
Clinical and Molecular Spectrum of Sporadic Vascular Malformations: A Single-Center Study

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Clinical Evaluation
2.3. Molecular Genetic Testing
3. Results
3.1. Patient Population
3.2. Sturge–Weber Syndrome
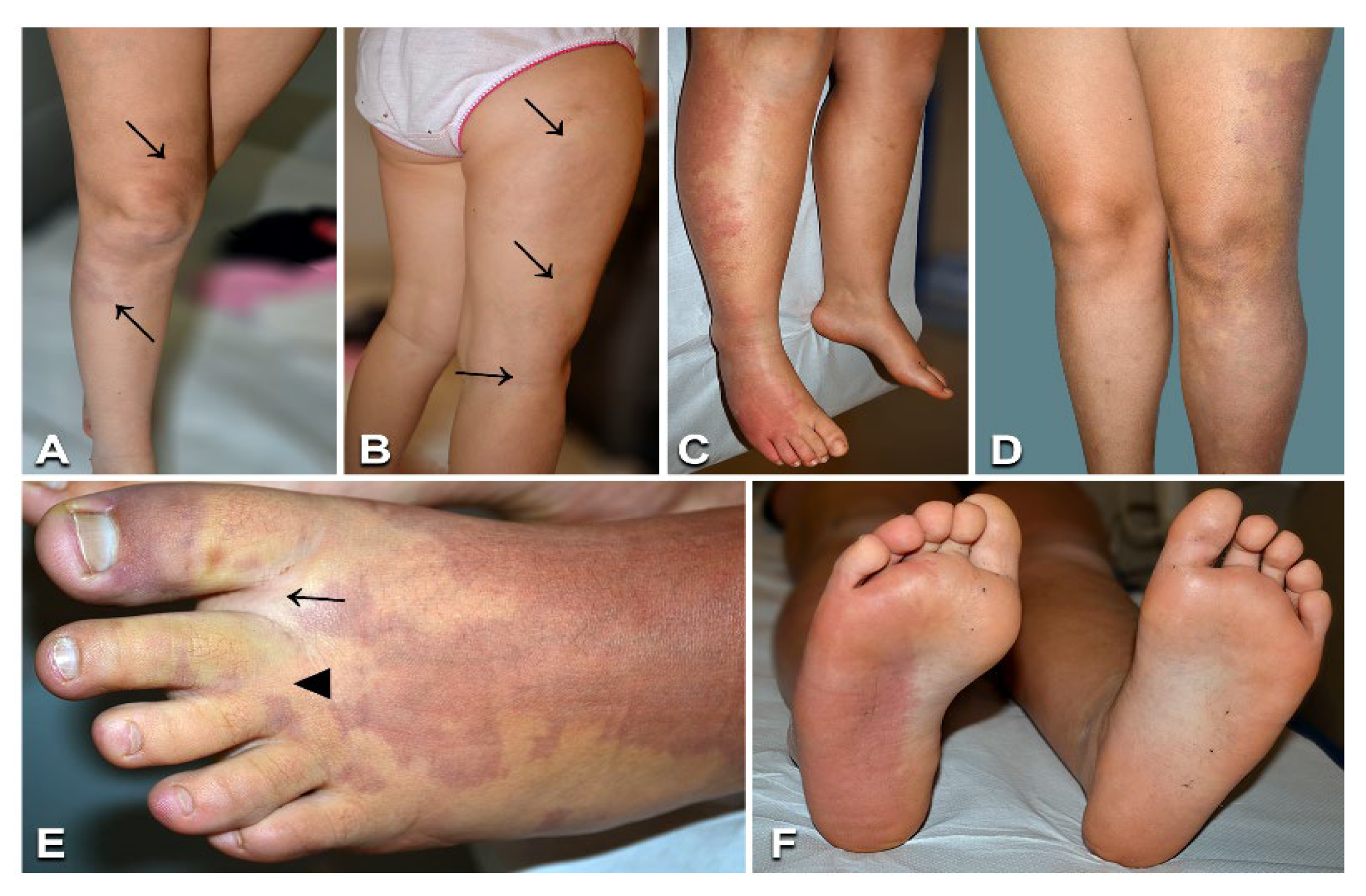
3.3. Capillary Malformation with Overgrowth
