
One Molecule for Mental Nourishment and More: Glucose Transporter Type 1—Biology and Deficiency Syndrome

 , ,
, ,  , , , , , and
, , , , , and
Abstract
:1. Introduction
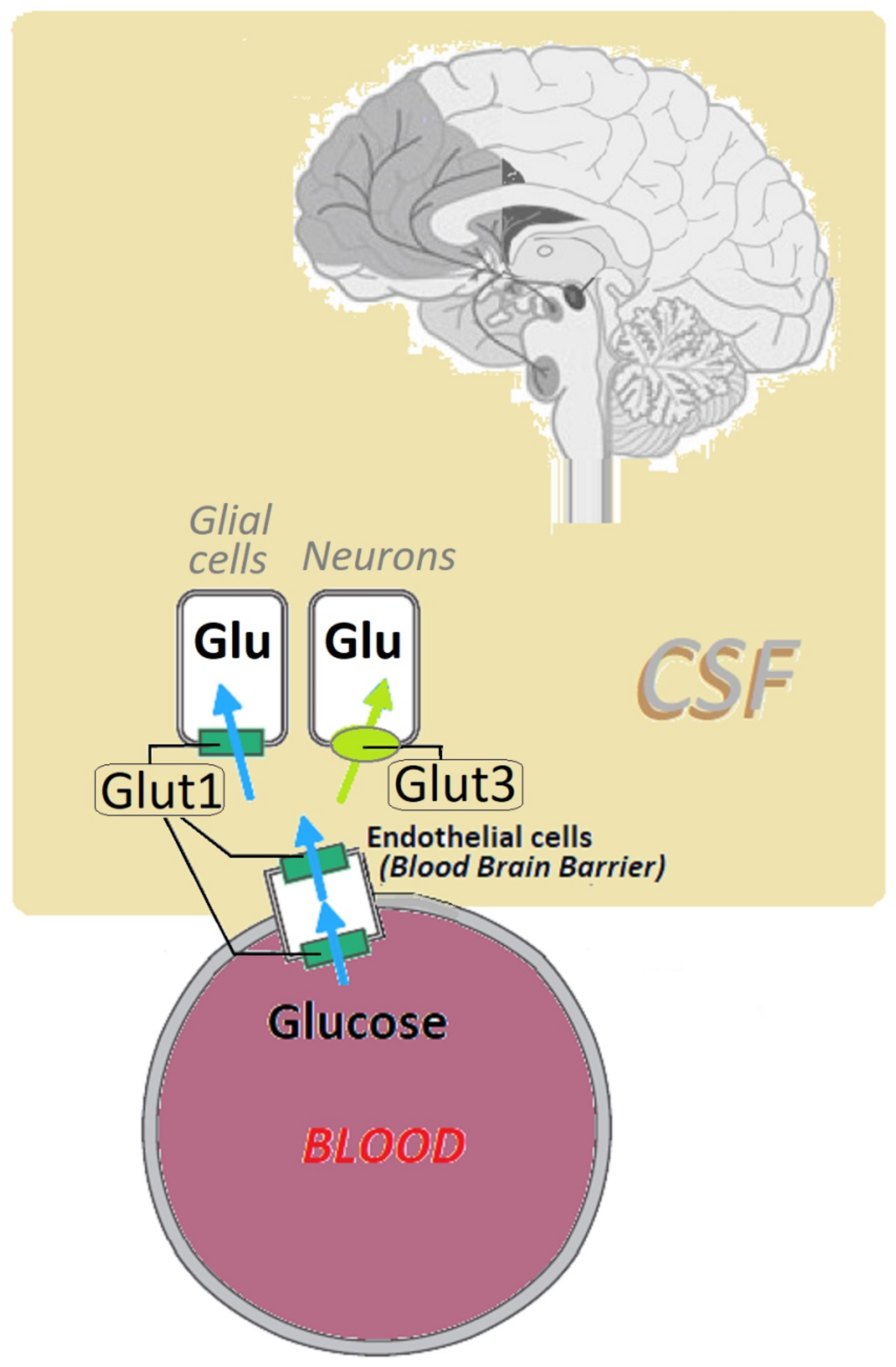
2. Glucose Transporter Protein Type 1 (Glut1): Structure and Function
3. Pathophysiology
4. Clinical Picture of Glut1 Deficiency Syndrome
5. Genetics and Metabolic Changes
6. Diagnosis
7. Treatment, Prognosis and Research
8. Glut-1 Deficiency in Other Tissues: Expanding the Clinical Phenotype?
8.1. Glut1 in Vessels
8.2. Glut1 in Retina
8.3. Glut 1 in Erythrocytes
8.4. Glut1 in Muscles
8.5. Glut1 in Immune Cells
9. Glut1 Inhibition in Other Settings
9.1. Glut1 in Keratinisation Disorders
9.2. Glut1 in Eye Diseases
9.3. Glut1 in Kidney Diseases
9.4. Glut1 in Placental Pathology
9.5. Glut1 in Heart Failure
9.6. Glut 1 in Crystal-Induced Inflammation
9.7. Glut1 in Viral Infections
9.8. Glut1 in Alzheimer’s Disease and Other Neurodegenerative Disorders
9.9. Glut1 in Cancers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell/Tissue | Glut1 | References |
|---|---|---|
| Vessels | Endothelial Glut1 is involved in vessel branching and migration in brain angiogenesis; Glut1 endothelial cell-specific haploinsufficiency was involved in triggering neuroinflammation | [24,68] |
| Retina | Glut1 depletion affects retinal angiogenesis and photoreceptor viability | [24] |
| Erythrocytes | Glut1 represents 5% of the erythrocyte membrane proteins In Glut1DS exercise may result in hemolytic anemia | [12,52] |
| Skin | Glut1 mediates glucose transport in keratinocytes, wound- and inflammation-associated keratinocyte proliferation | [68] |
| Muscle | Glut1 responds for 30–40% of skeletal muscle basal glucose uptake Glut1DS-associated muscle hypotonia may sometimes involve speech-associated muscles | [29,74,75] |
| Heart | Glut1—main glucose transporter in heart, but not critical for normal cardiac function | [91] |
| Placenta | Glut1 expressed in placenta, syncytiotrophoblast, cytotrophoblast, endothelial cells and villous stroma; Glut1 is decreased in chronic hypoxia and in preeclampsia, but not in intrauterine growth restriction | [8,90] |
| Kidneys | Glut1 expressed in glomerulus mainly in mesangial cells; Glut1 along with cytokines and growth factors favors diabetic glomerulosclerosis | [87,88] |
| Immune cells | Glut1 involved in macrophage plasticity and phenotype reprogramming in innate immune adaptations including in trained immunity; In gout interleukin-1 beta production depends on macrophage Glut1-mediated glucose uptake; Glut1 deficiency reduces T effector ability to induce inflammation, not affecting Tregs | [77,78,79,81,84] |
| Viral infections | Glut1 is a HTLV1 receptor molecule. The HCMV early protein IE72 downregulates GLUT1 to increase GLUT4 expression. In COVID19 Glut1 is critically involved, and a low Glut1/NPE-1 predicts COVID19 severity | [93,94,96] |
| Brain regions in Alzeimer’s disease | Glut1 and Glut3 are reduced in the hippocampus and cortex after β-amyloid deposition, resulting in reduced glucose uptake and metabolism | [97] |
| Cells in tumors | Glut1 is the predominant transporter in tumors, differentially required in different tumorigenesis stages. Blocking Glut1 inhibits tumorigenesis without disrupting normal cells. | [102,104] |
10. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Landowski, C.P.; Suzuki, Y.; Hediger, M.A. The Mammalian Transporter Families. In Seldin and Giebisch’s The Kidney: Physiology & Pathophysiology, 4th ed.; Alpern, R.J., Hebert, S.C., Eds.; Elsevier: Oxford, UK, 2008; pp. 91–146. [Google Scholar]
- Mueckler, M.; Caruso, C.; Baldwin, S.A.; Panico, M.; Blench, I.; Morris, H.R.; Allard, W.J.; Lienhard, G.E.; Lodish, H.F. Sequence and Structure of a Human Glucose Transporter. Science 1985, 229, 941–945. [Google Scholar] [CrossRef]
