
Hepatic Mitochondrial Dysfunction and Risk of Liver Disease in an Ovine Model of “PCOS Males”

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Tissue Collection
2.3. DNA, RNA, and Protein Extraction
2.4. RNA Sequencing Transcriptomic Analyses and Liver Protein Quantification
2.5. Bioinformatic Analysis
2.6. Quantitative RT-PCR
2.7. Mitochondrial DNA Content
2.8. Plasma Analyte Determination
2.9. Hepatic Lipids Determination
2.10. Hepatic Glycogen Determination
2.11. Hepatic ATP Determination
2.12. Hepatic Reactive Oxygen Species (ROS) Determination
2.13. Hepatic DNA Damage Determination
2.14. Hepatic Collagen Determination
2.15. Statistical Analysis
3. Results
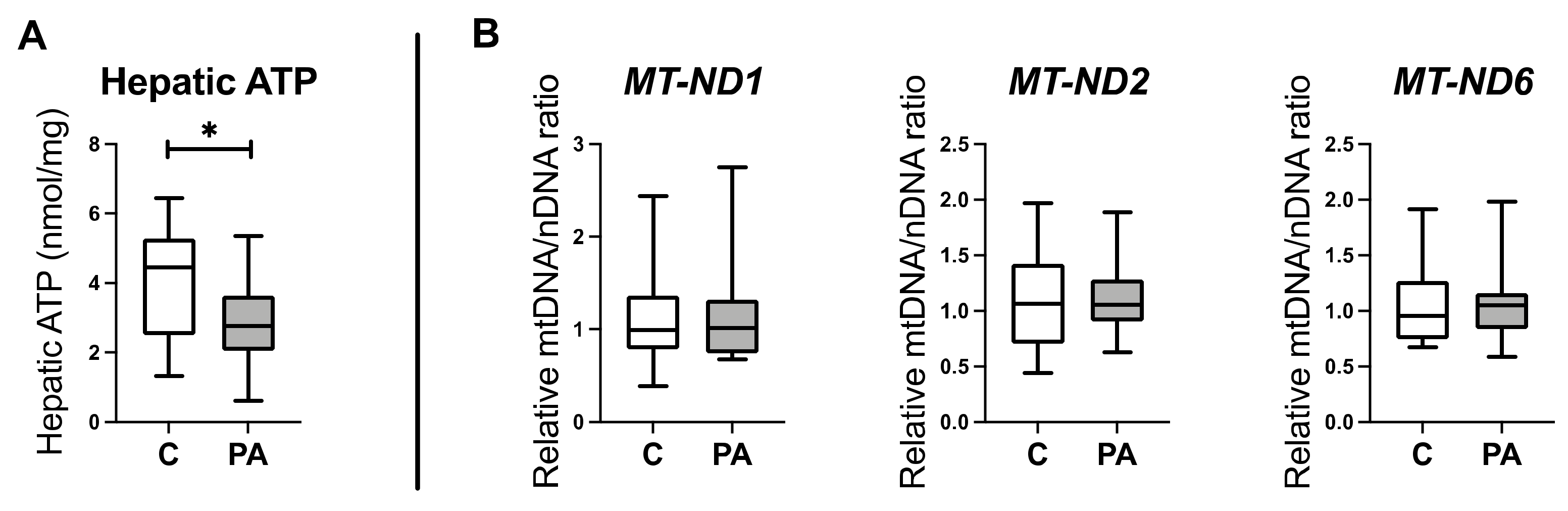
3.1. Altered Mitochondrial Function in Adolescent PA Males
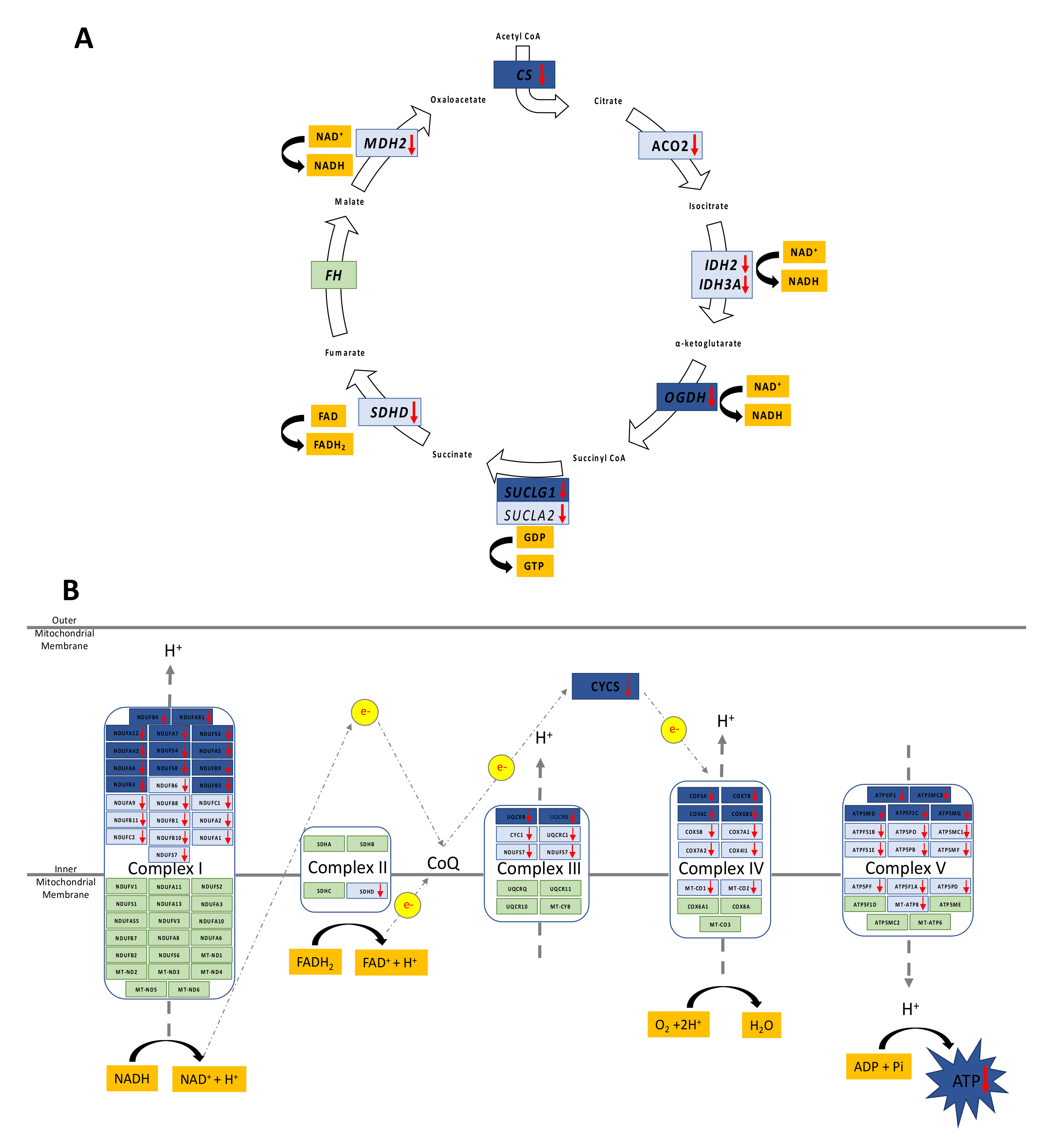
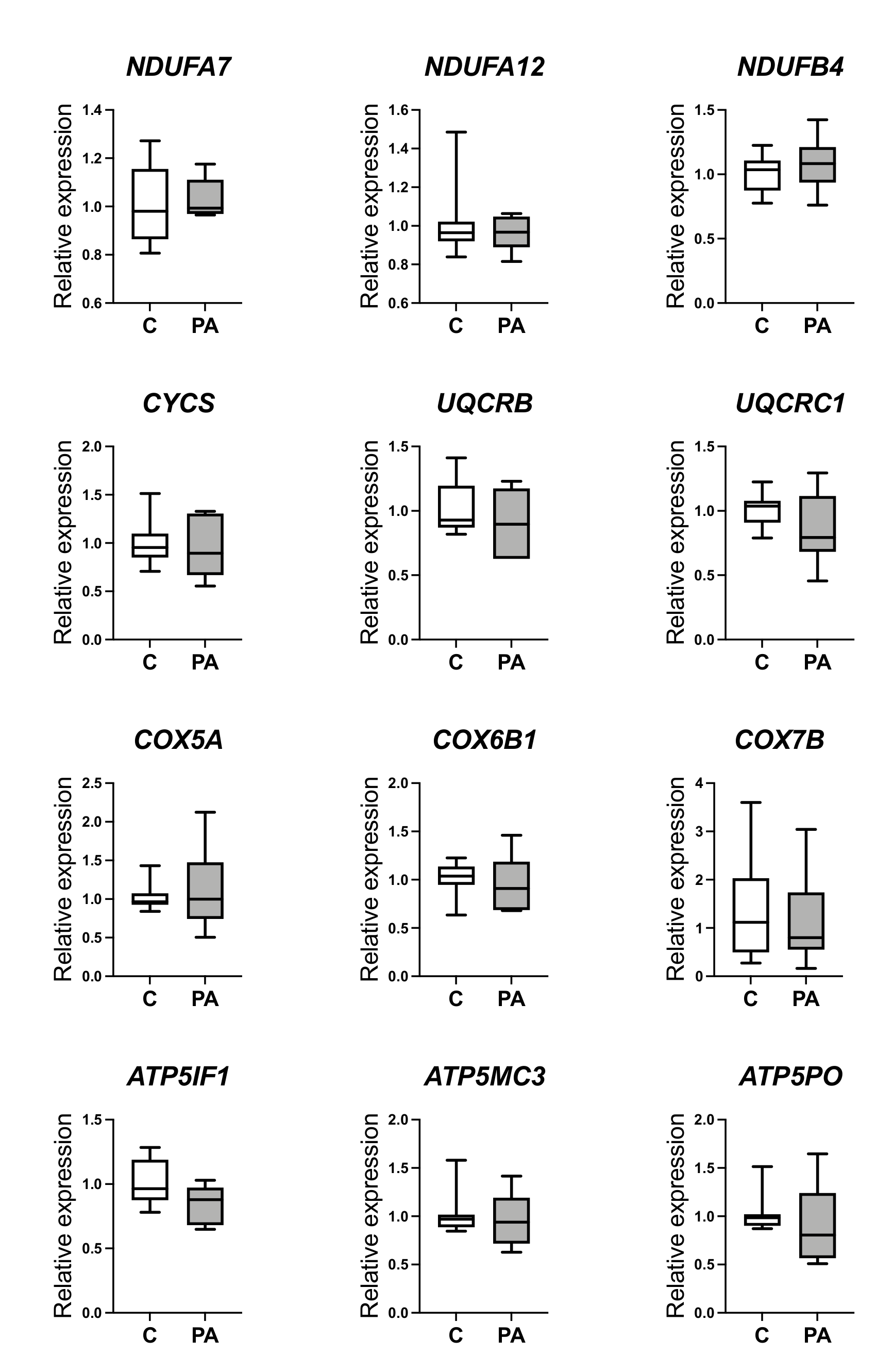
3.2. Decreased Oxidative Phosphorylation in Adolescent PA Males
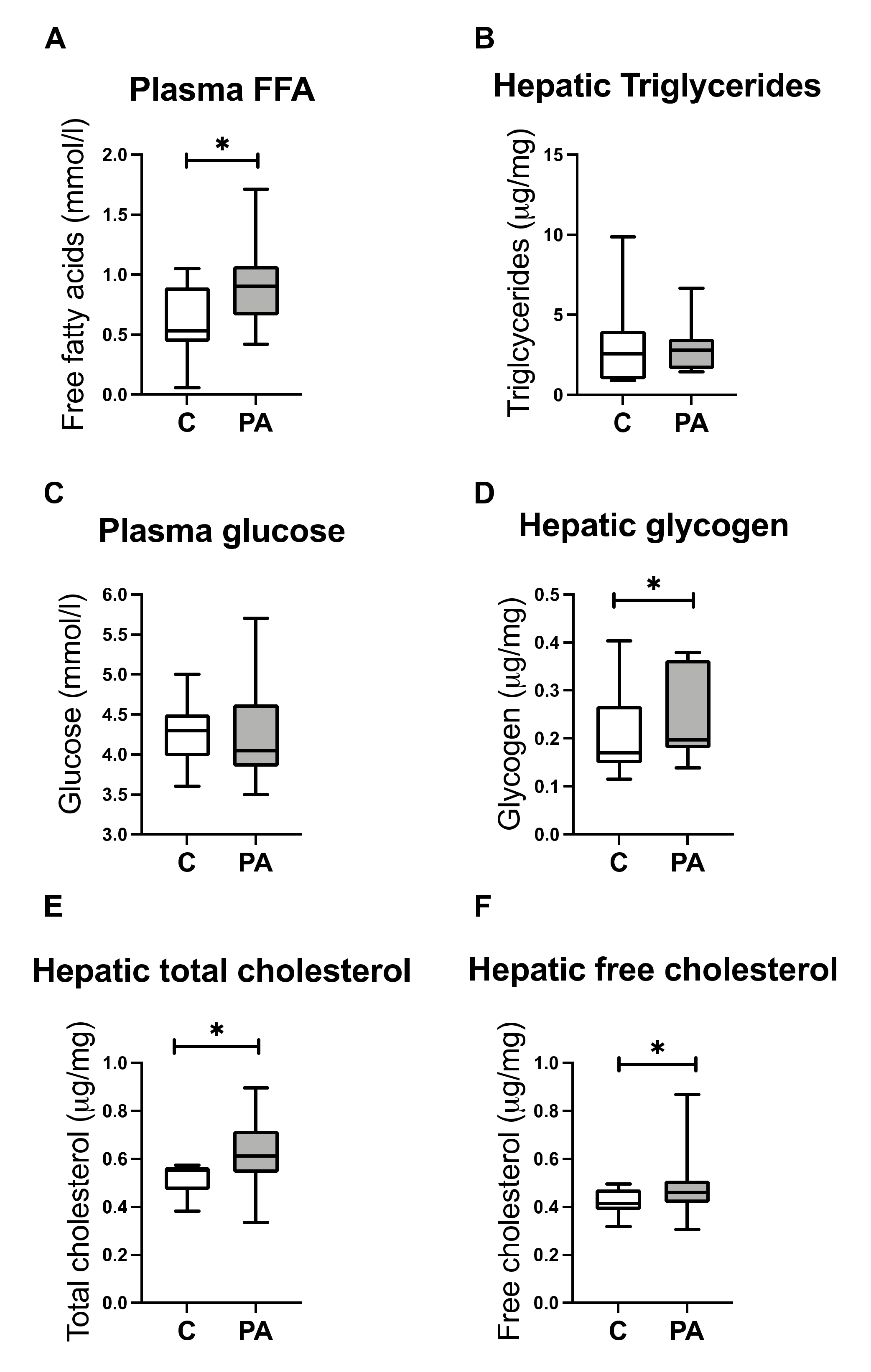
3.3. Increased FA Uptake into Mitochondria
3.4. Reduced Mitochondrial FA Utilization
3.5. Reduced Mitochondrial Glucose Utilization
3.6. Hepatic free Cholesterol Correlates with Mitochondrial Dysfunction
3.7. Reduced Capacity for Mitochondrial Rejuvenation
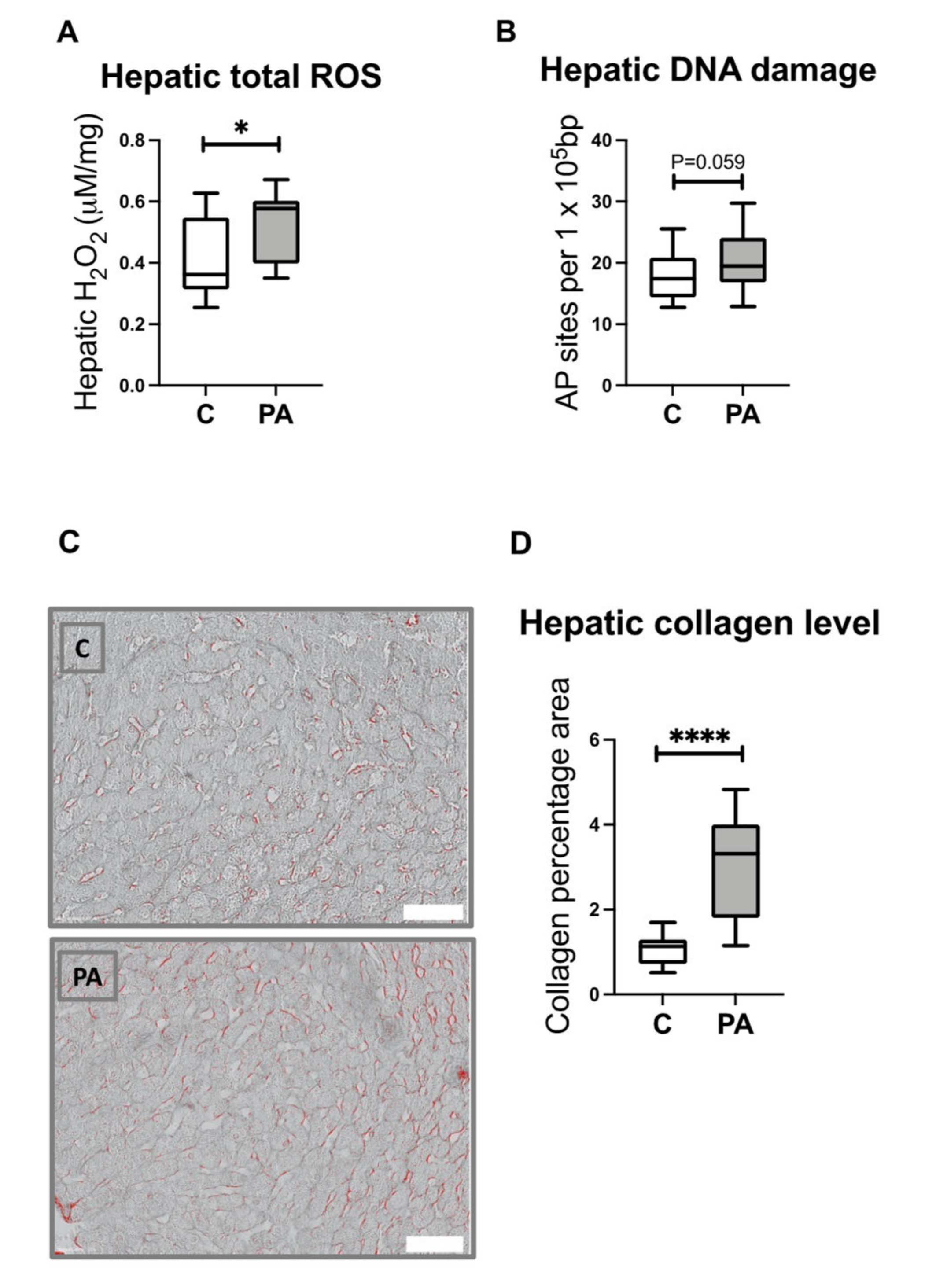
3.8. Increased Hepatic ROS Content in Adolescent PA Males
3.9. Increased Hepatic Collagen Deposition in PA Males
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fauser, B.C.J.M.; Tarlatzis, B.C.; Rebar, R.W.; Legro, R.S.; Balen, A.H.; Lobo, R.; Carmina, E.; Chang, J.; Yildiz, B.O.; Laven, J.S.E.; et al. Consensus on Women’s Health Aspects of Polycystic Ovary Syndrome (PCOS): The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil. Steril. 2011, 97, 28–38.e25. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.L.L.; Faria, L.C.; Guimarães, T.C.M.; Moreira, G.V.; Cândido, A.L.; Couto, C.A.; Reis, F.M. Non-Alcoholic Fatty Liver Disease in Women with Polycystic Ovary Syndrome: Systematic Review and Meta-Analysis. J. Endocrinol. Investig. 2017, 40, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Lunde, O.; Magnus, P.; Sandvik, L.; Høglo, S. Familial Clustering in the Polycystic Ovarian Syndrome. Gynecol. Obstet. Investig. 1989, 28, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Crisosto, N.; Sir-Petermann, T. Family Ties: Offspring Born to Women with Polycystic Ovary Syndrome. Curr. Opin. Endocr. Metab. Res. 2020, 12, 119–124. [Google Scholar] [CrossRef]
- Risal, S.; Pei, Y.; Lu, H.; Manti, M.; Fornes, R.; Pui, H.-P.; Zhao, Z.; Massart, J.; Ohlsson, C.; Lindgren, E.; et al. Prenatal Androgen Exposure and Transgenerational Susceptibility to Polycystic Ovary Syndrome. Nat. Med. 2019, 25, 1894–1904. [Google Scholar] [CrossRef]
- Cardoso, R.C.; Padmanabhan, V. Developmental Programming of PCOS Traits: Insights from the Sheep. Med. Sci. 2019, 7, 79. [Google Scholar] [CrossRef] [Green Version]
- Abbott, D.H.; Dumesic, D.A.; Levine, J.E. Hyperandrogenic Origins of Polycystic Ovary Syndrome—Implications for Pathophysiology and Therapy. Expert Rev. Endocrinol. Metab. 2019, 14, 131–143. [Google Scholar] [CrossRef]
- Daan, N.M.P.; Koster, M.P.H.; Steegers-Theunissen, R.P.; Eijkemans, M.J.C.; Fauser, B.C.J.M. Endocrine and Cardiometabolic Cord Blood Characteristics of Offspring Born to Mothers with and without Polycystic Ovary Syndrome. Fertil. Steril. 2017, 107, 261–268.e3. [Google Scholar] [CrossRef] [Green Version]
- Barrett, E.S.; Hoeger, K.M.; Sathyanarayana, S.; Abbott, D.H.; Redmon, J.B.; Nguyen, R.H.N.; Swan, S.H. Anogenital Distance in Newborn Daughters of Women with Polycystic Ovary Syndrome Indicates Fetal Testosterone Exposure. J. Dev. Orig. Health Dis. 2018, 9, 307–314. [Google Scholar] [CrossRef]