3.4. Diffuse Capillary Malformation with Overgrowth
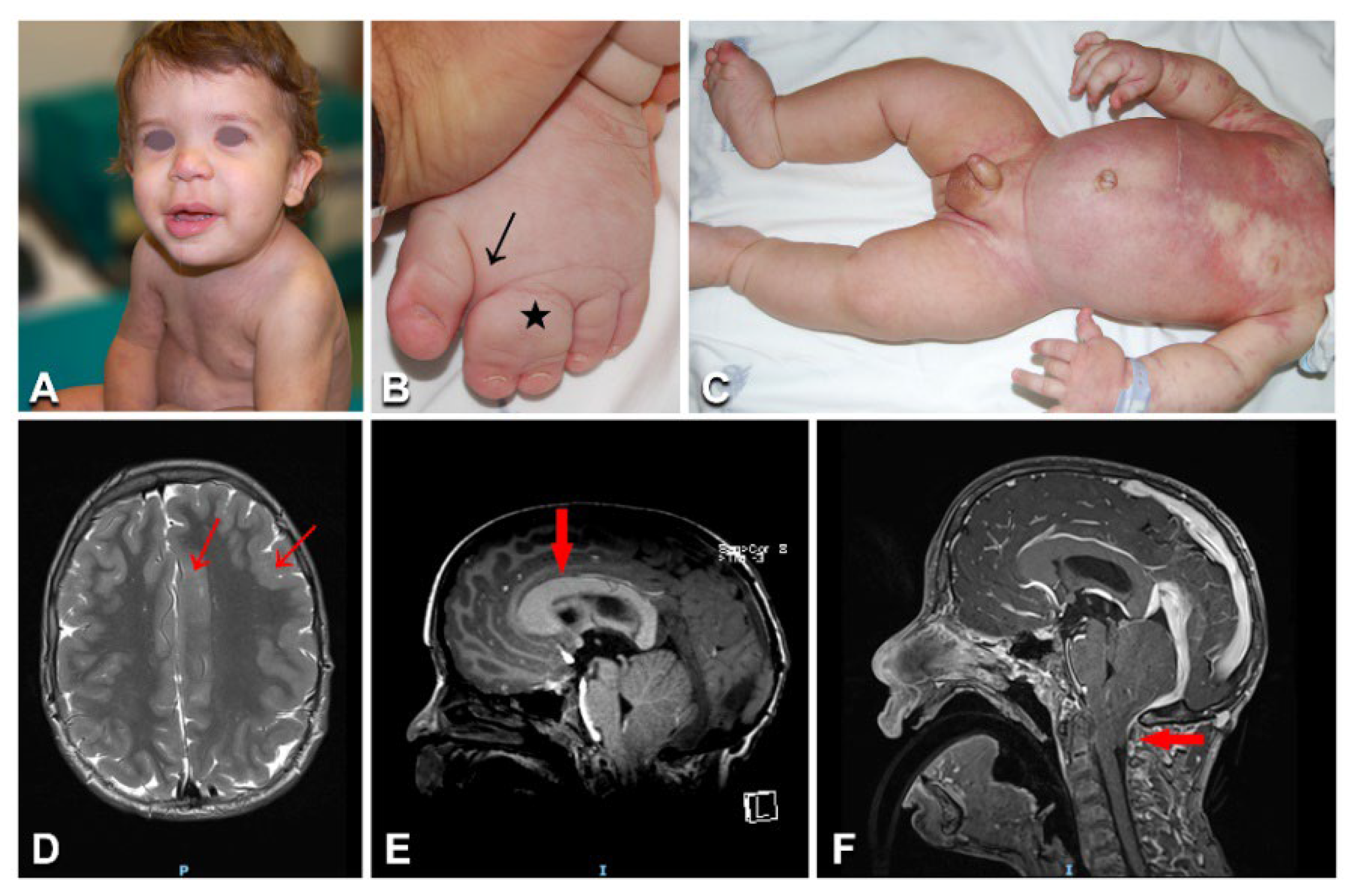
3.5. Megalencephaly–Capillary Malformation–Polymicrogyria Syndrome
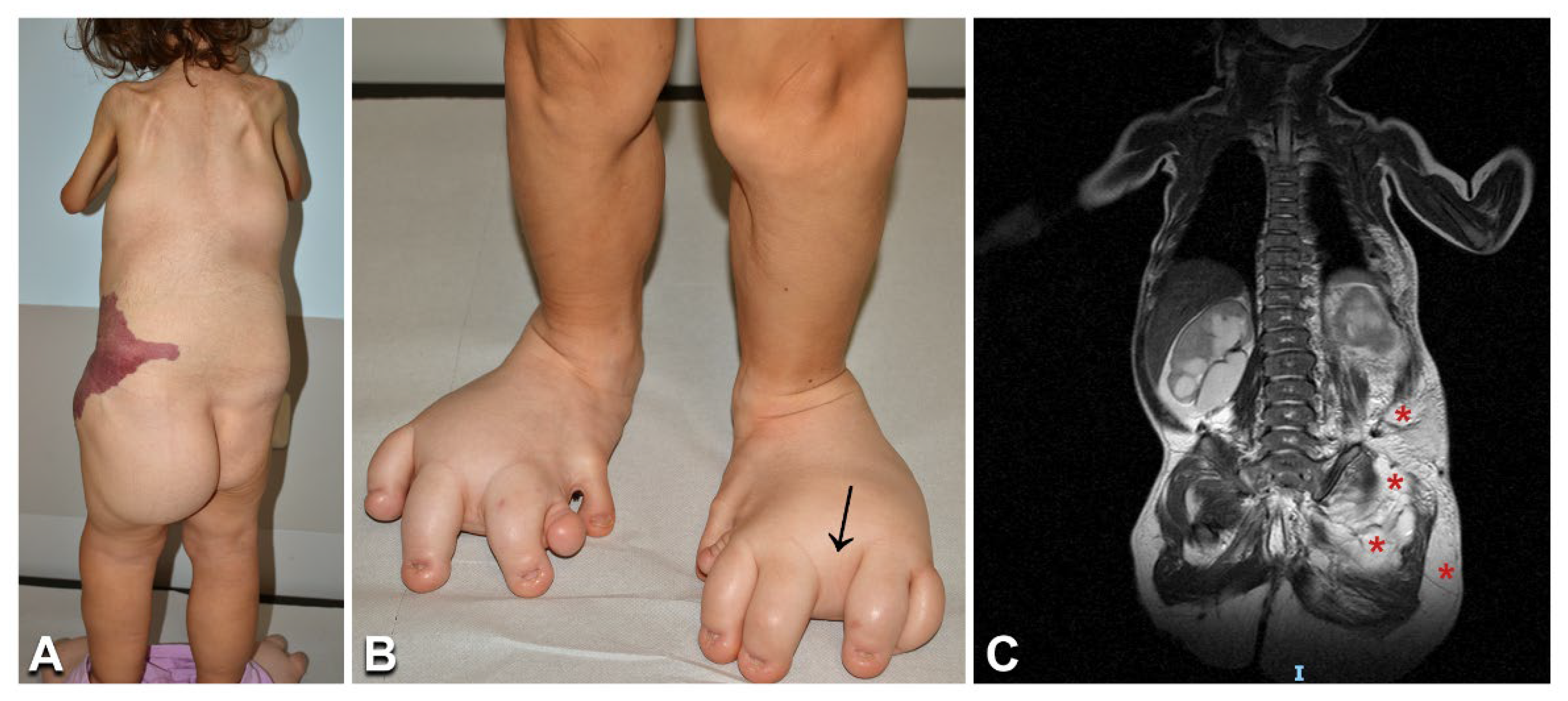
3.6. Congenital Lipomatous Overgrowth, Vascular Malformations, Epidermal Nevi, Scoliosis/Skeletal and Spinal (CLOVES) Syndrome, Klippel–Trenaunay Syndrome, and Combined Vascular Malformations
