
Naphthoquinone as a New Chemical Scaffold for Leishmanicidal Inhibitors of Leishmania GSK-3

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Procedures
2.1.1. Synthesis of Carbamate Derivatives 1–5
2.1.2. General Procedure for the Synthesis of Amines 6–13
2.1.3. Procedure for the Synthesis of 1-(tert-Butyl)-3-(3-chloro-1,4-naphthoquinone-2-yl)urea (14)
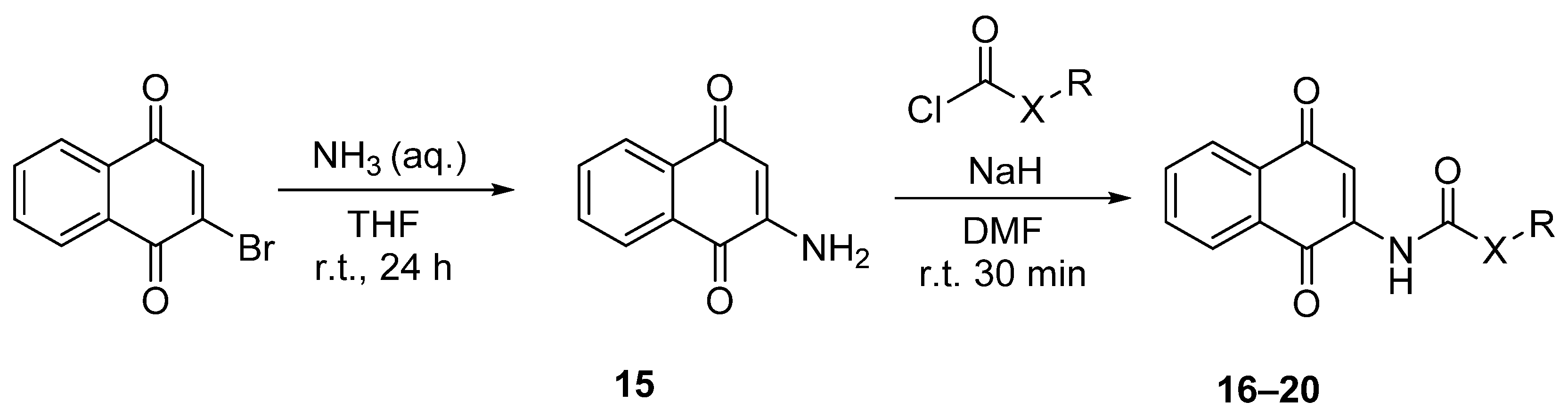
2.1.4. Procedure for the Synthesis of 2-Amino-1,4-naphthoquinone (15)
2.1.5. General Procedure for the Synthesis of Carbamates 16–17
2.1.6. General Procedure for the Synthesis of Amides 18–20
2.2. Computational Studies