- Homburg, R.; Gudi, A.; Shah, A.; Layton, A.M. A Novel Method to Demonstrate That Pregnant Women with Polycystic Ovary Syndrome Hyper-Expose Their Fetus to Androgens as a Possible Stepping Stone for the Developmental Theory of PCOS. A Pilot Study. Reprod. Biol. Endocrinol. 2017, 15, 61. [Google Scholar] [CrossRef]
- Baillargeon, J.P.; Carpentier, A.C. Brothers of Women with Polycystic Ovary Syndrome Are Characterised by Impaired Glucose Tolerance, Reduced Insulin Sensitivity and Related Metabolic Defects. Diabetologia 2007, 50, 2424–2432. [Google Scholar] [CrossRef] [Green Version]
- Sam, S.; Coviello, A.D.; Sung, Y.-A.; Legro, R.S.; Dunaif, A. Metabolic Phenotype in the Brothers of Women with Polycystic Ovary Syndrome. Diabetes Care 2008, 31, 1237–1241. [Google Scholar] [CrossRef] [Green Version]
- Recabarren, S.E.; Smith, R.; Rios, R.; Maliqueo, M.; Echiburú, B.; Codner, E.; Cassorla, F.; Rojas, P.; Sir-Petermann, T. Metabolic Profile in Sons of Women with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 1820–1826. [Google Scholar] [CrossRef]
- Kaushal, R.; Parchure, N.; Bano, G.; Kaski, J.; Nussey, S.S. Insulin Resistance and Endothelial Dysfunction in the Brothers of Indian Subcontinent Asian Women with Polycystic Ovaries. Clin. Endocrinol. 2004, 60, 322–328. [Google Scholar] [CrossRef]
- Recabarren, S.E.; Sir-Petermann, T.; Rios, R.; Maliqueo, M.; Echiburú, B.; Smith, R.; Rojas-García, P.; Recabarren, M.; Rey, R.A. Pituitary and Testicular Function in Sons of Women with Polycystic Ovary Syndrome from Infancy to Adulthood. J. Clin. Endocrinol. Metab. 2008, 93, 3318–3324. [Google Scholar] [CrossRef] [Green Version]
- Crisosto, N.; Echiburú, B.; Maliqueo, M.; Luchsinger, M.; Rojas, P.; Recabarren, S.; Sir-Petermann, T. Reproductive and Metabolic Features during Puberty in Sons of Women with Polycystic Ovary Syndrome. Endocr. Connect. 2017, 6, 607–613. [Google Scholar] [CrossRef] [Green Version]
- de Wilde, M.A.; Eising, J.B.; Gunning, M.N.; Koster, M.P.H.; Evelein, A.M.V.; Dalmeijer, G.W.; Uiterwaal, C.S.P.M.; Eijkemans, M.J.C.; van der Ent, C.K.; Meijboom, F.J.; et al. Cardiovascular and Metabolic Health of 74 Children from Women Previously Diagnosed with Polycystic Ovary Syndrome in Comparison With a Population-Based Reference Cohort. Reprod. Sci. 2018, 25, 1492–1500. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, K.; Tripathi, A.; Dabadghao, P. Familial Clustering of Metabolic Phenotype in Brothers of Women with Polycystic Ovary Syndrome. Gynecol. Endocrinol. 2019, 35, 601–603. [Google Scholar] [CrossRef]
- Zhu, J.; Pujol-Gualdo, N.; Wittemans, L.B.L.; Lindgren, C.M.; Laisk, T.; Hirschhorn, J.N.; Chan, Y.-M. Evidence from Men for Ovary-Independent Effects of Genetic Risk Factors for Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2022, 107, e1577–e1587. [Google Scholar] [CrossRef]
- Aversa, A.; Vignera, S.L.; Rago, R.; Gambineri, A.; Nappi, R.E.; Calogero, A.E.; Ferlin, A. Fundamental Concepts and Novel Aspects of Polycystic Ovarian Syndrome: Expert Consensus Resolutions. Front. Endocrinol. 2020, 11, 516. [Google Scholar] [CrossRef]
- Guardo, F.D.; Ciotta, L.; Monteleone, M.; Palumbo, M. Male Equivalent Polycystic Ovarian Syndrome: Hormonal, Metabolic and Clinical Aspects. Int. J. Fertil. Steril. 2020, 14, 79–83. [Google Scholar] [CrossRef]
- Joham, A.E.; Teede, H.J. PCOS—A Metabolic Condition with Health Impacts on Women and Men. Nat. Rev. Endocrinol. 2022, 18, 197–198. [Google Scholar] [CrossRef]