3.7. Blue Rubber Bleb Nevus Syndrome
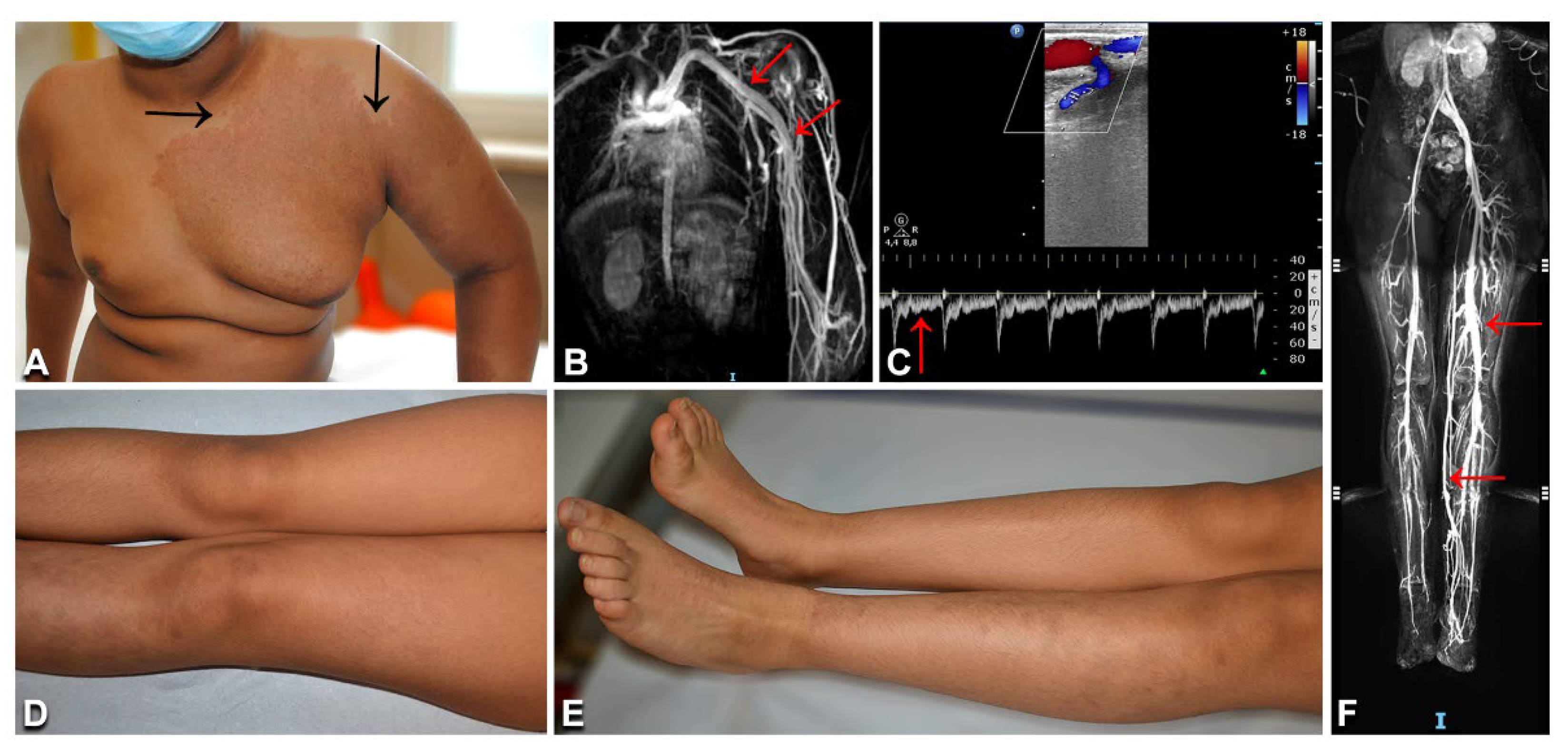
3.8. Parkes Weber Syndrome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ISSVA. Classification of Vascular Anomalies © 2018 International Society for the Study of Vascular Anomalies. Available online: https://www.issva.org/classification (accessed on 20 January 2022).
