
Comparative Profiling of TG2 and Its Effectors in Human Relapsing Remitting and Progressive Multiple Sclerosis



Abstract
:1. Introduction
2. Materials and Methods
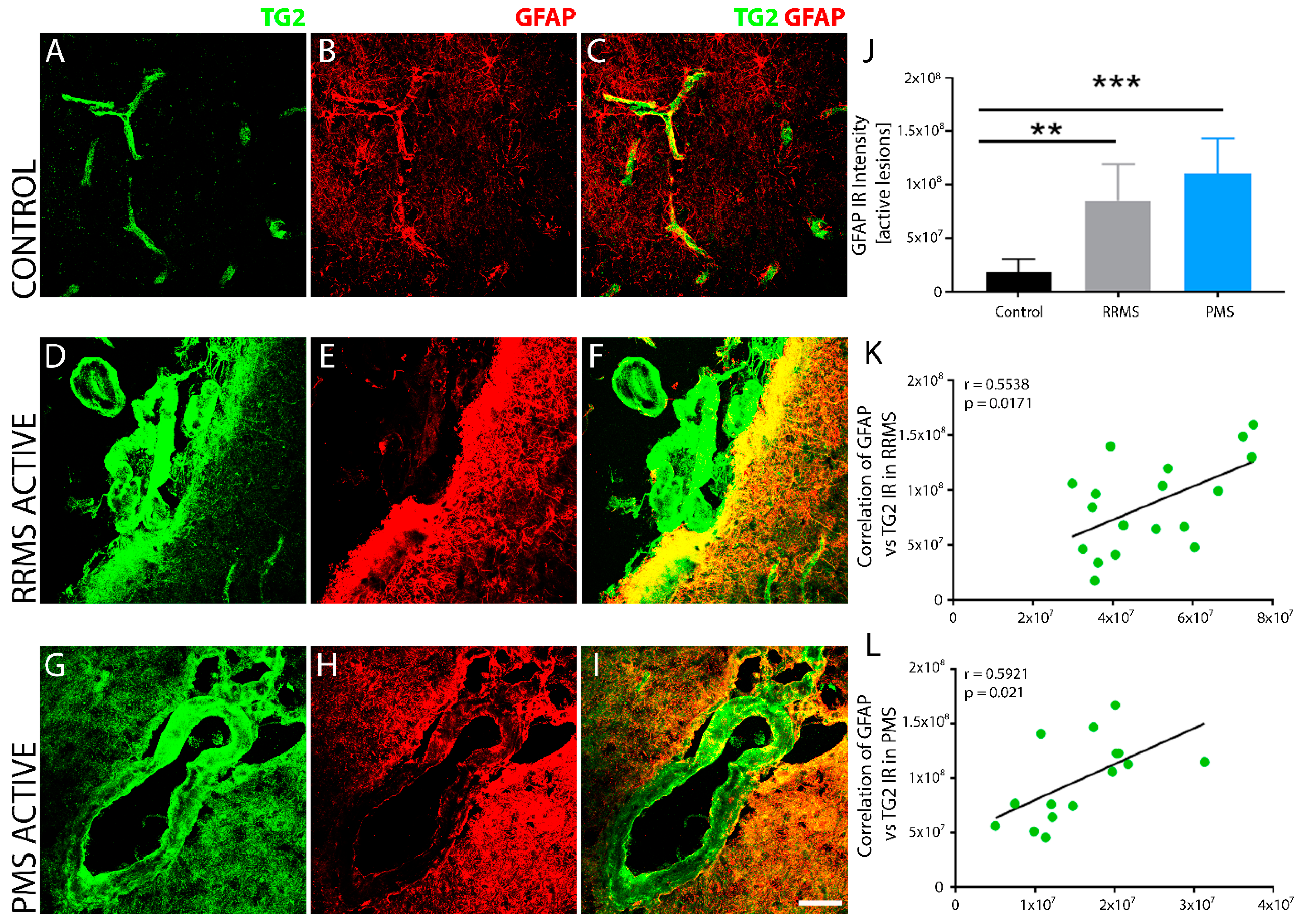
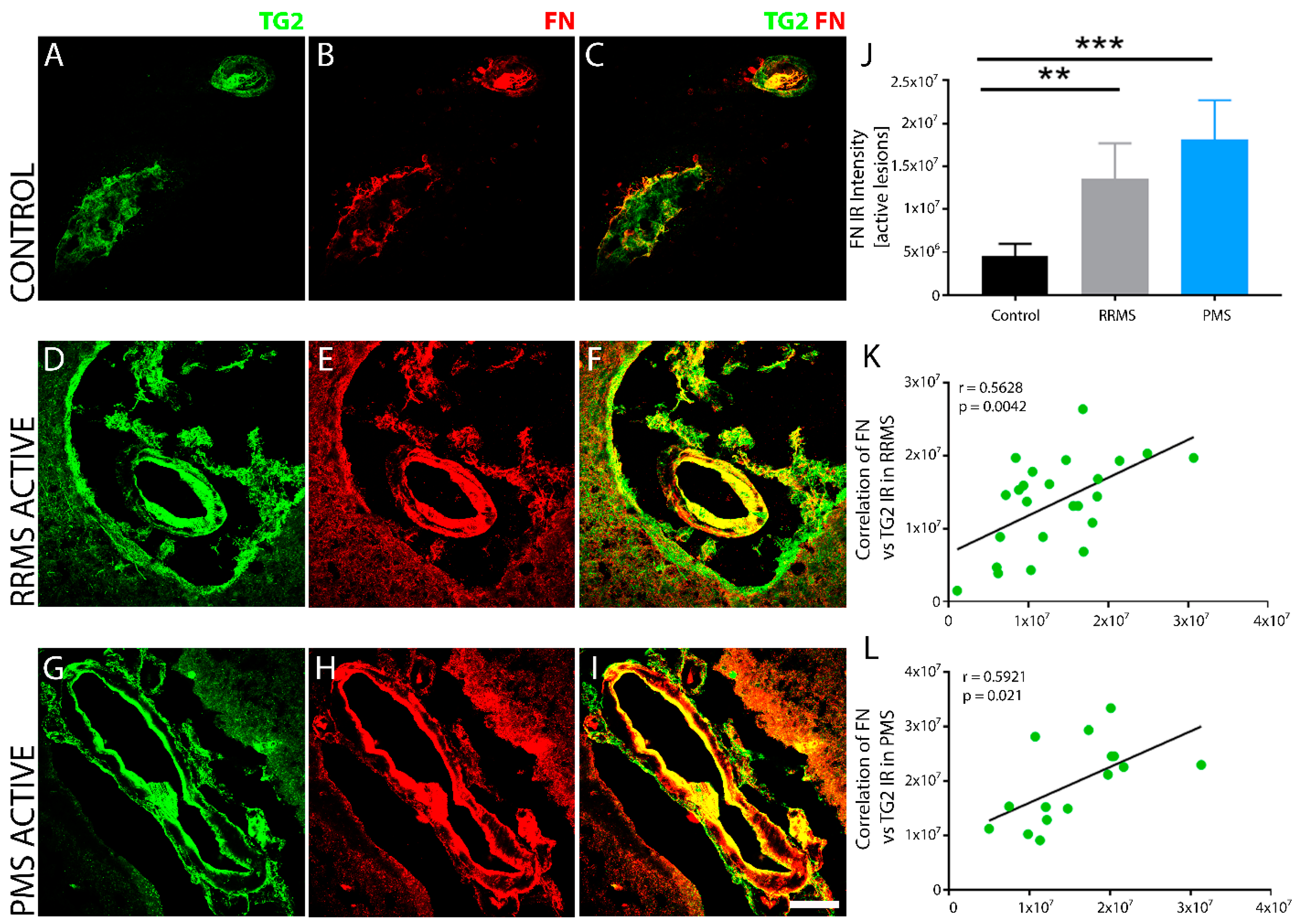
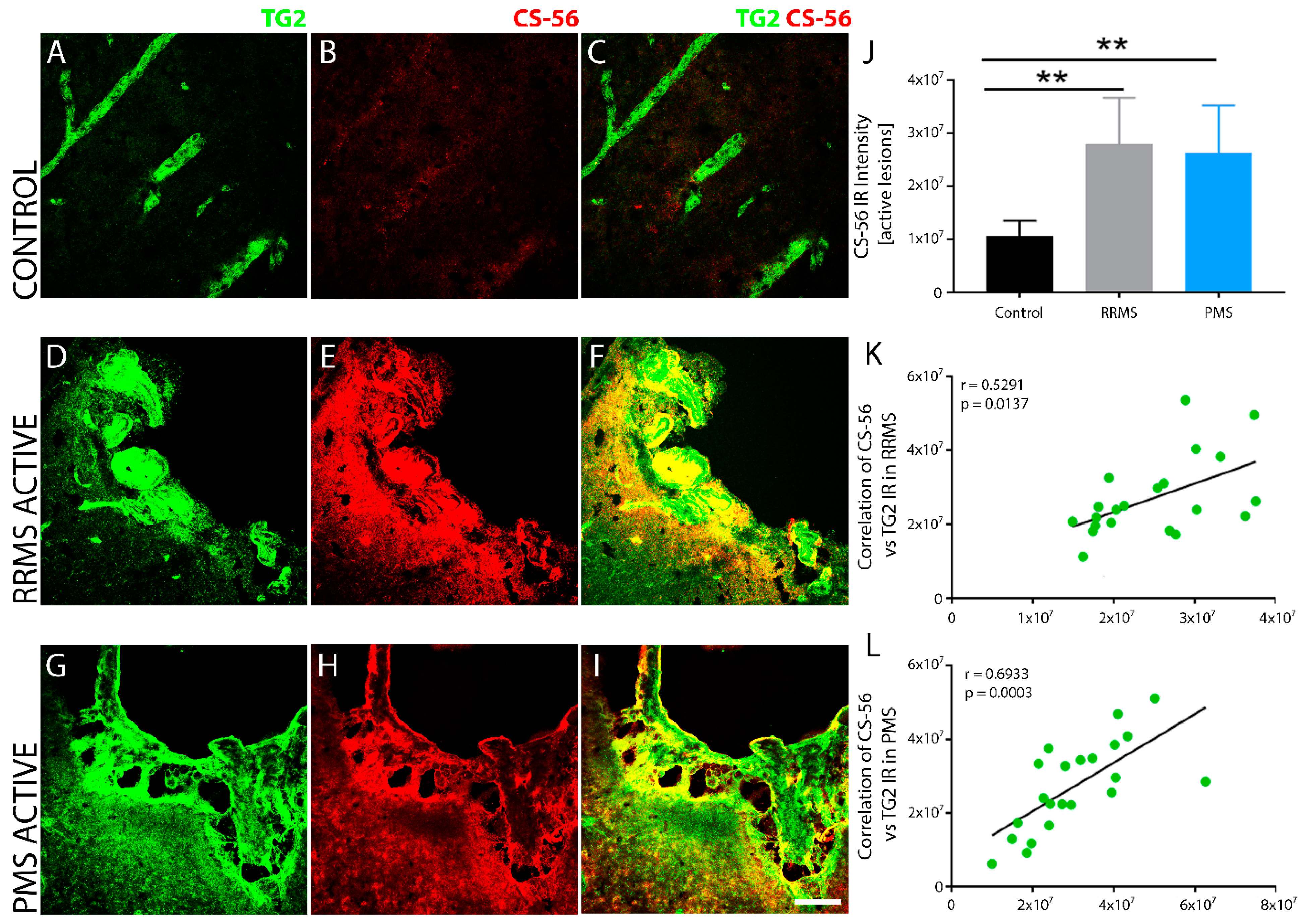
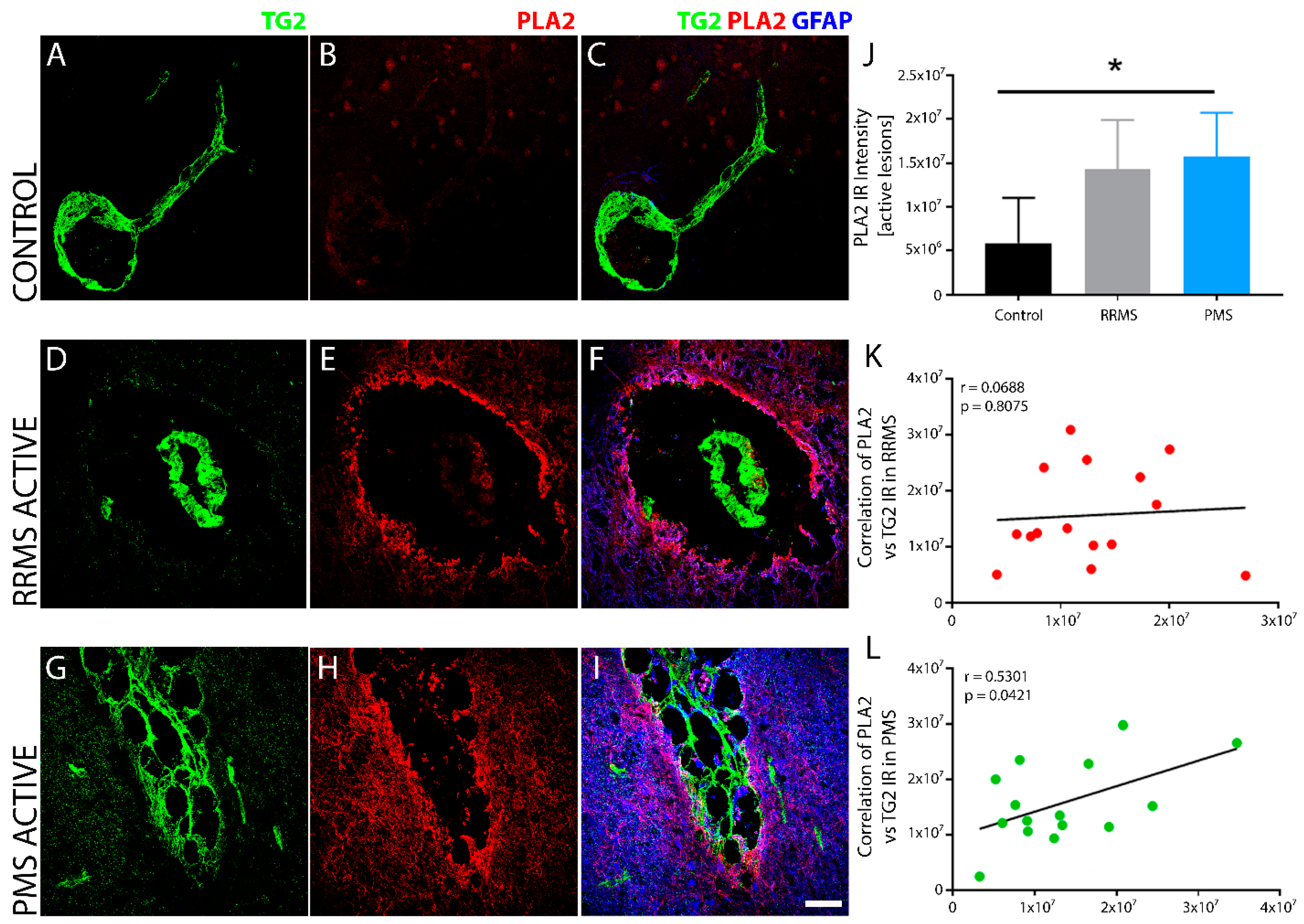
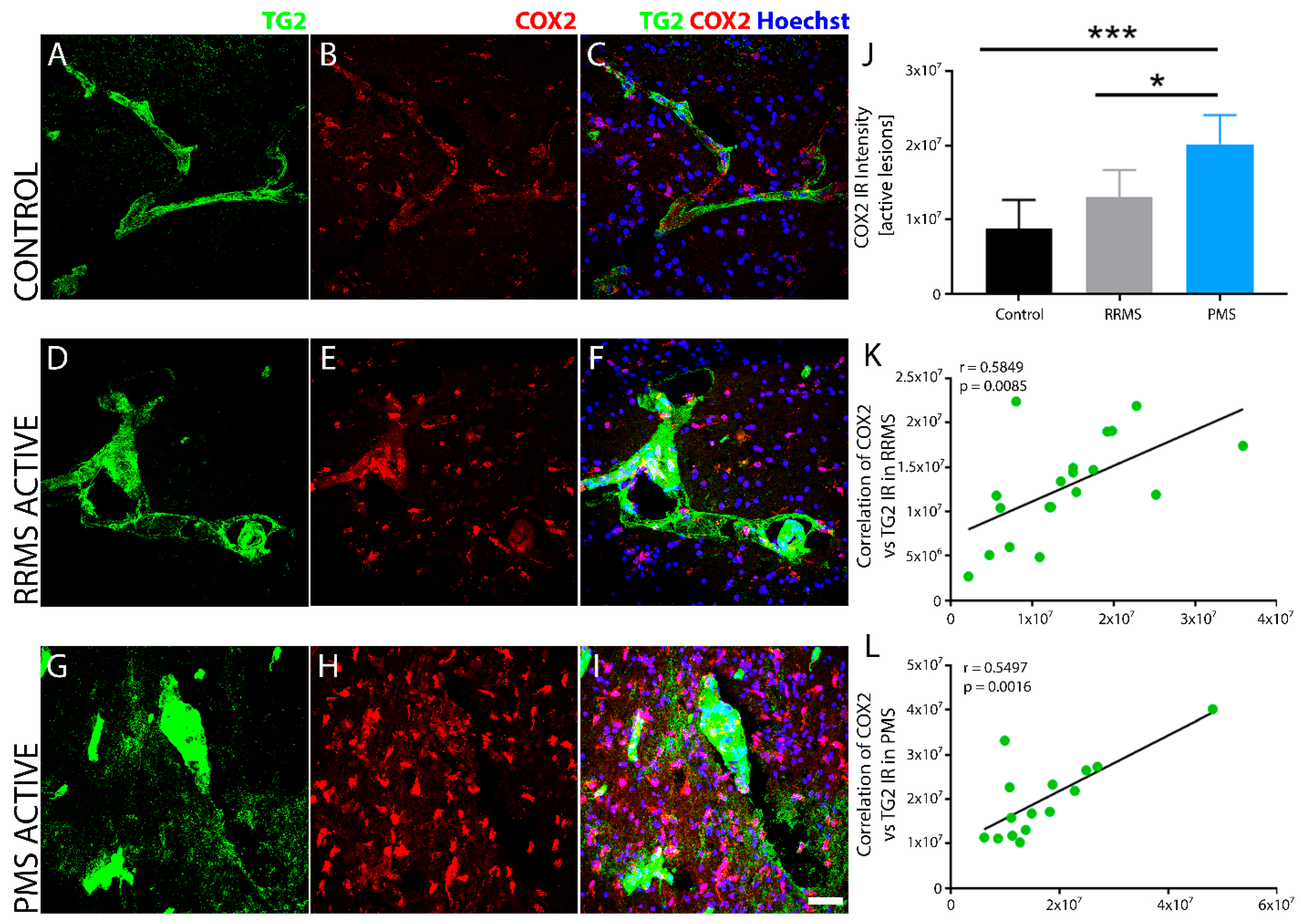
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Miehm, J.D.; Buonaccorsi, J.; Lim, J.; Sato, S.; Rajala, C.; Averill, J.; Khalighinejad, F.; Ionete, C.; Jones, S.L.; Kent, J.A.; et al. Sensorimotor function in progressive multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2020, 6, 2055217320934835. [Google Scholar] [CrossRef] [PubMed]
- Racosta, J.M.; Kimpinski, K.; Morrow, S.A.; Kremenchutzky, M. Autonomic dysfunction in multiple sclerosis. Auton. Neurosci. 2015, 193, 1–6. [Google Scholar] [CrossRef]
- Abbatemarco, J.R.; Fox, R.J.; Li, H.; Bermel, R.A.; Ontaneda, D. Vitamin D Levels and Visual System Measurements in Progressive Multiple Sclerosis: A Cross-sectional Study. Int. J. MS Care 2021, 23, 53–58. [Google Scholar] [CrossRef]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2017, 19, 1–10. [Google Scholar]
- Miller, D.H.; Leary, S.M. Primary-progressive multiple sclerosis. Lancet Neurol. 2007, 6, 903–912. [Google Scholar] [CrossRef]
- van Langelaar, J.; Rijvers, L.; Smolders, J.; van Luijn, M.M. B and T Cells Driving Multiple Sclerosis: Identity, Mechanisms and Potential Triggers. Front. Immunol. 2020, 11, 760. [Google Scholar] [CrossRef]
- Disano, K.D.; Linzey, M.R.; Royce, D.B.; Pachner, A.R.; Gilli, F. Differential neuro-immune patterns in two clinically relevant murine models of multiple sclerosis. J. Neuroinflammation 2019, 16, 109. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. Immune-cell crosstalk in multiple sclerosis. Nature 2018, 563, 194–195. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.F.; Alvarez, E. The immunopathophysiology of multiple sclerosis. Neurol. Clin. 2011, 29, 257–278. [Google Scholar] [CrossRef] [Green Version]
- Hemmer, B.; Kerschensteiner, M.; Korn, T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015, 14, 406–419. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H.G. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef]
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflammation 2017, 14, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minagar, A.; Alexander, J.S. Blood-brain barrier disruption in multiple sclerosis. Mult. Scler. 2003, 9, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Contributions of T cells in multiple sclerosis: What do we currently know? J. Neurol. 2021, 268, 4587–4593. [Google Scholar] [CrossRef]
- Lehmann-Horn, K.; Kronsbein, H.C.; Weber, M.S. Targeting B cells in the treatment of multiple sclerosis: Recent advances and remaining challenges. Ther. Adv. Neurol. Disord. 2013, 6, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends. Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2018, 9, 3116. [Google Scholar] [CrossRef]