2.2.1. Ligand Preparation
2.2.2. Protein Preparation
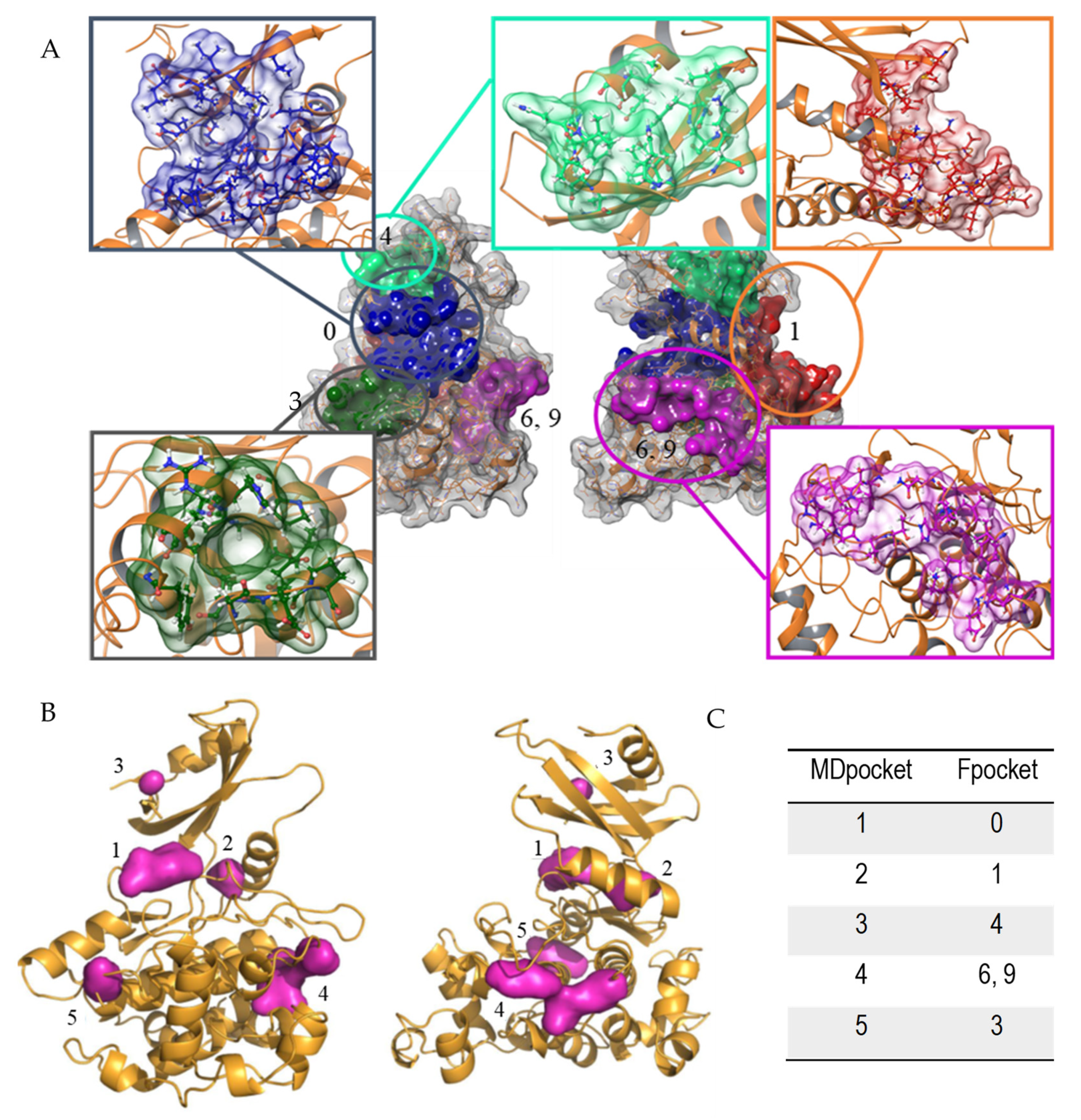
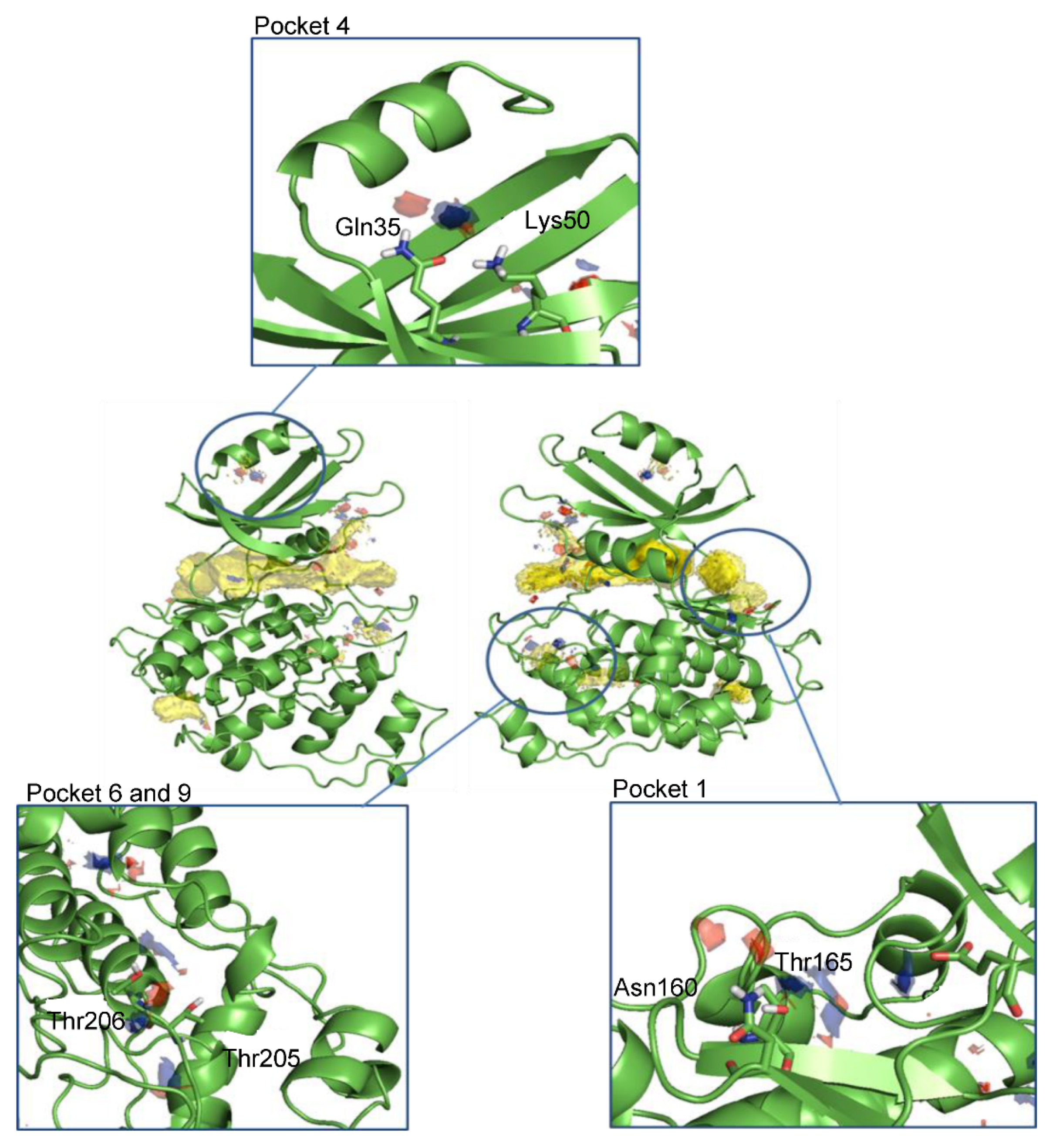
2.2.3. Cavity Detection Analysis
2.2.4. Hotspots Maps
2.2.5. Virtual Screening
2.3. Experimental Biology Procedures
2.3.1. Reagents
2.3.2. Assessment of Inhibition of GSK-3 with Kinase Glo®