- Siemienowicz, K.J.; Filis, P.; Shaw, S.; Douglas, A.; Thomas, J.; Mulroy, S.; Howie, F.; Fowler, P.A.; Duncan, W.C.; Rae, M.T. Fetal Androgen Exposure Is a Determinant of Adult Male Metabolic Health. Sci. Rep. 2019, 9, 20195. [Google Scholar] [CrossRef] [Green Version]
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 2019, 18, 2650–2666. [Google Scholar] [CrossRef]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Nobili, V.; Alisi, A.; Valenti, L.; Miele, L.; Feldstein, A.E.; Alkhouri, N. NAFLD in Children: New Genes, New Diagnostic Modalities and New Drugs. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 517–530. [Google Scholar] [CrossRef]
- Sanyal, A.J. Past, Present and Future Perspectives in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-Alcoholic Fatty Liver Disease and Dyslipidemia: An Update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef]
- Dowla, S.; Aslibekyan, S.; Goss, A.; Fontaine, K.; Ashraf, A.P. Dyslipidemia Is Associated with Pediatric Nonalcoholic Fatty Liver Disease. J. Clin. Lipidol. 2018, 12, 981–987. [Google Scholar] [CrossRef]
- Cai, J.; Wu, C.H.; Zhang, Y.; Wang, Y.Y.; Xu, W.D.; Lin, T.C.; Li, S.X.; Wang, L.H.; Zheng, J.; Sun, Y.; et al. High-Free Androgen Index Is Associated with Increased Risk of Non-Alcoholic Fatty Liver Disease in Women with Polycystic Ovary Syndrome, Independent of Obesity and Insulin Resistance. Int. J. Obes. 2017, 41, 1341–1347. [Google Scholar] [CrossRef]
- Morio, B.; Panthu, B.; Bassot, A.; Rieusset, J. Role of Mitochondria in Liver Metabolic Health and Diseases. Cell Calcium 2021, 94, 102336. [Google Scholar] [CrossRef] [PubMed]
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in Non-Alcoholic Fatty Liver Disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xie, Z.; Song, Q.; Li, J. Mitochondria Homeostasis: Biology and Involvement in Hepatic Steatosis to NASH. Acta Pharmacol. Sin. 2022, 43, 1141–1155. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Mukherjee, S. Mitochondrial Dysfunction: An Emerging Link in the Pathophysiology of Polycystic Ovary Syndrome. Mitochondrion 2020, 52, 24–39. [Google Scholar] [CrossRef]
- Rae, M.; Grace, C.; Hogg, K.; Wilson, L.M.; McHaffie, S.L.; Ramaswamy, S.; MacCallum, J.; Connolly, F.; McNeilly, A.S.; Duncan, C. The Pancreas Is Altered by in Utero Androgen Exposure: Implications for Clinical Conditions Such as Polycystic Ovary Syndrome (PCOS). PLoS ONE 2013, 8, e56263. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, S.; Grace, C.; Mattei, A.A.; Siemienowicz, K.; Brownlee, W.; MacCallum, J.; McNeilly, A.S.; Duncan, W.C.; Rae, M.T. Developmental Programming of Polycystic Ovary Syndrome (PCOS): Prenatal Androgens Establish Pancreatic Islet α/β Cell Ratio and Subsequent Insulin Secretion. Sci. Rep. 2016, 6, 27408. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of MtDNA/NDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Dabravolski, S.A.; Bezsonov, E.E.; Orekhov, A.N. The Role of Mitochondria Dysfunction and Hepatic Senescence in NAFLD Development and Progression. Biomed. Pharmacother. 2021, 142, 112041. [Google Scholar] [CrossRef]
- Martin, L.A.; Kennedy, B.E.; Karten, B. Mitochondrial Cholesterol: Mechanisms of Import and Effects on Mitochondrial Function. J. Bioenerg. Biomemb. 2014, 48, 137–151. [Google Scholar] [CrossRef]
- Musto, J.; Spengler, E.K.; German, M.N. Sexual Dimorphisms in Nonalcoholic Fatty Liver Disease. Curr. Hepatol. Rep. 2021, 20, 97–107. [Google Scholar] [CrossRef]