- Klepper, J.; Voit, T. Facilitated glucose transporter protein type 1 (GLUT1) deficiency syndrome: Impaired glucose transport into brain—A review. Eur. J. Pediatr. 2002, 161, 295–304. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, D.C.; Trifiletti, R.R.; Jacobson, R.I.; Ronen, G.M.; Behmand, R.A.; Harik, S.I. Defective Glucose Transport across the Blood-Brain Barrier as a Cause of Persistent Hypoglycorrhachia, Seizures, and Developmental Delay. N. Engl. J. Med. 1991, 325, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J.; Akman, C.; Armeno, M.; Auvin, S.; Cervenka, M.; Cross, H.J.; De Giorgis, V.; Della Marina, A.; Engelstad, K.; Heussinger, N.; et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 2020, 5, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Park, S.H.; De Vivo, D.C.; Monani, U.R. Therapeutic strategies for glucose transporter 1 deficiency syndrome. Ann. Clin. Transl. Neurol. 2019, 6, 1923–1932. [Google Scholar] [CrossRef] [Green Version]
- Galochkina, T.; Chong, M.N.F.; Challali, L.; Abbar, S.; Etchebest, C. New insights into GluT1 mechanics during glucose transfer. Sci. Rep. 2019, 9, 998. [Google Scholar] [CrossRef]
- Illsley, N.P.; Baumann, M.U. Human placental glucose transport in fetoplacental growth and metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1866, 165359. [Google Scholar] [CrossRef]
- Matsuo, S.; Hiasa, M.; Omote, H. Functional characterization and tissue localization of the facilitative glucose transporter GLUT12. J. Biochem. 2020, 168, 611–620. [Google Scholar] [CrossRef]
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflug. Arch. 2020, 472, 1273–1298. [Google Scholar] [CrossRef]
- Chen, L.Q.; Cheung, L.S.; Feng, L.; Tanner, W.; Frommer, W.B. Transport of sugars. Annu. Rev. Biochem. 2015, 84, 865–894. [Google Scholar] [CrossRef]
- Santer, R.; Klepper, J. Disorders of Glucose Transport in Inherited Metabolic Diseases, Diagnostic and Treatment, 6th ed.; Saudubray, J.M., Baumgartner, M., Waler, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 175–184. [Google Scholar]
- Pragallapati, S.; Manyam, R. Glucose transporter 1 in health and disease. J. Oral Maxillofac. Pathol. 2019, 23, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Gras, D.; Roze, E.; Caillet, S.; Méneret, A.; Doummar, D.; de Villemeur, T.B.; Vidailhet, M.; Mochel, F. GLUT1 deficiency syndrome: An update. Rev. Neurol. 2014, 170, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J. Glucose transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. Epilepsia 2008, 49, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Cheeseman, C. Structure of and functional insight into the GLUT family of membrane transporters. Cell Health Cytoskelet. 2015, 7, 167–183. [Google Scholar]
- Kasahara, M.; Hinkle, P.C. Reconstitution and purification of the D-glucose transporter from human erythrocytes. J. Biol. Chem. 1977, 252, 7384–7390. [Google Scholar] [CrossRef]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef]
- Custódio, T.F.; Paulsen, P.A.; Frain, K.M.; Pedersen, B.P. Structural comparison of GLUT1 to GLUT3 reveal transport regulation mechanism in sugar porter family. Life Sci. Alliance 2021, 4, e202000858. [Google Scholar] [CrossRef]
- Salas-Burgos, A.; Iserovich, P.; Zuniga, F.; Vera, J.C.; Fischbarg, J. Predicting the Three-Dimensional Structure of the Human Facilitative Glucose Transporter Glut1 by a Novel Evolutionary Homology Strategy: Insights on the Molecular Mechanism of Substrate Migration, and Binding Sites for Glucose and Inhibitory Molecules. Biophys. J. 2004, 87, 2990–2999. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Park, S.H.; Petri, S.; Yu, H.; Rueda, C.B.; Abel, E.D.; Kim, C.Y.; Hillman, E.M.; Li, F.; Lee, Y.; et al. An early endothelial cell–specific requirement for Glut1 is revealed in Glut1 deficiency syndrome model mice. JCI Insight 2021, 6, e145789. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Monani, U.R. Glut1 deficiency syndrome: New and emerging insights into a prototypical brain energy failure disorder. Neurosci. Insights 2021, 16, 26331055211011507. [Google Scholar] [CrossRef] [PubMed]