- Correale, J.; Gaitán, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkin, A.M. Extracellular TG2: Emerging functions and regulation. FEBS J. 2011, 278, 4704–4716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsukawa, H.; Furutani, Y.; Hitomi, K.; Kojima, S. Transglutaminase 2 has opposing roles in the regulation of cellular functions as well as cell growth and death. Cell Death. Dis. 2016, 7, e2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.D.; Tucholski, J.; Johnson, G.V. Transglutaminases in neurodegenerative disorders. Prog. Exp. Tumor. Res. 2005, 38, 139–157. [Google Scholar] [PubMed]
- Martin, A.; De Vivo, G.; Ricotta, M.; Iannuzzi, M.; Gentile, V. Transglutaminases as possible therapeutic targets in neurodegenerative diseases. Recent Pat. CNS Drug Discov. 2010, 5, 195–202. [Google Scholar] [CrossRef]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef] [Green Version]
- Nurminskaya, M.V.; Belkin, A.M. Cellular functions of tissue transglutaminase. Int. Rev. Cell Mol. Biol. 2012, 294, 1–97. [Google Scholar]
- Sestito, C.; Brevé, J.J.; Bol, J.G.; Wilhelmus, M.M.; Drukarch, B.; van Dam, A.-M. Tissue Transglutaminase contributes to myelin phagocytosis in interleukin-4-treated human monocyte-derived macrophages. Cytokine 2020, 128, 155024. [Google Scholar] [CrossRef]
- Sun, H.; Kaartinen, M.T. Transglutaminases in Monocytes and Macrophages. Med. Sci. 2018, 6, 115. [Google Scholar] [CrossRef] [Green Version]
- Giera, S.; Luo, R.; Ying, Y.; Ackerman, S.D.; Jeong, S.; Stoveken, H.M.; Folts, C.J.; Welsh, C.A.; Tall, G.G.; Stevens, B.; et al. Microglial transglutaminase-2 drives myelination and myelin repair via GPR56/ADGRG1 in oligodendrocyte precursor cells. eLife 2018, 7, e33385. [Google Scholar] [CrossRef] [PubMed]
- Ientile, R.; Curro, M.; Caccamo, D. Transglutaminase 2 and neuroinflammation. Amino Acids 2015, 47, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Tatsukawa, H.; Hitomi, K. Role of Transglutaminase 2 in Cell Death, Survival, and Fibrosis. Cells 2021, 10, 1842. [Google Scholar] [CrossRef]
- Keillor, J.W.; Johnson, G.V.W. Transglutaminase 2 as a therapeutic target for neurological conditions. Expert Opin. Targets 2021, 25, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Mastroberardino, P.G.; Piacentini, M. Type 2 transglutaminase in Huntington’s disease: A double-edged sword with clinical potential. J. Intern. Med. 2010, 268, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelmus, M.M.; de Jager, M.; Bakker, E.N.T.P.; Drukarch, B. Tissue transglutaminase in Alzheimer’s disease: Involvement in pathogenesis and its potential as a therapeutic target. J. Alzheimers Dis. 2014, 42 (Suppl. 3), S289–S303. [Google Scholar] [CrossRef]