- Queisser, A.; Seront, E.; Boon, L.M.; Vikkula, M. Genetic Basis and Therapies for Vascular Anomalies. Circ. Res. 2021, 129, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Lokhorst, M.M.; Horbach, S.E.R.; Waner, M.; Min-Jung O, T.; van der Vleuten, C.J.M.; Spuls, P.I.; van der Horst, C. Responsiveness of quality of life measures in children with peripheral vascular malformations: The OVAMA project. JPRAS Open 2021, 27, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.; Hao, M.; Luu, M. PIK3CA vascular overgrowth syndromes: An update. Curr. Opin. Pediatr. 2020, 32, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M.D.; Tang, H.; Gallione, C.J.; Baugher, J.D.; Frelin, L.P.; Cohen, B.; North, P.E.; Marchuk, D.A.; Comi, A.M.; Pevsner, J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med. 2013, 368, 1971–1979. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [Green Version]
- Mirzaa, G.; Timms, A.E.; Conti, V.; Boyle, E.A.; Girisha, K.M.; Martin, B.; Kircher, M.; Olds, C.; Juusola, J.; Collins, S.; et al. PIK3CA-associated developmental disorders exhibit distinct classes of mutations with variable expression and tissue distribution. JCI Insight 2016, 1, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kurek, K.C.; Luks, V.L.; Ayturk, U.M.; Alomari, A.I.; Fishman, S.J.; Spencer, S.A.; Mulliken, J.B.; Bowen, M.E.; Yamamoto, G.L.; Kozakewich, H.P.; et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am. J. Hum. Genet. 2012, 90, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Liang, M.G.; Mulliken, J.B. Diffuse capillary malformation with overgrowth: A clinical subtype of vascular anomalies with hypertrophy. J. Am. Acad. Dermatol. 2013, 69, 589–594. [Google Scholar] [CrossRef]
- Rafi, S.K.; Butler, M.G. The 15q11.2 BP1-BP2 Microdeletion (Burnside-Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Phenotypes. Int J. Mol. Sci. 2020, 21, 3296. [Google Scholar] [CrossRef]
- Rotunno, R.; Diociaiuti, A.; Pisaneschi, E.; Carnevale, C.; Dentici, M.; El Hachem, M. PI3KCA-related overgrowth with an uncommon phenotype. Ital. J. Pediatr. 2022, 48, 71. [Google Scholar] [CrossRef]
- Ranieri, C.; Di Tommaso, S.; Loconte, D.C.; Grossi, V.; Sanese, P.; Bagnulo, R.; Susca, F.C.; Forte, G.; Peserico, A.; De Luisi, A.; et al. In vitro efficacy of ARQ 092, an allosteric AKT inhibitor, on primary fibroblast cells derived from patients with PIK3CA-related overgrowth spectrum (PROS). Neurogenetics 2018, 19, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Soblet, J.; Kangas, J.; Natynki, M.; Mendola, A.; Helaers, R.; Uebelhoer, M.; Kaakinen, M.; Cordisco, M.; Dompmartin, A.; Enjolras, O.; et al. Blue Rubber Bleb Nevus (BRBN) Syndrome Is Caused by Somatic TEK (TIE2) Mutations. J. Investig. Dermatol. 2017, 137, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Shewchuk, L.M.; Hassell, A.M.; Ellis, B.; Holmes, W.D.; Davis, R.; Horne, E.L.; Kadwell, S.H.; McKee, D.D.; Moore, J.T. Structure of the Tie2 RTK domain: Self-inhibition by the nucleotide binding loop, activation loop, and C-terminal tail. Structure 2000, 8, 1105–1113. [Google Scholar] [CrossRef] [Green Version]
- Locascio, L.E.; Donoghue, D.J. KIDs rule: Regulatory phosphorylation of RTKs. Trends Biochem. Sci. 2013, 38, 75–84. [Google Scholar] [CrossRef]
- Du, Z.; Zheng, J.; Zhang, Z.; Wang, Y. Review of the endothelial pathogenic mechanism of TIE2-related venous malformation. J. Vasc. Surg. Venous Lymphat. Disord. 2017, 5, 740–748. [Google Scholar] [CrossRef]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef]
- Nakashima, M.; Miyajima, M.; Sugano, H.; Iimura, Y.; Kato, M.; Tsurusaki, Y.; Miyake, N.; Saitsu, H.; Arai, H.; Matsumoto, N. The somatic GNAQ mutation c.548G > A (p.R183Q) is consistently found in Sturge-Weber syndrome. J. Hum. Genet. 2014, 59, 691–693. [Google Scholar] [CrossRef]
- Frigerio, A.; Wright, K.; Wooderchak-Donahue, W.; Tan, O.T.; Margraf, R.; Stevenson, D.A.; Grimmer, J.F.; Bayrak-Toydemir, P. Genetic Variants Associated with Port-Wine Stains. PLoS ONE 2015, 10, e0133158. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M.; Carmignac, V.; Sorlin, A.; Kuentz, P.; Albuisson, J.; Borradori, L.; Bourrat, E.; Boute, O.; Bukvic, N.; Bursztejn, A.C.; et al. Reverse Phenotyping in Patients with Skin Capillary Malformations and Mosaic GNAQ or GNA11 Mutations Defines a Clinical Spectrum with Genotype-Phenotype Correlation. J. Investig. Dermatol. 2020, 140, 1106–1110.e1102. [Google Scholar] [CrossRef]