- Veys, K.; Fan, Z.; Ghobrial, M.; Bouché, A.; García-Caballero, M.; Vriens, K.; Conchinha, N.V.; Seuwen, A.; Schlegel, F.; Gorski, T.; et al. Role of the GLUT1 Glucose Transporter in Postnatal CNS Angiogenesis and Blood-Brain Barrier Integrity. Circ. Res. 2020, 127, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Tzadok, M.; Nissenkorn, A.; Porper, K.; Matot, I.; Marcu, S.; Anikster, Y.; Menascu, S.; Bercovich, D.; Ben Zeev, B. The Many Faces of Glut1 Deficiency Syndrome. J. Child Neurol. 2013, 29, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-Y.; Yang, H.; Klepper, J.; Fischbarg, J.; Wang, D.; De Vivo, D.C. Glucose Transporter Type 1 Deficiency Syndrome (Glut1DS): Methylxanthines Potentiate GLUT1 Haploinsufficiency In Vitro. Pediatr. Res. 2001, 50, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidner, G.; Alvarez, M.G.; Yeh, J.-I.; O’Driscoll, K.R.; Klepper, J.; Stump, T.S.; Wang, D.; Spinner, N.B.; Birnbaum, M.J.; De Vivo, D.C. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat. Genet. 1998, 18, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Pascual, J.M.; De Vivo, D. Glucose Transporter Type 1 Deficiency Syndrome. In GeneReviews® (Internet); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993; (Updated 1 March 2018); Available online: https://europepmc.org/article/NBK/nbk1430 (accessed on 20 April 2022).
- Pascual, J.M.; Wang, N.; Lecumberri, B.; Yang, H.; Mao, X.; Yang, R.; De Vivo, D.C. GLUT1 deficiency and other glucose transporter diseases. Eur. J. Endocrinol. 2004, 150, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Kelly, D.I.; Su, J.; Pascual, J.M. Clinical Aspects of Glucose Transporter Type 1 Deficiency. JAMA Neurol. 2017, 74, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Shen, Y.; Su, T.; Liu, Y.; Xu, S. Clinical and Genetic Characteristics of Chinese Children with GLUT1 Deficiency Syndrome: Case Report and Literature Review. Front. Genet. 2021, 12, 734481. [Google Scholar] [CrossRef]
- Castellotti, B.; Ragona, F.; Freri, E.; Solazzi, R.; Ciardullo, S.; Tricomi, G.; Venerando, A.; Salis, B.; Canafoglia, L.; Villani, F.; et al. Screening of SLC2A1 in a large cohort of patients suspected for Glut1 deficiency syndrome: Identification of novel variants and associated phenotypes. J. Neurol. 2019, 266, 1439–1448. [Google Scholar] [CrossRef]
- Winczewska-Wiktor, A.; Hoffman-Zacharska, D.; Starczewska, M.; Kaczmarek, I.; Badura-Stronka, M.; Steinborn, B. Variety of symptoms of GLUT1 deficiency syndrome in three-generation family. Epilepsy Behav. 2020, 106, 107036. [Google Scholar] [CrossRef]
- De Giorgis, V.; Varesio, C.; Baldassari, C.; Piazza, E.; Olivotto, S.; Macasaet, J.; Balottin, U.; Veggiotti, P. Atypical Manifestations in Glut1 Deficiency Syndrome. J. Child Neurol. 2016, 31, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.S.; Pons, R.; Engelstad, K.; Kane, S.A.; Goldberg, M.E.; De Vivo, D.C. Paroxysmal eye–head movements in Glut1 deficiency syndrome. Neurology 2017, 88, 1666–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Lee, J.S.; Lee, Y.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J.-H. Diagnostic Challenges Associated with GLUT1 Deficiency: Phenotypic Variabilities and Evolving Clinical Features. Yonsei Med. J. 2019, 60, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Pascual, J.M.; Yang, H.; Engelstad, K.; Jhung, S.; Sun, R.P.; De Vivo, D.C. Glut-1 deficiency syndrome: Clinical, genetic, and therapeutic aspects. Ann. Neurol. 2004, 57, 111–118. [Google Scholar] [CrossRef]
- Pong, A.W.; Geary, B.R.; Engelstad, K.M.; Natarajan, A.; Yang, H.; De Vivo, D.C. Glucose transporter type I deficiency syndrome: Epilepsy phenotypes and outcomes. Epilepsia 2012, 53, 1503–1510. [Google Scholar] [CrossRef]
- Leen, W.G.; Taher, M.; Verbeek, M.; Kamsteeg, E.J.; Van De Warrenburg, B.P.; Willemsen, M.A. GLUT1 deficiency syndrome into adulthood: A follow-up study. J. Neurol. 2014, 261, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Rotstein, M.; Bs, K.E.; Yang, H.; Wang, D.; Levy, B.; Chung, W.K.; De Vivo, D.C. Glut1 deficiency: Inheritance pattern determined by haploinsufficiency. Ann. Neurol. 2010, 68, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Symonds, J.; Zuberi, S.M.; Stewart, K.; McLellan, A.; O‘Regan, M.; MacLeod, S.; Jollands, A.; Joss, S.; Kirkpatrick, M.; Brunklaus, A.; et al. Incidence and phenotypes of childhood-onset genetic epilepsies: A prospective population-based national cohort. Brain 2019, 142, 2303–2318. [Google Scholar] [CrossRef] [Green Version]
- Coman, D.J.; Sinclair, K.G.; Burke, C.J.; Appleton, D.B.; Pelekanos, J.T.; O’Neil, C.M.; Wallace, G.; Bowling, F.G.; Wang, D.; De Vivo, D.C.; et al. Seizures, ataxia, developmental delay and the general paediatrician: Glucose transporter 1 deficiency syndrome. J. Paediatr. Child Health 2006, 42, 263–267. [Google Scholar] [CrossRef]