- Oono, M.; Okado-Matsumoto, A.; Shodai, A.; Ido, A.; Ohta, Y.; Abe, K.; Ayaki, T.; Ito, H.; Takahashi, R.; Taniguchi, N.; et al. Transglutaminase 2 accelerates neuroinflammation in amyotrophic lateral sclerosis through interaction with misfolded superoxide dismutase 1. J. Neurochem. 2014, 128, 403–418. [Google Scholar] [CrossRef] [Green Version]
- Pearse, D.D.; Otero, P.A.; Diaz, A.; Pan, X.; Ghosh, M. Neuronal and Endothelial Transglutaminase-2 Expression during Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Neuroscience 2021, 461, 140–154. [Google Scholar] [CrossRef]
- Sestito, C.; Leurs, C.E.; Steenwijk, M.D.; Brevé, J.J.; Twisk, J.W.; Wilhelmus, M.M.; Drukarch, B.; Teunissen, C.E.; van Dam, A.-M.; Killestein, J. Tissue Transglutaminase Expression Associates With Progression of Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e998. [Google Scholar] [CrossRef]
- Chrobok, N.L.; Bol, J.G.J.M.; Jongenelen, C.A.; Brevé, J.J.P.; Alaoui, S.E.; Wilhelmus, M.M.M.; Drukarch, B.; van Dam, A. Characterization of Transglutaminase 2 activity inhibitors in monocytes in vitro and their effect in a mouse model for multiple sclerosis. PLoS ONE 2018, 13, e0196433. [Google Scholar]
- Chrobok, N.L.; Bol, J.G.J.M.; Wilhelmus, M.M.M.; Drukarch, B.; Van Dam, A.-M. Tissue Transglutaminase Appears in Monocytes and Macrophages but Not in Lymphocytes in White Matter Multiple Sclerosis Lesions. J. Neuropathol. Exp. Neurol. 2019, 78, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Sestito, C.; Brevé, J.J.P.; van Eggermond, M.C.J.A.; Killestein, J.; Teunissen, C.E.; van Rossum, J.; Wilhelmus, M.M.M.; Drukarch, B.; van den Elsen, P.J.; van Dam, A. Monocyte-derived tissue transglutaminase in multiple sclerosis patients: Reflecting an anti-inflammatory status and function of the cells? J. Neuroinflammation 2017, 14, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soluri, M.F.; Boccafoschi, F.; Cotella, D.; Moro, L.; Forestieri, G.; Autiero, I.; Cavallo, L.; Oliva, R.; Griffin, M.; Wang, Z.; et al. Mapping the minimum domain of the fibronectin binding site on transglutaminase 2 (TG2) and its importance in mediating signaling, adhesion, and migration in TG2-expressing cells. FASEB J. 2019, 33, 2327–2342. [Google Scholar] [CrossRef] [Green Version]
- Furini, G.; Verderio, E.A.M. Spotlight on the Transglutaminase 2-Heparan Sulfate Interaction. Med. Sci. 2019, 7, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, P.; Zhang, X.; Li, J.P. Heparan Sulfate Proteoglycans as Relays of Neuroinflammation. J. Histochem. Cytochem. 2018, 66, 305–319. [Google Scholar] [CrossRef] [Green Version]
- Elahi, A.; Emerson, J.; Rudlong, J.; Keillor, J.W.; Salois, G.; Visca, A.; Girardi, P.; Johnson, G.V.; Pröschel, C. Deletion or Inhibition of Astrocytic Transglutaminase 2 Promotes Functional Recovery after Spinal Cord Injury. Cells 2021, 10, 2942. [Google Scholar] [CrossRef] [PubMed]