- Falzarano, C.; Lofton, T.; Osei-Ntansah, A.; Oliver, T.; Southward, T.; Stewart, S.; Andrisse, S. Nonalcoholic Fatty Liver Disease in Women and Girls with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2021, 107, 258–272. [Google Scholar] [CrossRef]
- Hogg, K.; Wood, C.; McNeilly, A.S.; Duncan, W.C. The in Utero Programming Effect of Increased Maternal Androgens and a Direct Fetal Intervention on Liver and Metabolic Function in Adult Sheep. PLoS ONE 2011, 6, e24877. [Google Scholar] [CrossRef] [Green Version]
- Siemienowicz, K.J.; Furmanska, K.; Filis, P.; Talia, C.; Thomas, J.; Fowler, P.A.; Rae, M.T.; Duncan, W.C. Pubertal FGF21 Deficit Is Central in the Metabolic Pathophysiology of an Ovine Model of Polycystic Ovary Syndrome. Mol. Cell. Endocrinol. 2021, 525, 111196. [Google Scholar] [CrossRef]
- Suiter, C.; Singha, S.K.; Khalili, R.; Shariat-Madar, Z. Free Fatty Acids: Circulating Contributors of Metabolic Syndrome. Cardiovasc. Hematol. Agents Med. Chem. 2018, 16, 20–34. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, Y.; Xu, C.; Hong, Y.; Lu, H.; Wu, J.; Chen, Y. Association between Serum Free Fatty Acid Levels and Nonalcoholic Fatty Liver Disease: A Cross-Sectional Study. Sci. Rep. 2014, 4, 5832. [Google Scholar] [CrossRef] [Green Version]
- Mashek, D.G. Hepatic Fatty Acid Trafficking: Multiple Forks in the Road. Adv. Nutr. Int. Rev. J. 2013, 4, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Hagenfeldt, L.; Wahren, J.; Pernow, B.; Räf, L. Uptake of Individual Free Fatty Acids by Skeletal Muscle and Liver in Man. J. Clin. Investig. 1972, 51, 2324–2330. [Google Scholar] [CrossRef]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular Mechanisms and the Role of Saturated Fatty Acids in the Progression of Non-Alcoholic Fatty Liver Disease. Prog. Lipid. Res. 2013, 52, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride Accumulation Protects against Fatty Acid-Induced Lipotoxicity. Proc. National. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular Mechanism of PPARα Action and Its Impact on Lipid Metabolism, Inflammation and Fibrosis in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, I.R.; Joshi, M. CPT1A-Mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [Green Version]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.B.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine Transport and Fatty Acid Oxidation. BBA Mol. Cell Res. 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Islam, S.M.T.; Won, J.; Khan, M.; Chavin, K.D.; Singh, I. Peroxisomal Footprint in the Pathogenesis of Nonalcoholic Steatohepatitis. Ann. Hepatol. 2020, 19, 466–471. [Google Scholar] [CrossRef]
- Randle, P.J. Regulatory Interactions between Lipids and Carbohydrates: The Glucose Fatty Acid Cycle after 35 Years. Diabetes Metab. Rev. 1998, 14, 263–283. [Google Scholar] [CrossRef]
- Von Wilamowitz-Moellendorff, A.; Hunter, R.W.; García-Rocha, M.; Kang, L.; López-Soldado, I.; Lantier, L.; Patel, K.; Peggie, M.W.; Martínez-Pons, C.; Voss, M.; et al. Glucose-6-Phosphate–Mediated Activation of Liver Glycogen Synthase Plays a Key Role in Hepatic Glycogen Synthesis. Diabetes 2013, 62, 4070–4082. [Google Scholar] [CrossRef] [Green Version]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The Interaction of Hepatic Lipid and Glucose Metabolism in Liver Diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [Green Version]