- Jafry, M.; Sidbury, R. RASopathies. Clin. Dermatol. 2020, 38, 455–461. [Google Scholar] [CrossRef]
- Nikolaev, S.I.; Vetiska, S.; Bonilla, X.; Boudreau, E.; Jauhiainen, S.; Rezai Jahromi, B.; Khyzha, N.; DiStefano, P.V.; Suutarinen, S.; Kiehl, T.R.; et al. Somatic Activating KRAS Mutations in Arteriovenous Malformations of the Brain. N. Engl. J. Med. 2018, 378, 250–261. [Google Scholar] [CrossRef]
- Al-Olabi, L.; Polubothu, S.; Dowsett, K.; Andrews, K.A.; Stadnik, P.; Joseph, A.P.; Knox, R.; Pittman, A.; Clark, G.; Baird, W.; et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J. Clin. Investig. 2018, 128, 1496–1508. [Google Scholar] [CrossRef]
- Chang, C.A.; Perrier, R.; Kurek, K.C.; Estrada-Veras, J.; Lehman, A.; Yip, S.; Hendson, G.; Diamond, C.; Pinchot, J.W.; Tran, J.M.; et al. Novel findings and expansion of phenotype in a mosaic RASopathy caused by somatic KRAS variants. Am. J. Med. Genet. A 2021, 185, 2829–2845. [Google Scholar] [CrossRef]
- Siegel, D.H.; Cottrell, C.E.; Streicher, J.L.; Schilter, K.F.; Basel, D.G.; Baselga, E.; Burrows, P.E.; Ciliberto, H.M.; Vigh-Conrad, K.A.; Eichenfield, L.F.; et al. Analyzing the Genetic Spectrum of Vascular Anomalies with Overgrowth via Cancer Genomics. J. Investig. Dermatol. 2018, 138, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Couto, J.A.; Ayturk, U.M.; Konczyk, D.J.; Goss, J.A.; Huang, A.Y.; Hann, S.; Reeve, J.L.; Liang, M.G.; Bischoff, J.; Warman, M.L.; et al. A somatic GNA11 mutation is associated with extremity capillary malformation and overgrowth. Angiogenesis 2017, 20, 303–306. [Google Scholar] [CrossRef]
- Goss, J.A.; Konczyk, D.J.; Smits, P.; Sudduth, C.L.; Bischoff, J.; Liang, M.G.; Greene, A.K. Diffuse capillary malformation with overgrowth contains somatic PIK3CA variants. Clin. Genet. 2020, 97, 736–740. [Google Scholar] [CrossRef]
- Keppler-Noreuil, K.M.; Sapp, J.C.; Lindhurst, M.J.; Parker, V.E.; Blumhorst, C.; Darling, T.; Tosi, L.L.; Huson, S.M.; Whitehouse, R.W.; Jakkula, E.; et al. Clinical delineation and natural history of the PIK3CA-related overgrowth spectrum. Am. J. Med. Genet. A 2014, 164A, 1713–1733. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Shin, C.H.; Yoo, W.J.; Cho, T.J.; Kim, M.J.; Seong, M.W.; Park, S.S.; Lee, J.H.; Sim, N.S.; Ko, J.M. Detailed analysis of phenotypes and genotypes in megalencephaly-capillary malformation-polymicrogyria syndrome caused by somatic mosaicism of PIK3CA mutations. Orphanet J. Rare Dis. 2020, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Kuentz, P.; St-Onge, J.; Duffourd, Y.; Courcet, J.B.; Carmignac, V.; Jouan, T.; Sorlin, A.; Abasq-Thomas, C.; Albuisson, J.; Amiel, J.; et al. Molecular diagnosis of PIK3CA-related overgrowth spectrum (PROS) in 162 patients and recommendations for genetic testing. Genet. Med. 2017, 19, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouillard, P.; Schlogel, M.J.; Homayun Sepehr, N.; Helaers, R.; Queisser, A.; Fastre, E.; Boutry, S.; Schmitz, S.; Clapuyt, P.; Hammer, F.; et al. Non-hotspot PIK3CA mutations are more frequent in CLOVES than in common or combined lymphatic malformations. Orphanet J. Rare Dis. 2021, 16, 267. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Lopez-Knowles, E.; Luis, N.M.; Toll, A.; Baselga, E.; Fernandez-Casado, A.; Hernandez, S.; Ribe, A.; Mentzel, T.; Stoehr, R.; et al. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc. Natl. Acad. Sci. USA 2007, 104, 13450–13454. [Google Scholar] [CrossRef] [Green Version]
- Revencu, N.; Boon, L.M.; Mendola, A.; Cordisco, M.R.; Dubois, J.; Clapuyt, P.; Hammer, F.; Amor, D.J.; Irvine, A.D.; Baselga, E.; et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum. Mutat. 2013, 34, 1632–1641. [Google Scholar] [CrossRef]
- Macmurdo, C.F.; Wooderchak-Donahue, W.; Bayrak-Toydemir, P.; Le, J.; Wallenstein, M.B.; Milla, C.; Teng, J.M.; Bernstein, J.A.; Stevenson, D.A. RASA1 somatic mutation and variable expressivity in capillary malformation/arteriovenous malformation (CM/AVM) syndrome. Am. J. Med. Genet. A 2016, 170, 1450–1454. [Google Scholar] [CrossRef]
- Revencu, N.; Fastre, E.; Ravoet, M.; Helaers, R.; Brouillard, P.; Bisdorff-Bresson, A.; Chung, C.W.T.; Gerard, M.; Dvorakova, V.; Irvine, A.D.; et al. RASA1 mosaic mutations in patients with capillary malformation-arteriovenous malformation. J. Med. Genet. 2020, 57, 48–52. [Google Scholar] [CrossRef]