- Yunes-Medina, L.; Paciorkowski, A.; Nuzbrokh, Y.; Johnson, G.V. Depletion of transglutaminase 2 in neurons alters expression of extracellular matrix and signal transduction genes and compromises cell viability. Mol. Cell Neurosci. 2018, 86, 72–80. [Google Scholar] [CrossRef]
- Yoo, J.-O.; Lim, Y.-C.; Kim, Y.-M.; Ha, K.-S. Transglutaminase 2 promotes both caspase-dependent and caspase-independent apoptotic cell death via the calpain/Bax protein signaling pathway. J. Biol. Chem. 2012, 287, 14377–14388. [Google Scholar] [CrossRef] [Green Version]
- Führmann, T.; Ghosh, M.; Otero, A.; Goss, B.; Dargaville, T.R.; Pearse, D.D.; Dalton, P.D. Peptide-functionalized polymeric nanoparticles for active targeting of damaged tissue in animals with experimental autoimmune encephalomyelitis. Neurosci. Lett. 2015, 602, 126–132. [Google Scholar] [CrossRef]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflammation 2016, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Shiryaev, S.A.; Chernov, A.V.; Kim, Y.; Shubayev, I.; Remacle, A.G.; Baranovskaya, S.; Golubkov, V.S.; Strongin, A.Y.; Shubayev, V.I. Immunodominant fragments of myelin basic protein initiate T cell-dependent pain. J. Neuroinflammation 2012, 9, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currò, M.; Ferlazzo, N.; Risitano, R.; Condello, S.; Vecchio, M.; Caccamo, D.; Ientile, R. Transglutaminase 2 and phospholipase A(2) interactions in the inflammatory response in human Thp-1 monocytes. Amino Acids 2014, 46, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Østerlund, E.C.; Stamnaes, J.; Iversen, R.; Andersen, J.T.; Jørgensen, T.J.D.; Sollid, L.M. Dissecting the interaction between transglutaminase 2 and fibronectin. Amino Acids 2017, 49, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Kalyvas, A.; David, S. Cytosolic phospholipase A2 plays a key role in the pathogenesis of multiple sclerosis-like disease. Neuron 2004, 41, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Marusic, S.; Leach, M.W.; Pelker, J.W.; Azoitei, M.L.; Uozumi, N.; Cui, J.; Shen, M.W.; Declercq, C.M.; Miyashiro, J.S.; Carito, B.A.; et al. Cytosolic phospholipase A2 alpha-deficient mice are resistant to experimental autoimmune encephalomyelitis. J. Exp. Med. 2005, 202, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, C. Cyclooxygenase-2 in synaptic signaling. Curr. Pharm. Des. 2008, 14, 1443–1451. [Google Scholar] [CrossRef] [Green Version]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; van de Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, S.S.; Wood, E.G.; Hassan, S.-U.; Bolton, C. Cyclooxygenase expression and prostaglandin levels in central nervous system tissues during the course of chronic relapsing experimental autoimmune encephalomyelitis (EAE). Inflamm. Res. 2011, 60, 919–928. [Google Scholar] [CrossRef]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Sobel, R.A. The extracellular matrix in multiple sclerosis: An update. Braz. J. Med. Biol. Res. 2001, 34, 603–609. [Google Scholar] [CrossRef] [PubMed]