- Allende, D.S.; Gawrieh, S.; Cummings, O.W.; Belt, P.; Wilson, L.; Natta, M.V.; Behling, C.A.; Carpenter, D.; Gill, R.M.; Kleiner, D.E.; et al. Glycogenosis Is Common in Nonalcoholic Fatty Liver Disease and Is Independently Associated with Ballooning, but Lower Steatosis and Lower Fibrosis. Liver Int. 2021, 41, 996–1011. [Google Scholar] [CrossRef] [PubMed]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V. Lipid Droplets and Liver Disease: From Basic Biology to Clinical Implications. Nat Rev Gastroentero 2017, 14, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Meroni, M.; Paolini, E.; Macchi, C.; Dongiovanni, P. Mitochondrial Dynamics and Nonalcoholic Fatty Liver Disease (NAFLD): New Perspectives for a Fairy-Tale Ending? Metabolism 2021, 117, 154708. [Google Scholar] [CrossRef] [PubMed]
- Orlicky, D.J.; Libby, A.E.; Bales, E.S.; McMahan, R.H.; Monks, J.; Rosa, F.G.; McManaman, J.L. Perilipin-2 Promotes Obesity and Progressive Fatty Liver Disease in Mice through Mechanistically Distinct Hepatocyte and Extra-hepatocyte Actions. J. Physiol. 2019, 597, 1565–1584. [Google Scholar] [CrossRef] [Green Version]
- Asimakopoulou, A.; Engel, K.M.; Gassler, N.; Bracht, T.; Sitek, B.; Buhl, E.M.; Kalampoka, S.; Pinoé-Schmidt, M.; van Helden, J.; Schiller, J.; et al. Deletion of Perilipin 5 Protects against Hepatic Injury in Nonalcoholic Fatty Liver Disease via Missing Inflammasome Activation. Cells 2020, 9, 1346. [Google Scholar] [CrossRef]
- Fujii, H.; Ikura, Y.; Arimoto, J.; Sugioka, K.; Iezzoni, J.C.; Park, S.H.; Naruko, T.; Itabe, H.; Kawada, N.; Caldwell, S.H.; et al. Expression of Perilipin and Adipophilin in Nonalcoholic Fatty Liver Disease; Relevance to Oxidative Injury and Hepatocyte Ballooning. J. Atheroscler. Thromb. 2010, 16, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Carr, R.M.; Ahima, R.S. Pathophysiology of Lipid Droplet Proteins in Liver Diseases. Exp. Cell Res. 2016, 340, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Najt, C.P.; Senthivinayagam, S.; Aljazi, M.B.; Fader, K.A.; Olenic, S.D.; Brock, J.R.L.; Lydic, T.A.; Jones, A.D.; Atshaves, B.P. Liver-Specific Loss of Perilipin 2 Alleviates Diet-Induced Hepatic Steatosis, Inflammation, and Fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 310, G726–G738. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, A.R.; Sztalryd, C. Perilipin 5, a Lipid Droplet Protein Adapted to Mitochondrial Energy Utilization. Curr. Opin. Lipidol. 2014, 25, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Pawella, L.M.; Hashani, M.; Eiteneuer, E.; Renner, M.; Bartenschlager, R.; Schirmacher, P.; Straub, B.K. Perilipin Discerns Chronic from Acute Hepatocellular Steatosis. J. Hepatol. 2014, 60, 633–642. [Google Scholar] [CrossRef]
- Horn, C.L.; Morales, A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol. Commun. 2022, 6, 12–35. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic Lipotoxicity and the Pathogenesis of Nonalcoholic Steatohepatitis: The Central Role of Nontriglyceride Fatty Acid Metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Landis, C.S.; Jin, G.; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated with Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef]
- Endo-Umeda, K.; Makishima, M. Liver X Receptors Regulate Cholesterol Metabolism and Immunity in Hepatic Nonparenchymal Cells. Int. J. Mol. Sci. 2019, 20, 5045. [Google Scholar] [CrossRef] [Green Version]
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High Cholesterol/Low Cholesterol: Effects in Biological Membranes: A Review. Cell Biochem. Biophys. 2017, 75, 369–385. [Google Scholar] [CrossRef]
- Serviddio, G.; Blonda, M.; Bellanti, F.; Villani, R.; Iuliano, L.; Vendemiale, G. Oxysterols and Redox Signaling in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Free Radic. Res. 2013, 47, 881–893. [Google Scholar] [CrossRef]