- Boccara, O.; Eyries, M.; Pannier, S.; Ariche-Maman, S.; Hadj-Rabia, S.; Coulet, F. Parkes-Weber syndrome related to RASA1 mosaic mutation. Clin. Genet. 2021, 99, 330–331. [Google Scholar] [CrossRef]
- Gordo, G.; Rodriguez-Laguna, L.; Agra, N.; Mendez, P.; Feito, M.; Lapunzina, P.; Lopez-Gutierrez, J.C.; Martinez-Glez, V. Constitutional mosaicism in RASA1-related capillary malformation-arteriovenous malformation. Clin. Genet. 2019, 95, 516–519. [Google Scholar] [CrossRef]
- Schmidt, V.F.; Wieland, I.; Wohlgemuth, W.A.; Ricke, J.; Wildgruber, M.; Zenker, M. Mosaic RASopathy due to KRAS variant G12D with segmental overgrowth and associated peripheral vascular malformations. Am. J. Med. Genet. A 2021, 185, 3122–3128. [Google Scholar] [CrossRef]
- Eng, W.; Sudduth, C.L.; Konczyk, D.J.; Smits, P.J.; Taghinia, A.H.; Fishman, S.J.; Alomari, A.; Adams, D.M.; Greene, A.K. Parkes Weber syndrome with lymphedema caused by a somatic KRAS variant. Cold Spring Harb. Mol. Case Stud. 2021, 7, a006118. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt. n. | Age (y)/Sex | Diagnosis | Clinical Features | Imaging Findings | Gene | Variant(s) * | VAF (Tissue **/ Blood) |
|---|---|---|---|---|---|---|---|
| 1 | 8/M | CM | Right lower face up to lower eyelid CM | Brain MRI: normal | GNAQ | c.548G > A (p.Arg183Gln) | 17%/nd |
| 2 | 0.8/F | SWS | Bilateral face, trunk, and limb CM; right eye glaucoma; seizures | Brain CT: subcortical calcifications; brain MRI: leptomeningeal angiomatosis | GNAQ | c.548G > A (p.Arg183Gln) | 3%/nd |
| 3 | 8/F | SWS | Left face and upper limb CM; left thorax and scapular prominent veins; left upper limb and thorax hypertrophy; left eye glaucoma; headache; epistaxis | Brain MRI: left cerebral hypotrophy; leptomeningeal angiomatosis | GNAQ | c.548G > A (p.Arg183Gln) | 2%/nd |
| 4 | 21/M | SWS | Right facial and hemibody CM; ipsilateral lip, upper and lower limb hypertrophy; right eye glaucoma; seizures | Brain MRI: leptomeningeal angiomatosis | GNAQ | c.548G > A (p.Arg183Gln) | 5%/nd |
| 5 | 4/F | CMO | Left chest, shoulder, and upper limb partly reticulate CM; ipsilateral limb overgrowth | DU: no AVF | GNAQ | c.548G > A (p.Arg183Gln) | 3%/nd |
| 6 | 15/M | CMO | Left lower limb CM and overgrowth; leg length discrepancy | DU: no AVF | GNAQ | c.548G > A (p.Arg183Gln) | 3%/nd |
| 7 | 12/F | CMO | Right chest, shoulder, and upper limb partly reticulate CM; ipsilateral limb overgrowth | DU: no AVF | GNAQ | c.548G > A (p.Arg183Gln) | 5%/nd |
| 8 | 11/M | CMO | Right lower limb CM and overgrowth; leg length discrepancy | DU: no AVF | GNAQ | c.548G > A (p.Arg183Gln) | 2%/nd |
| 9 | 14/F | CMO | Left lumbar and lower limb CM; ipsilateral limb overgrowth; leg length discrepancy (surgically treated) | DU: no AVF | GNAQ | c.548G > A (p.Arg183Gln) | 5%/nd |
| 10 | 14/F | CMO | Right chest and upper limb partly reticulate CM; ipsilateral limb overgrowth | DU: no AVF | GNAQ | c.548G > A (p.Arg183Gln) | 3%/nd |
| 11 | 7/F | CMO | Right lower limb CM and prominent veins, warmth to palpation; ipsilateral limb overgrowth; leg length discrepancy; proximal toe syndactyly | DU: no detectable AVF; MRI: right lower limb ectatic, tortuous superficial and intramuscular veins | KRAS | c.35G > T (p.Gly12Val) | 3%/nd |
| 12 | 4/F | CMO | Left lower limb CM and overgrowth | DU: no AVF | PIK3CA | c.1636C > A (p.Gln546Lys) | 3%/nd |
| 13 | 17/F | CMO | Left lower limb CM and overgrowth; sandal gap; proximal toe syndactyly; leg length discrepancy | DU: no AVF | PIK3CA | c.353G > A (p.Gly118Asp) | 3%/nd |
| 14 | 1/F | DCMO | Lower limb, trunk, and face reticulate CM with vermillion stain and prominent left face veins; left lower limb overgrowth | DU: no AVF | PIK3CA | c.1090G > A (p.Gly364Arg) | 7%/1% |
| 15 | 11/M | DCMO | Limb, trunk, and head reticulate CM; right finger and toe macrodactyly | DU: no AVF | PIK3CA | c.1133G > A (p.Cys378Tyr) | 13.5%/nd |
| 16 | 1/M | DCMO | Limb and trunk reticulate CM with centrofacial stain ***; left upper limb overgrowth; sandal gap; triangular foot | DU: no AVF; brain MRI: normal | PIK3CA | c.1093G > A (p.Glu365Lys) | 7%/nd |
| 17 | 6/F | DCMO | Trunk, left thigh, and cheek reticulate CM; abdominal wall prominent veins; overgrowth of left cheek, palatine tonsil, parotid gland, and paraumbilical and lumbosacral regions; sandal gap; proximal toe syndactyly | DU: no AVF; Brain MRI: temporal and occipital lobes asymmetry with left predominance | PIK3CA | c.1357G > A (p.Glu453Lys) | 7%/nd |