- van Strien, M.E.; Brevé, J.J.P.; Fratantoni, S.; Schreurs, M.W.J.; Bol, J.G.J.M.; Jongenelen, C.A.M.; Drukarch, B.; van Dam, A. Astrocyte-derived tissue transglutaminase interacts with fibronectin: A role in astrocyte adhesion and migration? PLoS ONE 2011, 6, e25037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelea, V.; Nakano, Y.; Kaartinen, M.T. Size distribution and molecular associations of plasma fibronectin and fibronectin crosslinked by transglutaminase 2. Protein. J. 2008, 27, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Stoffels, J.M.J.; de Jonge, J.C.; Stancic, M.; Nomden, A.; van Strien, M.E.; Ma, D.; Šišková, Z.; Maier, O.; Ffrench-Constant, C.; Franklin, R.J.M.; et al. Fibronectin aggregation in multiple sclerosis lesions impairs remyelination. Brain 2013, 136, 116–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šišková, Z.; Baron, W.; de Vries, H.; Hoekstra, D. Fibronectin impedes "myelin" sheet-directed flow in oligodendrocytes: A role for a beta 1 integrin-mediated PKC signaling pathway in vesicular trafficking. Mol. Cell Neurosci. 2006, 33, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Park, H.-B.; Seo, M.W.; Byoun, O.-J.; Lee, D.-S. Transglutaminase 2 exacerbates experimental autoimmune encephalomyelitis through positive regulation of encephalitogenic T cell differentiation and inflammation. Clin. Immunol. 2012, 145, 122–132. [Google Scholar] [CrossRef] [PubMed]
- van Strien, M.E.; de Vries, H.E.; Chrobok, N.L.; Bol, J.G.J.M.; Breve, J.J.P.; van der Pol, S.M.P.; Kooij, G.; van Buul, J.D.; Karpuj, M.; Steinman, L.; et al. Tissue Transglutaminase contributes to experimental multiple sclerosis pathogenesis and clinical outcome by promoting macrophage migration. Brain Behav. Immun. 2015, 50, 141–154. [Google Scholar] [CrossRef]
- de Jong, J.M.; Wang, P.; Oomkens, M.; Baron, W. Remodeling of the interstitial extracellular matrix in white matter multiple sclerosis lesions: Implications for remyelination (failure). J. Neurosci. Res. 2020, 98, 1370–1397. [Google Scholar] [CrossRef] [Green Version]
- Yahn, S.L.; Li, J.; Goo, I.; Gao, H.; Brambilla, R.; Lee, J.K. Fibrotic scar after experimental autoimmune encephalomyelitis inhibits oligodendrocyte differentiation. Neurobiol. Dis. 2020, 134, 104674. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, M.J.; Sand, J.M.; Henriksen, K.; Genovese, F.; Bay-Jensen, A.-C.; Smith, V.; Adamkewicz, J.I.; Christiansen, C.; Leeming, D.J. Extracellular matrix remodeling: The common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev. Technol. 2013, 11, 70–92. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.W.; Keough, M.B.; Haylock-Jacobs, S.; Cua, R.; Döring, A.; Sloka, S.; Stirling, D.P.; Rivest, S.; Yong, V.W. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Ann. Neurol. 