- Larsen, J.; Johannesen, K.M.; Ek, J.; Tang, S.; Marini, C.; Blichfeldt, S.; Kibaek, M.; von Spiczak, S.; Weckhuysen, S.; Frangu, M.; et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia 2015, 56, e203–e208. [Google Scholar] [CrossRef] [PubMed]
- Kolic, I.; Nisevic, J.R.; Cicvaric, I.V.; Ahel, I.B.; Tomulic, K.L.; Segulja, S.; Dekanic, K.B.; Serifi, S.; Ovuka, A.; Prpic, I. GLUT1 Deficiency Syndrome—Early Treatment Maintains Cognitive Development? (Literature Review and Case Report). Genes 2021, 12, 1379. [Google Scholar] [CrossRef] [PubMed]
- Raja, M.; Kinne, R.K.H. Mechanistic Insights into Protein Stability and Self-Aggregation in GLUT1 genetic variants causing GLUT1-deficiency Syndrome. J. Membr. Biol. 2020, 253, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayorga, L.; Gamboni, B.; Mampel, A.; Roqué, M. A frame-shift deletion in the PURA gene associates with a new clinical finding: Hypoglycorrhachia. Is GLUT1 a new PURA target? Mol. Genet. Metab. 2018, 123, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Lijarcio, O.; Yubero, D.; Leal, F.; Couce, M.L.; Gutiérrez-Solana, L.G.; López-Laso, E.; García-Cazorla, À.; Pías-Peleteiro, L.; Brea, B.A.; Ibáñez-Micó, S.; et al. The clinical and biochemical hallmarks generally associated with GLUT1DS may be caused by defects in genes other than SLC2A1. Clin. Genet. 2022. [Google Scholar] [CrossRef] [PubMed]
- Zschocke, J.; Hoffman, G. Vademecum metabolicum, Diagnosis and Treatment of Inherited Metabolic Disorders; Thieme: Stuttgart, Germany, 2020; p. 229. [Google Scholar]
- Klepper, J. Absence of SLC2A1 Mutations Does Not Exclude Glut1 Deficiency Syndrome. Neuropediatrics 2013, 44, 235–236. [Google Scholar] [CrossRef]
- Yang, H.; Wang, D.; Ms, K.E.; Bagay, L.; Wei, Y.; Rotstein, M.; Aggarwal, V.; Levy, B.; Ma, L.; Chung, W.K.; et al. Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann. Neurol. 2011, 70, 996–1005. [Google Scholar] [CrossRef]
- Gras, D.; Cousin, C.; Kappeler, C.; Fung, C.-W.; Auvin, S.; Essid, N.; Chung, B.H.; Da Costa, L.; Hainque, E.; Luton, M.-P.; et al. A simple blood test expedites the diagnosis of glucose transporter type 1 deficiency syndrome. Ann. Neurol. 2017, 82, 133–138. [Google Scholar] [CrossRef]
- Soliani, L.; Martorell, L.; Yubero, D.; Verges, C.; Petit, V.; Ortigoza-Escobar, J.D. Paroxysmal Non-Kinesigenic Dyskinesia: Utility of the Quantification of GLUT1 in Red Blood Cells. Mov. Disord. Clin. Pract. 2021, 9, 252–254. [Google Scholar] [CrossRef]
- Pearson, T.S.; Akman, C.; Hinton, V.J.; Engelstad, K.; De Vivo, D.C. Phenotypic Spectrum of Glucose Transporter Type 1 Deficiency Syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 2013, 13, 342. [Google Scholar] [CrossRef]
- Kass, H.R.; Winesett, S.P.; Bessone, S.K.; Turner, Z.; Kossoff, E.H. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure 2016, 35, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daci, A.; Bozalija, A.; Jashari, F.; Krasniqi, S. Individualizing Treatment Approaches for Epileptic Patients with Glucose Transporter Type1 (GLUT-1) Deficiency. Int. J. Mol. Sci. 2018, 19, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandu, C.; Burloiu, C.M.; Barca, D.G.; Magureanu, S.A.; Craiu, D.C. Ketogenic Diet in Patients with GLUT1 Deficiency Syndrome. Maedica 2019, 14, 93–97. [Google Scholar] [CrossRef] [PubMed]
- De Amicis, R.; Leone, A.; Lessa, C.; Foppiani, A.; Ravella, S.; Ravasenghi, S.; Trentani, C.; Ferraris, C.; Veggiotti, P.; De Giorgis, V.; et al. Long-Term Effects of a Classic Ketogenic Diet on Ghrelin and Leptin Concentration: A 12-Month Prospective Study in a Cohort of Italian Children and Adults with GLUT1-Deficiency Syndrome and Drug Resistant Epilepsy. Nutrients 2019, 11, 1716. [Google Scholar] [CrossRef] [Green Version]
- Tagliabue, A.; Ferraris, C.; Uggeri, F.; Trentani, C.; Bertoli, S.; De Giorgis, V.; Veggiotti, P.; Elli, M. Short-term impact of a classical ketogenic diet on gut microbiota in GLUT1 Deficiency Syndrome: A 3-month prospective observational study. Clin. Nutr. ESPEN 2016, 17, 33–37. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, D.C.; Bohan, T.P.; Coulter, D.L.; Dreifuss, F.E.; Greenwood, R.S.; Nordli, D.R.; Shields, W.D.; Stafstrom, C.E.; Tein, I. l-Carnitine Supplementation in Childhood Epilepsy: Current Perspectives. Epilepsia 1998, 39, 1216–1225. [Google Scholar] [CrossRef]