2.3.3. Cell
2.3.4. Cell Harvesting
2.3.5. Leishmanicidal and Cytotoxicity Assays of the Different Compounds
2.3.6. Cytotoxicity against Intracellular Amastigotes
2.3.7. Mitochondrial Membrane Depolarization
2.3.8. Oxygen Consumption Rate
2.3.9. Measurement of Sub G0/G1 Population
3. Results and Discussion
3.1. Computational Analysis of Leishmania GSK-3 Structure
3.2. Searching for Novel Inhibitors Using Virtual Screening
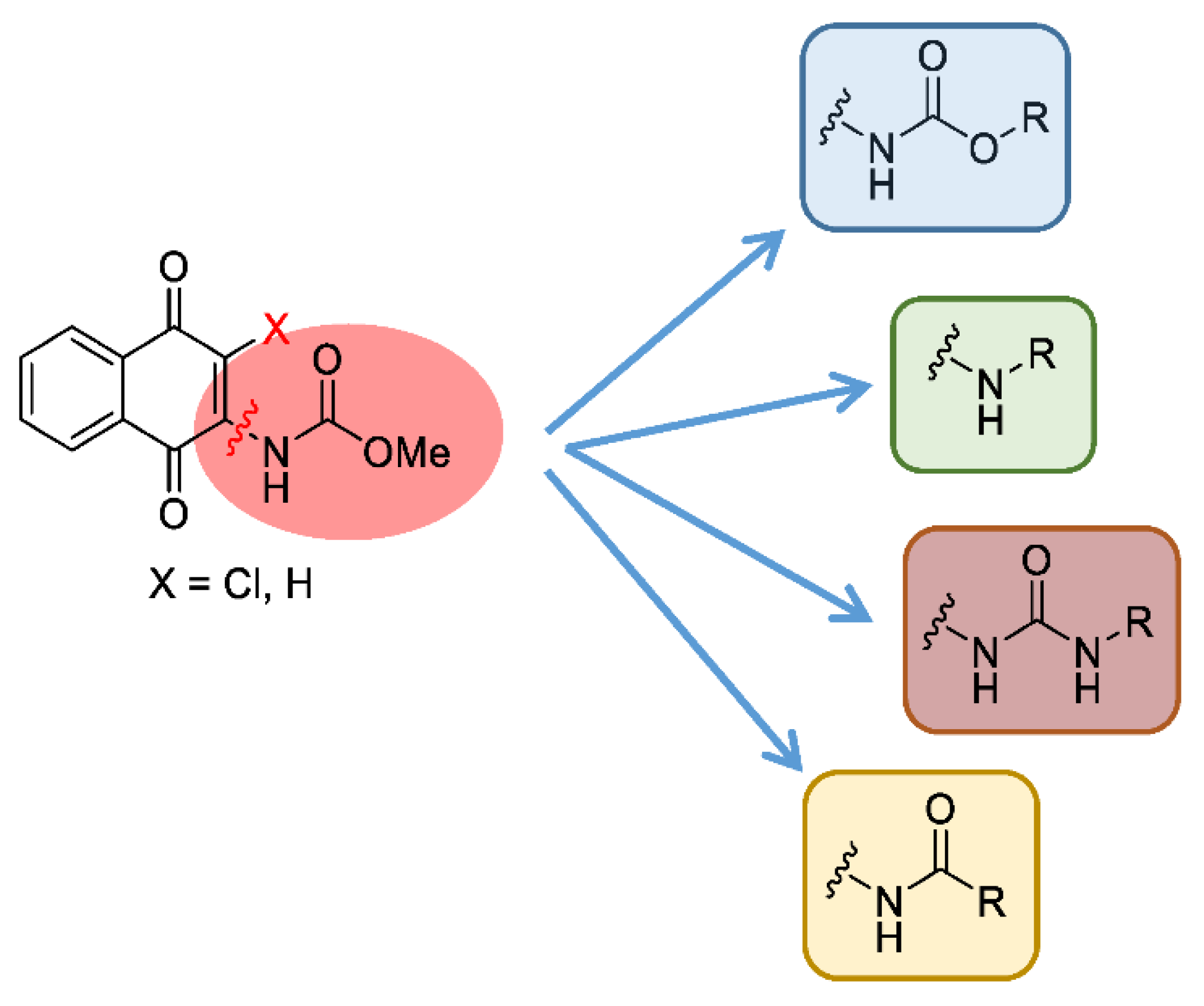
3.3. Design, Synthesis, Biological Evaluation, and SAR Analysis of a Second Generation of Naphthoquinone Derivatives
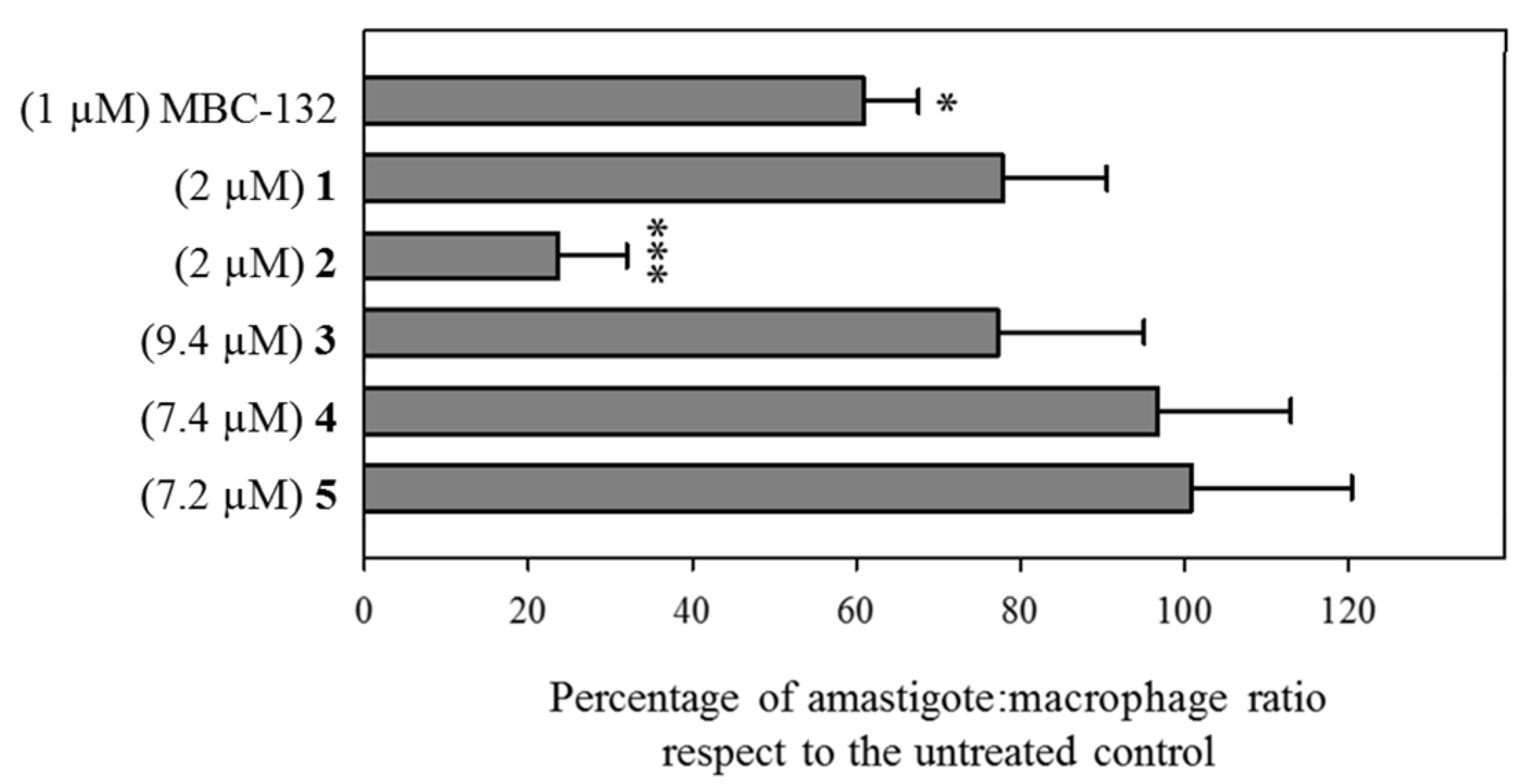