2012, 72, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Sobel, R.A.; Ahmed, A.S. White matter extracellular matrix chondroitin sulfate/dermatan sulfate proteoglycans in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2001, 60, 1198–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteagudo, A.; Feola, J.; Natola, H.; Ji, C.; Pröschel, C.; Johnson, G.V. Depletion of astrocytic transglutaminase 2 improves injury outcomes. Mol. Cell Neurosci. 2018, 92, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Lord, M.S.; Chuang, C.Y.; Melrose, J.; Davies, M.J.; Iozzo, R.V.; Whitelock, J.M. The role of vascular-derived perlecan in modulating cell adhesion, proliferation and growth factor signaling. Matrix Biol. 2014, 35, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.; Kahle, M.P.; Bix, G.J. Perlecan and the blood-brain barrier: Beneficial proteolysis? Front. Pharm. 2012, 3, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y. Traffic lights for axon growth: Proteoglycans and their neuronal receptors. Neural. Regen. Res. 2014, 9, 356–361. [Google Scholar] [CrossRef] [PubMed]
- van Horssen, J.; Bö, L.; Dijkstra, C.D.; de Vries, H.E. Extensive extracellular matrix depositions in active multiple sclerosis lesions. Neurobiol. Dis. 2006, 24, 484–491. [Google Scholar] [CrossRef]
- Beckouche, N.; Bignon, M.; Lelarge, V.; Mathivet, T.; Pichol-Thievend, C.; Berndt, S.; Hardouin, J.; Garand, M.; Ardidie-Robouant, C.; Barret, A.; et al. The interaction of heparan sulfate proteoglycans with endothelial transglutaminase-2 limits VEGF165-induced angiogenesis. Sci. Signal. 2015, 8, ra70. [Google Scholar] [CrossRef]
- Lengfeld, J.; Cutforth, T.; Agalliu, D. The role of angiogenesis in the pathology of multiple sclerosis. Vasc. Cell 2014, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Hang, J.; Zemskov, E.A.; Lorand, L.; Belkin, A.M. Identification of a novel recognition sequence for fibronectin within the NH2-terminal beta-sandwich domain of tissue transglutaminase. J. Biol. Chem. 2005, 280, 23675–23683. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSB# Patient ID | Age (years) | Gender | Microscopic Description of Lesion from the Neuropathological Report | Post-Mortem Interval (hours) | Clinical Diagnosis |
|---|---|---|---|---|---|
| 3422 | 62 | Male | Section of periventricular white matter showed demyelination characterized by a variably decreased axonal density (80–100%) and complete demyelination. There was severe associated gliosis and oligodendrocyte loss but no associated macrophage activity or perivascular lymphocytic cuffing. Presence of chronic MS plaque formation. | 11.75 | MS/RRMS |
| 3891 | 53 | Male | Section of periventricular white matter showed plaque formation with up to 100% axonal loss and 100% demyelination, near complete loss of oligodendrocytes, and associated gliosis was prominent along with scattered evidence of macrophage activity and perivascular lymphocytic cuffing Presence of chronic/active multiple sclerosis plaque formation and active multiple sclerosis plaque formation. | 25.3 | MS/CPMS, depression, post-traumatic stress disorder, degenerative spine disease |