- Konrad, D.; Somwar, R.; Sweeney, G.; Yaworsky, K.; Hayashi, M.; Ramlal, T.; Klip, A. The Antihyperglycemic Drug α-Lipoic Acid Stimulates Glucose Uptake via Both GLUT4 Translocation and GLUT4 Activation. Diabetes 2001, 50, 1464–1471. [Google Scholar] [CrossRef] [Green Version]
- Herrero, J.R.; Villarroya, E.C.; Gutiérrez-Solana, L.G.; Alcolea, B.G.; Fernández, B.G.; Macfarland, L.P.; Pedrón-Giner, C. Classic Ketogenic Diet and Modified Atkins Diet in SLC2A1 Positive and Negative Patients with Suspected GLUT1 Deficiency Syndrome: A Single Center Analysis of 18 Cases. Nutrients 2021, 13, 840. [Google Scholar] [CrossRef]
- Mochel, F.; Hainque, E.; Gras, D.; Adanyeguh, I.M.; Caillet, S.; Héron, B.; Roubertie, A.; Kaphan, E.; Valabregue, R.; Rinaldi, D.; et al. Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J. Neurol. Neurosurg. Psychiatry 2015, 87, 550–553. [Google Scholar] [CrossRef]
- Mochel, F. Triheptanoin for the treatment of brain energy deficit: A 14-year experience. J. Neurosci. Res. 2017, 95, 2236–2243. [Google Scholar] [CrossRef] [Green Version]
- Almuqbil, M.; Go, C.; Nagy, L.L.; Pai, N.; Mamak, E.; Mercimek-Mahmutoglu, S. New Paradigm for the Treatment of Glucose Transporter 1 Deficiency Syndrome: Low Glycemic Index Diet and Modified High Amylopectin Cornstarch. Pediatr. Neurol. 2015, 53, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Logel, S.N.; Connor, E.L.; Hsu, D.A.; Fenske, R.J.; Paloian, N.J.; De Vivo, D.C. Exploring diazoxide and continuous glucose monitoring as treatment for Glut1 deficiency syndrome. Ann. Clin. Transl. Neurol. 2021, 8, 2205–2209. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009, 31, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zi, Z.; Lee, E.E.; Zhao, J.; Contreras, D.C.; South, A.P.; Abel, E.D.; Chong, B.F.; Vandergriff, T.; Hosler, G.A.; et al. Differential glucose requirement in skin homeostasis and injury identifies a therapeutic target for psoriasis. Nat. Med. 2018, 24, 617–627. [Google Scholar] [CrossRef]
- North, P.E.; Waner, M.; Mizeracki, A.; Mihm, M.C., Jr. GLUT1: A newly discovered immunohistochemical marker for juvenile hemangiomas. Hum. Pathol. 2000, 31, 11–22. [Google Scholar] [CrossRef]
- Carmona-Fontaine, C.; Bucci, V.; Akkari, L.; Deforet, M.; Joyce, J.A.; Xavier, J.B. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc. Natl. Acad. Sci. USA 2013, 110, 19402–19407. [Google Scholar] [CrossRef] [Green Version]
- Bozkurt, T.; Alanay, Y.; Isik, U.; Sezerman, U. Re-analysis of whole-exome sequencing data reveals a novel splicing variant in the SLC2A1 in a patient with GLUT1 Deficiency Syndrome 1 accompanied by hemangioma: A case report. BMC Med. Genom. 2021, 14, 197. [Google Scholar] [CrossRef]
- Henry, M.; Kitchens, J.; Pascual, J.M.; Maldonado, R.S. GLUT1 deficiency: Retinal detrimental effects of gliovascular modulation. Neurol. Genet. 2020, 6, e472. [Google Scholar] [CrossRef]
- Aït-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clérin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-Derived Cone Viability Factor Promotes Cone Survival by Stimulating Aerobic Glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef] [Green Version]
- Andrisse, S.; Patel, G.D.; Chen, J.E.; Webber, A.M.; Spears, L.D.; Koehler, R.M.; Robinson-Hill, R.M.; Ching, J.K.; Jeong, I.; Fisher, J.S. ATM and GLUT1-S490 Phosphorylation Regulate GLUT1 Mediated Transport in Skeletal Muscle. PLoS ONE 2013, 8, e66027. [Google Scholar] [CrossRef] [Green Version]
- Zanaboni, M.; Pasca, L.; Villa, B.; Faggio, A.; Grumi, S.; Provenzi, L.; Varesio, C.; De Giorgis, V. Characterization of Speech and Language Phenotype in GLUT1DS. Children 2021, 8, 344. [Google Scholar] [CrossRef]