| 18 | 10/F | DCMO | Right hemibody reticulate CM; right lower limb prominent veins; upper limb and focal trunk overgrowth with toe macrodactyly; sandal gap; proximal toe syndactyly | DU: no AVF; brain MRI: normal | PIK3CA | c.1357G > A (p.Glu453Lys) | 2%/nd |
| 19 | 1/F | DCMO | Trunk and lower limb reticulate CM with centrofacial stain ***; right lower limb overgrowth | DU: no AVF | PIK3CA | c.2740G > A (p.Gly914Arg) | 26%/nd |
| 20 | 19/F | DCMO | Right hemibody partly reticulate CM; right upper and lower limb overgrowth; leg-length discrepancy | DU: no AVF; MRI and CT angiography: left internal carotid artery hypoplasia | GNA11 | c.547C > T (p.Arg183Cys) | 3%/nd |
| 21 | 11/M | DCMO | Limb, trunk, and face reticulate CM; left upper limb and right lower limb overgrowth; leg length discrepancy | DU: no AVF; brain MRI: normal | GNA11 | c.547C > T (p.Arg183Cys) | 20%/1% |
| 22 | 10/F | DCMO | Limb, trunk, and face reticulate CM; right lower limb and left cheek overgrowth; leg length discrepancy | DU: no AVF | GNA11 | c.548G > A (p.Arg183His) | 3%/1% |
| 23 | 10/F | DCMO | Limb, trunk, and face reticulate CM; left hemihypertrophy and macrodactyly; sandal gap; proximal toe syndactyly | DU: no AVF | PIK3CA | c.1133G > A (p.Cys378Tyr) | 11.5%/nd |
| 24 | 6/M | DCMO | Face, trunk, and limb partly reticulate CM; right hemihypertrophy with macrodactyly; leg length discrepancy; proximal toe syndactyly; sandal gap; psychomotor delay **** | DU: no AVF; brain MRI: thick corpus callosum | PIK3CA | c.311C > T (p.Pro104Leu) | 6%/nd |
| 25 | 14/M | MCAP | Face, trunk, and limb partly reticulate CM; macrocephaly; left face, upper and lower limb, and right fingers hypertrophy; bilateral toe syndactyly; psychomotor delay | Brain MRI: left hemimegalencephaly; polymicrogyria; thick corpus callosum; Chiari malformation type I and obstructive ventricular dilation; cerebellar vein ectasia | PIK3CA | c.353G > A (p.Gly118Asp) | 14%/nd |
| 26 | 5/F | MCAP | Left hemibody partly reticulate CM; macrocephaly; overgrowth of left face, teeth, lower limb, and toes bilaterally; leg length discrepancy; sandal gap | Brain MRI: left hemimegalencephaly; thick corpus callosum | PIK3CA | c.2176G > A (p.Glu726Lys) | 19%/4% |
| 27 | 11/M | MCAP | Trunk, limb, and face partly reticulate CM; face and left lower limb prominent veins; macrocephaly; lower limb hypertrophy; upper limb hypotrophy; leg length discrepancy; trunk focally thick subcutaneous tissue; scoliosis; sandal gap; proximal toe syndactyly; psychomotor delay | Brain MRI: left hemimegalencephaly; thick corpus callosum | PIK3CA | c. 3132T > G (p.Asn1044Lys) | 21%/nd |
| 28 | 19/M | MCAP | Trunk, limb, and face partly reticulate CM; limb prominent veins; macrocephaly; left hemihypertrophy with macrodactyly; trunk focal thick subcutaneous tissue; sandal gap; proximal toe syndactyly; scoliosis; epidermal nevus; psychomotor delay | Brain MRI: left hemimegalencephaly; thick corpus callosum; Chiari malformation type I; hydrocephalus; cervical syringomyelia | PIK3CA | c.241G > A (p.Glu81Lys) | 3%/nd |
| 29 | 3/F | MCAP | Trunk and face *** partly reticulate CM; macrocephaly; left lower limb hypertrophy; sandal gap; toe syndactyly | Brain MRI: left hemimegalencephaly | PIK3CA | c.2176G > A (p.Glu726Lys) | 16%/nd |
| 30 | 4/F | MCAP | Centrofacial ***, trunk, and limb partly reticulate CM; macrocephaly; lower limb overgrowth; toe syndactyly; joint hypermobility; blaschkoid hypochromic macules on the thighs; psychomotor delay | Brain MRI: megalencephaly; polymicrogyria | PIK3CA | c.344G > C (p.Arg115Pro) | 15%/12% |
| 31 | 6/F | CLOVES | Left flank combined capillary–lymphatic–venous malformation; abdominal phlebectasia; dorsal lipomas; lower limb and left buttock overgrowth; upper limb and shoulder girdle hypotrophy; scoliosis; left foot hexadactyly; toe macrodactyly; widened web spaces; urogenital malformations; psychomotor delay | MRI: trunk and buttock lipomatous overgrowth; flank subcutaneous combined vascular malformation extending to the pelvis and retroperitoneum; terminal filum lipoma | PIK3CA | c.3140 A > G (p. His1047Arg) | 16.1%/nd |
| 32 | 1/M | CLOVES | Dorsal, abdominal, and flank CM and prominent veins; left flank and lumbosacral lipomas; left lower limb and buttock overgrowth; proximal toe syndactyly; sandal gap; triangular feet | DU: back lipomatous overgrowth | PIK3CA | c.3073A > G (p.Thr1025Ala) | 5%/nd |