| 5053 | 67 | Male | Section of periventricular white matter showed plaque formation with up to 100% axonal loss and 100% demyelination There was near complete oligodendrocyte loss and prominent associated gliosis. There was no evidence of macrophage activity or perivascular lymphocytic cuffing Evidence of chronic multiple sclerosis plaque formation. | 24.9 | MS/secondary progressive MS, migraine, history of stroke, transient ischemic attack (TIA), Chronic Obstructive Pulmonary Disease (COPD), pneumonia, aspiration, meningitis, ataxia, chronic UTI |
| 5095 | 60 | Male | Section of periventricular white matter showed irregular but fairly well-defined area of extensive myelin loss and relative preservation of axons with scant perivascular mononuclear infiltrates. There was associated gliosis at the periphery of the lesion. | 8.1 | MS, RRMS, sclerosis, heart attack |
| 5102 | 60 | Female | Section of periventricular white matter showed fairly well-defined area of demyelination and relative preservation of axons with scattered macrophages. | 14.7 | MS, RRMS, depression, celiac disease, headache. Neurogenic bladder, chronic UTI, osteoporosis, insomnia |
| 5123 | 79 | Female | Section of periventricular white matter displayed somewhat ill-defined areas of myelin pallor with relative axonal preservation. | 24.8 | MS/RRMS, congestive heart failure, neuralgia, trigeminal, asthma |
| 5139 | 81 | Male | Section of periventricular white matter showed myelin loss with relative axonal preservation consistent with chronic inactive plaque of multiple sclerosis. Evidence of demyelinating lesions consistent with clinical history of multiple sclerosis. | 36.8 | MS/RRMS, dysphagia, paraplegic, neurogenic bladder, diabetes type 11, hypertension, hyperlipidemia, myocardial infarction (Ml), atherosclerosis |
| 5154 | 63 | Male | Section of periventricular white matter showed irregular but fairly well-defined area of extensive myelin loss and relative preservation of axons with rare perivascular mononuclear infiltrates. There was associated mild gliosis at the periphery of the lesion. | 12.6 | MS/RRMS, optic neuritis |
| 5160 | 82 | Male | Sections of periventricular white matter showed plaque formation with up to 100% axonal loss and 100% demyelination with near-complete oligodendrocyte loss and associated gliosis. There was no evidence of macrophage activity or perivascular lymphocytic cuffing. Chronic multiple sclerosis plaque formation. | 9.8 | MS/RRMS, stroke/CVA, pneumonia, aspiration, MRSA (methicillin-resistant Staphylococcus aureus), chronic UTI |
| 5170 | 52 | Female | Sections from the lateral angle of the lateral ventricle showed an old demyelination plaque surrounded by hypercellular white matter. Presence of old demyelination plaque surrounded by hypercellular white matter. | 25.0 | MS, CPMS, optic neuritis, depression |