- Evans, P.L.; McMillin, S.L.; Weyrauch, L.A.; Witczak, C.A. Regulation of Skeletal Muscle Glucose Transport and Glucose Metabolism by Exercise Training. Nutrients 2019, 11, 2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavakis, T. Immunometabolism: Where Immunology and Metabolism Meet. J. Innate Immun. 2021, 14, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Kolliniati, O.; Ieronymaki, E.; Vergadi, E.; Tsatsanis, C. Metabolic Regulation of Macrophage Activation. J. Innate Immun. 2021, 14, 51–68. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekkering, S.; Domínguez-Andrés, J.; Joosten, L.A.; Riksen, N.P.; Netea, M.G. Trained Immunity: Reprogramming Innate Immunity in Health and Disease. Annu. Rev. Immunol. 2021, 39, 667–693. [Google Scholar] [CrossRef] [PubMed]
- Badii, M.; Gaal, O.; Popp, R.A.; Crișan, T.O.; Joosten, L.A. Trained immunity and inflammation in rheumatic diseases. Jt. Bone Spine 2022, 89, 105364. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.M.; Martinez, G.J.; Chung, Y.; Dong, C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 13064–13069. [Google Scholar] [CrossRef] [Green Version]
- Kunisawa, J.; Sugiura, Y.; Wake, T.; Nagatake, T.; Suzuki, H.; Nagasawa, R.; Shikata, S.; Honda, K.; Hashimoto, E.; Suzuki, Y.; et al. Mode of Bioenergetic Metabolism during B Cell Differentiation in the Intestine Determines the Distinct Requirement for Vitamin B1. Cell Rep. 2015, 13, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; DeOliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [Green Version]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Z.-P.; Zhang, Y.-L.; Shi, K.; Shi, L.; Zhang, Y.-Z.; Wang, C.-Y. Suppression of diabetic retinopathy with Glut-1 siRNA. Sci. Rep. 2017, 7, 7437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewko, B.; Maryn, A.; Latawiec, E.; Daca, A.; Rybczynska, A. Angiotensin II Modulates Podocyte Glucose Transport. Front. Endocrinol. 2018, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Kao, W.L.; Steffes, M.W.; Gambir, T.; Brancati, F.L.; Heilig, C.W.; Shuldiner, A.R.; Boerwinkle, E.A.; Coresh, J. Genetic variation of Glucose Transporter-1 (GLUT1) and albuminuria in 10,278 European Americans and African Americans: A case-control study in the Atherosclerosis Risk in Communities (ARIC) Study. BMC Med. Genet. 2011, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Cassis, P.; Locatelli, M.; Cerullo, D.; Corna, D.; Buelli, S.; Zanchi, C.; Villa, S.; Morigi, M.; Remuzzi, G.; Benigni, A.; et al. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight 2018, 3, e98720. [Google Scholar] [CrossRef] [Green Version]
- Lüscher, B.P.; Marini, C.; Joerger-Messerli, M.S.; Huang, X.; Hediger, M.A.; Albrecht, C.; Baumann, M.U.; Surbek, D.V. Placental glucose transporter (GLUT)-1 is down-regulated in preeclampsia. Placenta 2017, 55, 94–99. [Google Scholar] [CrossRef]
- Pereira, R.O.; Wende, A.R.; Olsen, C.; Soto, J.; Rawlings, R.; Zhu, Y.; Riehle, C.; Abel, E.D. GLUT 1 deficiency in cardiomyocytes does not accelerate the transition from compensated hypertrophy to heart failure. J. Mol. Cell Cardiol. 2014, 72, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Renaudin, F.; Orliaguet, L.; Castelli, F.; Fenaille, F.; Prignon, A.; Alzaid, F.; Combes, C.; Delvaux, A.; Adimy, Y.; Cohen-Solal, M.; et al. Gout and pseudo-gout-related crystals promote GLUT1-mediated glycolysis that governs NLRP3 and interleukin-1β activation on macrophages. Ann. Rheum. Dis. 2020, 79, 1506–1514. [Google Scholar] [CrossRef]
- Girdhar, K.; Powis, A.; Raisingani, A.; Chrudinová, M.; Huang, R.; Tran, T.; Sevgi, K.; Dogus Dogru, Y.; Altindis, E. Viruses and Me-tabolism: The Effects of Viral Infections and Viral Insulins on Host Metabolism. Annu. Rev. Virol. 2021, 8, 373–391. [Google Scholar] [CrossRef]
- Thaker, S.K.; Ch’Ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef]
- Blonz, E.R. Zika virus and GLUT1. Lancet Infect. Dis. 2016, 16, 642. [Google Scholar] [CrossRef] [Green Version]
- Mustroph, J.; Hupf, J.; Hanses, F.; Evert, K.; Baier, M.J.; Evert, M.; Meindl, C.; Wagner, S.; Hubauer, U.; Pietrzyk, G.; et al. Decreased GLUT1/NHE1 RNA expression in whole blood predicts disease severity in patients with COVID-19. ESC Heart Fail. 2020, 8, 309–316. [Google Scholar] [CrossRef]
- Kyrtata, N.; Emsley, H.C.A.; Sparasci, O.; Parkes, L.M.; Dickie, B.R. A Systematic Review of Glucose Transport Alterations in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 626636. [Google Scholar] [CrossRef]