- Elustondo, P.; Martin, L.A.; Karten, B. Mitochondrial Cholesterol Import. BBA Mol. Cell Biol. Lipids 2017, 1862, 90–101. [Google Scholar] [CrossRef]
- Coll, O.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Sensitivity of the 2-oxoglutarate Carrier to Alcohol Intake Contributes to Mitochondrial Glutathione Depletion. Hepatology 2003, 38, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial Free Cholesterol Loading Sensitizes to TNF- and Fas-Mediated Steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nuñez, S.; Nuño, N.; Enrich, C.; García-Ruiz, C.; Fernández-Checa, J.C. Cholesterol Enrichment in Liver Mitochondria Impairs Oxidative Phosphorylation and Disrupts the Assembly of Respiratory Supercomplexes. Redox Biol. 2019, 24, 101214. [Google Scholar] [CrossRef]
- Bellanti, F.; Villani, R.; Tamborra, R.; Blonda, M.; Iannelli, G.; di Bello, G.; Facciorusso, A.; Poli, G.; Iuliano, L.; Avolio, C.; et al. Synergistic Interaction of Fatty Acids and Oxysterols Impairs Mitochondrial Function and Limits Liver Adaptation during Nafld Progression. Redox Biol. 2018, 15, 86–96. [Google Scholar] [CrossRef]
- Domínguez-Pérez, M.; Simoni-Nieves, A.; Rosales, P.; Nuño-Lámbarri, N.; Rosas-Lemus, M.; Souza, V.; Miranda, R.U.; Bucio, L.; Carvajal, S.U.; Marquardt, J.U.; et al. Cholesterol Burden in the Liver Induces Mitochondrial Dynamic Changes and Resistance to Apoptosis. J. Cell. Physiol. 2019, 234, 7213–7223. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Haddad, A.; Osme, A.; Kim, C.; Borzou, A.; Ilchenko, S.; Allende, D.; Dasarathy, S.; McCullough, A.J.; Sadygov, R.G.; et al. Hepatic Mitochondrial Defects in a Mouse Model of NAFLD Are Associated with Increased Degradation of Oxidative Phosphorylation Subunits. Mol. Cell. Proteom 2018, 17, 2371–2386. [Google Scholar] [CrossRef] [Green Version]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in Liver ATP Homeostasis in Human Nonalcoholic Steatohepatitis: A Pilot Study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D. Mitochondrial Abnormalities in Non-Alcoholic Steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell–Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic Steatohepatitis: Association of Insulin Resistance and Mitochondrial Abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Portincasa, P.; Grattagliano, I.; Lauterburg, B.H.; Palmieri, V.O.; Palasciano, G.; Stellaard, F. Liver Breath Tests Non-Invasively Predict Higher Stages of Non-Alcoholic Steatohepatitis. Clin. Sci. 2006, 111, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banasch, M.; Ellrichmann, M.; Tannapfel, A.; Schmidt, W.E.; Goetze, O. The Non-Invasive (13)C-Methionine Breath Test Detects Hepatic Mitochondrial Dysfunction as a Marker of Disease Activity in Non-Alcoholic Steatohepatitis. Eur. J. Med. Res. 2011, 16, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Carreras, M.; Hoyo, P.D.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective Hepatic Mitochondrial Respiratory Chain in Patients with Nonalcoholic Steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. MTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol. Cell 2017, 67, 922–935.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Zhang, D.; Long, B.; An, T.; Zhang, J.; Zhou, L.-Y.; Liu, C.-Y.; Li, P.-F. NFAT4-Dependent MiR-324-5p Regulates Mitochondrial Morphology and Cardiomyocyte Cell Death by Targeting Mtfr1. Cell Death Dis. 2015, 6, e2007. [Google Scholar] [CrossRef] [Green Version]