| 33 | 7/M | CLOVES | Right upper limb CM, phlebectasia and lipomatous overgrowth; right thorax combined capillary–lymphatic–venous malformation; right hand hexadactyly and finger syndactyly; trunk lipomatous overgrowth | MRI: right hand lipomatous overgrowth; right upper limb venous malformation; thoracic and abdominal venous–lymphatic malformation; CT scan: multiple lung nodules | PIK3CA | c.3140A > G (p.His1047Arg) | 46.5%/nd |
| 34 | 10/F | KTS | Left lower limb and abdominal combined capillary–venous–lymphatic malformation; left lower limb overgrowth and pain; leg length discrepancy | DU, CT, and phlebography: abdominal and pelvic phlebectasias; left lower limb deep vein partial agenesis and embryonal superficial veins; portosystemic shunt through umbilical vein remnant | PIK3CA | c.325_327delGAA (p.Glu109del) | 2.7%/nd |
| 35 | 3/F | KTS | Left buttock and lower limb (including toes) combined capillary–venous–lymphatic malformation; left buttock and lower limb overgrowth and pain; leg length discrepancy | DU and CT: pelvic and left lower limb vein ectasia with persistent embryonic superficial veins; left leg deep vein agenesis | PIK3CA | c.3140A > T (p.His1047Leu) | 6%/nd |
| 36 | 1/M | Combined CLM | Right flank CM with overlying vesicles | DU: no deep anomalies | PIK3CA | c.1633G > A (p.Glu545Lys) | 3%/nd **** |
| 37 | 5/M | Combined CVM with overgrowth | Right flank CM with prominent veins and mild overgrowth; proximal toe syndactyly | DU and MRI: abdominal vein ectasia; no AVF | PIK3CA | c.325_327delGAA (p.Glu109del) | 4%/nd |
| 38 | 6/M | Microcystic LM | Grouped vesicles on intergluteal and right popliteal folds; gluteal swelling | MRI: subcutaneous and intramuscular microcystic lymphatic malformation extending from the buttocks to the right lower limb | PIK3CA | c.3140A > G (p.His1047Arg) | 2%/nd |
| 39 | 2/M | Combined VLM with overgrowth | Left upper limb normal-colored papules and nodules, bluish vesicles; prominent veins; left upper limb overgrowth | DU: left upper limb vein ectasia with thrombosis; lymphatic microcysts; thickened subcutaneous tissue | PIK3CA | c.1633G > A (p.Glu545Lys) | 1%/nd **** |
| 40 | 42/F | BRBNS | Diffuse (>100) bluish papules and nodules also on palmoplantar surfaces; violaceous congenital sacral plaque; recurrent gastrointestinal bleeding | MRI: cerebral, hepatic, and osseous vascular nodules; Video capsule endoscopy: numerous gastrointestinal vascular nodules | TEK | c.2690A > G (p.Tyr897Cys); c.2800T > C (p.Ser934Pro) | 1%/nd **** |
| 41 | 9/M | BRBNS | A few bluish papules of the limbs and right plant; violaceous congenital scalp plaque; prominent ipsilateral facial veins | Video capsule endoscopy: a few jejunal and ileal vascular nodules | TEK | c.2690A > T (p.Tyr897Phe); c.2743C > A (p.Arg915Ser) | 1%/nd |
| 42 | 11/M | PWS | Left upper limb and thorax reddish patch with pale halo, warmth to palpation; limb hypertrophy and prominent veins; several red-brownish macules on trunk, neck, and upper limbs | DU: left upper limb enlarged arteries with high flow, humeral vein arterialization; MRI: humeral and subclavian vein ectasia, early vein enhancement | RASA1 | c.1570_1571insTA (p.Cys525fs*19) | 3%/nd |
| 43 | 12/F | PWS | Left lower limb brownish patches, warmth to palpation; left lower limb overgrowth; pain; leg length discrepancy; trunk macules | DU: arteriolovenulous shunts; vein ectasia without arterialization MRI: vein ectasia | KRAS | c.183A > T (p.Gln61His) | 4%/nd |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diociaiuti, A.; Rotunno, R.; Pisaneschi, E.; Cesario, C.; Carnevale, C.; Condorelli, A.G.; Rollo, M.; Di Cecca, S.; Quintarelli, C.; Novelli, A.; et al. Clinical and Molecular Spectrum of Sporadic Vascular Malformations: A Single-Center Study. Biomedicines 2022, 10, 1460. https://doi.org/10.3390/biomedicines10061460
Diociaiuti A, Rotunno R, Pisaneschi E, Cesario C, Carnevale C, Condorelli AG, Rollo M, Di Cecca S, Quintarelli C, Novelli A, et al. Clinical and Molecular Spectrum of Sporadic Vascular Malformations: A Single-Center Study. Biomedicines. 2022; 10(6):1460. https://doi.org/10.3390/biomedicines10061460
Chicago/Turabian StyleDiociaiuti, Andrea, Roberta Rotunno, Elisa Pisaneschi, Claudia Cesario, Claudia Carnevale, Angelo Giuseppe Condorelli, Massimo Rollo, Stefano Di Cecca, Concetta Quintarelli, Antonio Novelli, and et al. 2022. "Clinical and Molecular Spectrum of Sporadic Vascular Malformations: A Single-Center Study" Biomedicines 10, no. 6: 1460. https://doi.org/10.3390/biomedicines10061460