- Mullins, R.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 118. [Google Scholar] [CrossRef]
- Gejl, M.; Brock, B.; Egefjord, L.; Vang, K.; Rungby, J.; Gjedde, A. Blood-Brain Glucose Transfer in Alzheimer’s disease: Effect of GLP-1 Analog Treatment. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Guedj, E.; Varrone, A.; Boellaard, R.; Albert, N.L.; Barthel, H.; van Berckel, B.; Brendel, M.; Cecchin, D.; Ekmekcioglu, O.; Garibotto, V.; et al. EANM procedure guidelines for brain PET imaging using [18F] FDG, version 3. Eur. J. Pediatr. 2022, 49, 632–651. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liow, J.-S.; Zhang, Z.; Li, J.; Long, T.; Li, Y.; Tang, B.; Hu, S. The Evaluation of Dynamic FDG-PET for Detecting Epileptic Foci and Analyzing Reduced Glucose Phosphorylation in Refractory Epilepsy. Front. Neurosci. 2019, 12, 993. [Google Scholar] [CrossRef]
- Wellberg, E.A.; Johnson, S.; Finlay-Schultz, J.; Lewis, A.S.; Terrell, K.L.; Sartorius, C.A.; Abel, E.D.; Muller, W.J.; Anderson, S.M. The glucose transporter GLUT1 is required for ErbB2-induced mammary tumorigenesis. Breast Cancer Res. 2016, 18, 131. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Lecerf, C.; Briand, M.; Louis, M.-H.; Dutoit, S.; Jebahi, A.; Giffard, F.; Fournier, C.B.; Batalla, A.; Poulain, L.; et al. 18F-FDG Is a Surrogate Marker of Therapy Response and Tumor Recovery after Drug Withdrawal during Treatment with a Dual PI3K/mTOR Inhibitor in a Preclinical Model of Cisplatin-Resistant Ovarian Cancer. Transl. Oncol. 2013, 6, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.A.; Cockfield, J.A.; Pape, D.J.; Meissner, H.; Sokolowski, M.T.; White, T.C.; López, J.C.V.; Liu, J.; Liu, X.; Martínez-Reyes, I.; et al. SGK1 signaling promotes glucose metabolism and survival in extracellular matrix detached cells. Cell Rep. 2021, 34, 108821. [Google Scholar] [CrossRef]
- Rastogi, S.; Banerjee, S.; Chellappan, S.; Simon, G.R. Glut-1 antibodies induce growth arrest and apoptosis in human cancer cell lines. Cancer Lett. 2007, 257, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Smith, L. Ketogenic Diet in Glut 1 Deficiency Through the Life Cycle: Pregnancy to Neonate to Preschooler. Child Neurol. Open 2021, 8, 2329048X211034655. [Google Scholar] [CrossRef] [PubMed]
- Dakic, T.; Jevdjovic, T.; Lakic, I.; Djurasevic, S.F.; Djordjevic, J.; Vujovic, P. Food For Thought: Short-Term Fasting Upregulates Glucose Transporters in Neurons and Endothelial Cells, But Not in Astrocytes. Neurochem. Res. 2018, 44, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Ye, Z.; Shi, Y.; Zhou, L.; Hua, Y. Curcumin improves diabetes mellitus-associated cerebral infarction by increasing the expression of GLUT1 and GLUT3. Mol. Med. Rep. 2018, 17, 1963–1969. [Google Scholar] [CrossRef] [Green Version]





| Transporter | Main Substrate | Location | Main Properties | Type of Transport |
|---|---|---|---|---|
| Glut1 | Glucose, galactose, mannose, glucosamine | RBC, kidney, colon, retina, placenta, myocardium, adipose tissue, brain, blood-brain barrier, blood-tissue barrier, many fetal tissues | Glucose uptake in most of cells, expression is age-related | Passive transport, sodium-independent transporters |
| Glut2 | Glucose, galactose, fructose, mannose, glucosamine | Serosal surface of intestinal cells, liver, beta cells of pancreas, kidney | Low affinity; glucose uptake in liver; glucose sensor in pancreatic beta cells | |
| Glut3 | Glucose, galactose, mannose, xylose | Brain (neurons membrane), testis | High affinity; transports glucose into brain cells | |
| Glut4 | Glucose, glucosamine | Skeletal and cardiac muscle, adipose tissue [white and brown] | Insulin mediated glucose uptake, expression is age-related | |
| Glut5 | Fructose | Small intestine, kidney | Poor ability to transport glucose; is mainly a fructose transporter | |
| Glut6 | Glucose | Spleen, leucocytes, brain | Glucose transport | |
| Glut7 | Glucose, fructose | Liver endoplasmic reticulum, small intestine, colon, testis, prostate | Glucose transport from ER to cytoplasm | |
| Glut8 | Glucose, fructose, galactose | Testis, brain, blastocyst, adrenal gland, liver, spleen, muscle, brown adipose tissue, lung [intracellular] | Glucose/(Fructose) transport | |