- Palma, E.; Ma, X.; Riva, A.; Iansante, V.; Dhawan, A.; Wang, S.; Ni, H.-M.; Sesaki, H.; Williams, R.; Ding, W.-X.; et al. Dynamin-1–Like Protein Inhibition Drives Megamitochondria Formation as an Adaptive Response in Alcohol-Induced Hepatotoxicity. Am. J. Pathol. 2019, 189, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Parone, P.A.; Cruz, S.D.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.-C. Preventing Mitochondrial Fission Impairs Mitochondrial Function and Leads to Loss of Mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin Is Recruited Selectively to Impaired Mitochondria and Promotes Their Autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 Acts in Parallel to the PINK1/Parkin Pathway to Control Mitochondrial Function and Autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin Is Phosphorylated by PINK1 to Activate Parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 Causes Mitochondrial Functional Defects and Increased Sensitivity to Oxidative Stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Zhi, L.; Qin, Q.; Muqeem, T.; Seifert, E.L.; Liu, W.; Zheng, S.; Li, C.; Zhang, H. Loss of PINK1 Causes Age-Dependent Decrease of Dopamine Release and Mitochondrial Dysfunction. Neurobiol. Aging 2019, 75, 1–10. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The Mitochondrial Deubiquitinase USP30 Opposes Parkin-Mediated Mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. Two Different Axes CALCOCO2-RB1CC1 and OPTN-ATG9A Initiate PRKN-Mediated Mitophagy. Autophagy 2020, 16, 2105–2107. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy Receptor FUNDC1 Regulates Mitochondrial Dynamics and Mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) Repression of PGC-1α Contributes to Neurodegeneration in Parkinson’s Disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional Control of Mitochondrial Biogenesis: The Central Role of PGC-1α. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.-P.; Liu, X.-J.; Xie, L.; Shen, X.-Z.; Wu, J. Impaired Mitophagy Triggers NLRP3 Inflammasome Activation during the Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis. Lab. Investig. 2019, 99, 749–763. [Google Scholar] [CrossRef]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 Inhibition Attenuates Non-Alcoholic Fatty Liver Disease via Reversing Parkin-Related Mitophagy. Redox Biol. 2019, 21, 101120. [Google Scholar] [CrossRef]
- Yamada, T.; Murata, D.; Adachi, Y.; Itoh, K.; Kameoka, S.; Igarashi, A.; Kato, T.; Araki, Y.; Huganir, R.L.; Dawson, T.M.; et al. Mitochondrial Stasis Reveals P62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018, 28, 588–604.e5. [Google Scholar] [CrossRef] [Green Version]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Bockowska, S.B.; Lebensztejn, D.M. Pediatric Non-Alcoholic Steatohepatitis: The First Report on the Ultrastructure of Hepatocyte Mitochondria. World J. Gastroenterol. 2014, 20, 4335–4340. [Google Scholar] [CrossRef]
- Shami, G.J.; Cheng, D.; Verhaegh, P.; Koek, G.; Wisse, E.; Braet, F. Three-Dimensional Ultrastructure of Giant Mitochondria in Human Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2021, 11, 3319. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Auger, C.; Alhasawi, A.; Contavadoo, M.; Appanna, V.D. Dysfunctional Mitochondrial Bioenergetics and the Pathogenesis of Hepatic Disorders. Front. Cell Dev. Biol. 2015, 3, 40. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.; Park, G.-S.; Shin, S.-M.; Park, C.E.; Kim, S.; Han, S.J.; Pham, H.Q.; Shin, J.-H.; Lee, D.-W. Mitochondrial Mutations in Cholestatic Liver Disease with Biliary Atresia. Sci. Rep. 2018, 8, 905. [Google Scholar] [CrossRef]
