee, M.Y.; Mun, J.; Lim, G.; Oh, K.-S. CDI Exerts Anti-Tumor Effects by Blocking the FoxM1-DNA Interaction. Biomedicines 2022, 10, 1671. https://doi.org/10.3390/biomedicines10071671
Jang WD, Lee MY, Mun J, Lim G, Oh K-S. CDI Exerts Anti-Tumor Effects by Blocking the FoxM1-DNA Interaction. Biomedicines. 2022; 10(7):1671. https://doi.org/10.3390/biomedicines10071671
Chicago/Turabian StyleJang, Woo Dae, Mi Young Lee, Jihye Mun, Gyutae Lim, and Kwang-Seok Oh. 2022. "CDI Exerts Anti-Tumor Effects by Blocking the FoxM1-DNA Interaction" Biomedicines 10, no. 7: 1671. https://doi.org/10.3390/biomedicines10071671