| 5270 | 38 | Female | Old demyelination plaques were seen in the sections of posterior cingulate gyrus and pons. Presence of old demyelination plaques. | 17.5 | MS, CPMS, asthma, depression, pain, paraplegic, ataxia (cerebellar) |
| 4307 | 84 | Male | Normal Appearing White Matter (NAWM) | 11.8 | CA, stomach, renal failure, acute, COPD (control, non-MS) |
| 4308 | 70 | Male | NAWM | 11.8 | Coronary heart disease, leukemia, diabetes type I, myocardial infarction (control, non-MS) |
| 4294 | 80 | Male | NAWM | 19.2 | CA, pancreas, hypertension (control, non-MS) |
| 4631 | 59 | Male | NAWM | 20.2 | COPD, pulmonary emphysema, congestive heart failure, tobacco abuse, atrial fibrillation, hypertension (control, non-MS) |
| 5072 | 83 | Male | NAWM | 19.5 | COPD, seizure disorder (clinical only), atrial fibrillation (control, non-MS) |
| 4615 | 49 | Male | NAWM | 15 | CA, colon with metastasis to liver, depression (control, non-MS) |
| Primary Antibody | Manufacturer | Catalog Number | Antibody Host | Dilution Used |
|---|---|---|---|---|
| TGM2 pAb (CUB 7402) | Thermo Fisher | MA5-12739 | Mouse | 1:100 |
| Rb pAb to Transglutaminase-2 | Abcam | ab421 | Rabbit | 1:100 |
| Anti IBA1, Rabbit (for ICC) | Wako/Fuji | 019-19741 | Rabbit | 1:1000 |
| Chicken Polyclonal to IBA1 | Encor | CPCA-IBA1 | Chicken | 1:1000 |
| Goat pAb to IBA1 | Abcam | ab5076 | Goat | 1:500 |
| Myelin Basic Protein | Encor | CPCA-MBP | Chicken | 1:2500 |
| Degraded Myelin Basic Protein | Millipore | AB5864 | Rabbit | 1:1000 |
| Glial Fibrillary Acidic Protein | Dako | Z0334 | Rabbit | 1:500 |
| Anti-Glial Fibrillary Acidic Protein | Millipore | AB5541 | Chicken | 1:250 |
| Anti-Fibronectin antibody | Sigma | F3648 | Rabbit | 1:200 |
| Monoclonal Anti-Chondroitin Sulfate (Clone CS-56) | Sigma | C8035 (SAB4200696) | Mouse | 1:200 |
| Anti-Heparan Sulfate Proteoglycan, (Perlecan), clone 5D7-2E4 | Sigma | MABT 12 | Mouse | 1:200 |
| Anti-phospho-c-PLA2 (pSer505) | Sigma | SAB4503812 | Rabbit | 1:100 |
| Anti-COX-2/Cyclooxygenase 2 | Abcam | ab15191 | Rabbit | 1:100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pearse, D.D.; Hefley, A.B.; Morales, A.A.; Ghosh, M. Comparative Profiling of TG2 and Its Effectors in Human Relapsing Remitting and Progressive Multiple Sclerosis. Biomedicines 2022, 10, 1241. https://doi.org/10.3390/biomedicines10061241
Pearse DD, Hefley AB, Morales AA, Ghosh M. Comparative Profiling of TG2 and Its Effectors in Human Relapsing Remitting and Progressive Multiple Sclerosis. Biomedicines. 2022; 10(6):1241. https://doi.org/10.3390/biomedicines10061241
Chicago/Turabian StylePearse, Damien D., Andrew B. Hefley, Alejo A. Morales, and Mousumi Ghosh. 2022. "Comparative Profiling of TG2 and Its Effectors in Human Relapsing Remitting and Progressive Multiple Sclerosis" Biomedicines 10, no. 6: 1241. https://doi.org/10.3390/biomedicines10061241