- Karkucinska-Wieckowska, A.; Simoes, I.C.M.; Kalinowski, P.; Lebiedzinska-Arciczewska, M.; Zieniewicz, K.; Milkiewicz, P.; Górska-Ponikowska, M.; Pinton, P.; Malik, A.N.; Krawczyk, M.; et al. Mitochondria, Oxidative Stress and Non-Alcoholic Fatty Liver Disease: A Complex Relationship. Eur. J. Clin. Investig. 2021, 52, e13622. [Google Scholar] [CrossRef]
- Marí, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 909. [Google Scholar] [CrossRef]
- Klisic, A.; Kavaric, N.; Ninic, A.; Kotur-Stevuljevic, J. Oxidative Stress and Cardiometabolic Biomarkers in Patients with Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2021, 11, 18455. [Google Scholar] [CrossRef]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic Markers of Lipid Peroxidation and Antioxidants in Patients with Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef]
- Nishida, N.; Yada, N.; Hagiwara, S.; Sakurai, T.; Kitano, M.; Kudo, M. Unique Features Associated with Hepatic Oxidative DNA Damage and DNA Methylation in Non-alcoholic Fatty Liver Disease. J. Gastroen. Hepatol. 2016, 31, 1646–1653. [Google Scholar] [CrossRef]
- Tanaka, S.; Miyanishi, K.; Kobune, M.; Kawano, Y.; Hoki, T.; Kubo, T.; Hayashi, T.; Sato, T.; Sato, Y.; Takimoto, R.; et al. Increased Hepatic Oxidative DNA Damage in Patients with Nonalcoholic Steatohepatitis Who Develop Hepatocellular Carcinoma. J. Gastroenterol. 2013, 48, 1249–1258. [Google Scholar] [CrossRef]
- Read, A.D.; Bentley, R.E.T.; Archer, S.L.; Dunham-Snary, K.J. Mitochondrial Iron–Sulfur Clusters: Structure, Function, and an Emerging Role in Vascular Biology. Redox Biol. 2021, 47, 102164. [Google Scholar] [CrossRef]
- Tana, C.; Ballestri, S.; Ricci, F.; Vincenzo, A.D.; Ticinesi, A.; Gallina, S.; Giamberardino, M.A.; Cipollone, F.; Sutton, R.; Vettor, R.; et al. Cardiovascular Risk in Non-Alcoholic Fatty Liver Disease: Mechanisms and Therapeutic Implications. Int. J. Environ. Res. Public Health 2019, 16, 3104. [Google Scholar] [CrossRef] [Green Version]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and Cardiovascular Diseases: A Clinical Review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | p-Value | Direction | Molecules |
|---|---|---|---|
| Mitochondrial Function | |||
| Mitochondrial dysfunction | 5.43 × 10−11 | NP | 31 genes |
| 2.85 × 10−12 | NP | 20 proteins | |
| Oxidative phosphorylation | 3.75 × 10−9 | ↓↓ | 22 genes |
| 1.02 × 10−7 | ↓↓ | 11 proteins | |
| TCA cycle | 1.13 × 10−7 | ↓ | 8 proteins |
| Mitochondrial respiratory chain deficiency | 3.36 × 10−7 | NP | 10 proteins |
| Assembly of respiratory chain complex | 3.37 × 10−4 | NP | 5 proteins |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siemienowicz, K.J.; Filis, P.; Thomas, J.; Fowler, P.A.; Duncan, W.C.; Rae, M.T. Hepatic Mitochondrial Dysfunction and Risk of Liver Disease in an Ovine Model of “PCOS Males”. Biomedicines 2022, 10, 1291. https://doi.org/10.3390/biomedicines10061291
Siemienowicz KJ, Filis P, Thomas J, Fowler PA, Duncan WC, Rae MT. Hepatic Mitochondrial Dysfunction and Risk of Liver Disease in an Ovine Model of “PCOS Males”. Biomedicines. 2022; 10(6):1291. https://doi.org/10.3390/biomedicines10061291
Chicago/Turabian StyleSiemienowicz, Katarzyna J., Panagiotis Filis, Jennifer Thomas, Paul A. Fowler, W. Colin Duncan, and Mick T. Rae. 2022. "Hepatic Mitochondrial Dysfunction and Risk of Liver Disease in an Ovine Model of “PCOS Males”" Biomedicines 10, no. 6: 1291. https://doi.org/10.3390/biomedicines10061291