| Glut9 | Urate (glucose, fructose) | Liver, kidney, small intestine, placenta, lung, leukocytes | Glucose/Fructose transport, not galactose | |
| Glut10 | Glucose, galactose | Heart, lung, brain, liver, skeletal muscle, pancreas, placenta, kidney, mitochondria of smooth muscle cells | Facilitates DHAA, import into mitochondria of smooth muscle cells and insulin stimulated adipocytes; protects cells against oxidative stress, connects mitochondrial function to TGF-β signaling | |
| Glut11 | Glucose, fructose | Heart, kidney, skeletal muscle, adipose tissue and pancreas | The 3 Glut11 variants are differentially expressed; primary physiological substrates have not been definitively identified | |
| Glut12 | Glucose; also transports α-methyl-D-glucopyranoside | Heart, renal tubules, digestive tube epithelium, prostate, adipose tissue, liver, skeletal muscle, placenta, thyroid, adrenal and pituitary glands | The role in glucose homeostasis under normal or pathophysiological conditions is not fully understood; but insulin has been reported to acutely stimulate the translocation of Glut12 from intracellular membrane compartments to the plasma membrane in human skeletal muscle | |
| Glut13 (also called HMIT] | Myo- inositol | Muscle, thyroid, adrenal and pituitary glands, kidney, white and brown adipose tissue; brain (both in neurons and glial cells): highly expressed in the hippocampus, hypothalamus, cerebellum, brainstem | In neurons is present in intracellular vesicles involved in increasing myo-inositol uptake. Possible role in regulating processes such as membrane recycling, growth cone dynamics and synaptic vesicle exocytosis (requiring high levels of myo-inositol or its derivatives). | |
| Glut14 | Testis | The role is not fully understood; his gene (SLC2A14) shares 95% sequence identity with the Glut3 gene and, therefore, appears to be encoded by a gene duplication. | ||
| SGLT (sodium-dependent transporters] | SGLT1 in intestine, in kidney | Co-transport; from lumen into cells. | Active transport | |
| SGLT2 in kidney | ||||
| SWEETs mediate mainly the efflux of glucose in humans and are ubiquitous in human body | They have the highest expression in the oviduct, epididymis and intestine; also are localized in pancreatic beta cells. Further studies are required to discover SWEET physiology in humans. | SWEETs may function as uniporters, although this hypothesis remains unproven. Have the ability to transport various mono- and disaccharides, the ability to mediate both cellular uptake and efflux, and have typically low affinities for sugars. | Passive transport, sodium-independent transporters |
| Diet/Treatment | AED Indicated | Drugs Not Recommended in Association with KD | References |
|---|---|---|---|
| Ketogenic diet (KD) | Acetazolamide | Valproate | [5,12,56] |
| Modified Atkins Diet | Topiramate | Zonisamide | [56] |
| Medium chain Triglycerides | Zonisamide | Acetazolamide | [5,12,56] |
| Low glycemic index treatment | Phenytoin | Topiramate | [5,56] |
| Triheptanoin | Carbamazepine | - | [56,64] |
| α-lipoic acid (under investigation) | - | - | [5,12,56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vulturar, R.; Chiș, A.; Pintilie, S.; Farcaș, I.M.; Botezatu, A.; Login, C.C.; Sitar-Taut, A.-V.; Orasan, O.H.; Stan, A.; Lazea, C.; et al. One Molecule for Mental Nourishment and More: Glucose Transporter Type 1—Biology and Deficiency Syndrome. Biomedicines 2022, 10, 1249. https://doi.org/10.3390/biomedicines10061249
Vulturar R, Chiș A, Pintilie S, Farcaș IM, Botezatu A, Login CC, Sitar-Taut A-V, Orasan OH, Stan A, Lazea C, et al. One Molecule for Mental Nourishment and More: Glucose Transporter Type 1—Biology and Deficiency Syndrome. Biomedicines. 2022; 10(6):1249. https://doi.org/10.3390/biomedicines10061249
Chicago/Turabian StyleVulturar, Romana, Adina Chiș, Sebastian Pintilie, Ilinca Maria Farcaș, Alina Botezatu, Cristian Cezar Login, Adela-Viviana Sitar-Taut, Olga Hilda Orasan, Adina Stan, Cecilia Lazea, and et al. 2022. "One Molecule for Mental Nourishment and More: Glucose Transporter Type 1—Biology and Deficiency Syndrome" Biomedicines 10, no. 6: 1249. https://doi.org/10.3390/biomedicines10061249