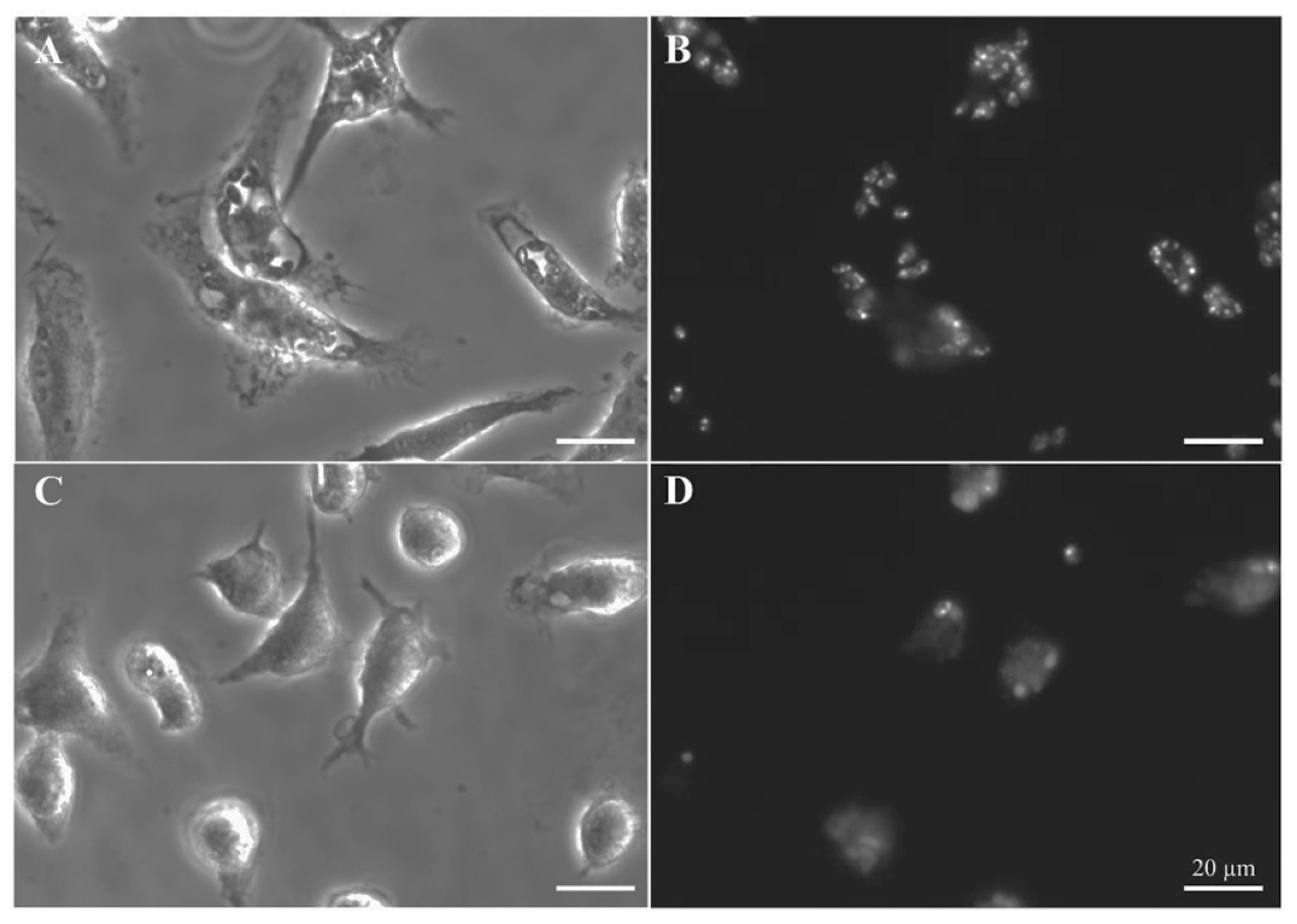
3.4. Leishmanicidal Activity on Intracellular Amastigotes of LdGSK-3s Quinone Inhibitors
3.5. Energy Metabolism of Leishmania as an Off-Target Effect of LdGSK-3s Inhibitors
3.5.1. Inhibition of the Electrochemical Potential of the Leishmania Mitochondrion (ΔΨm)
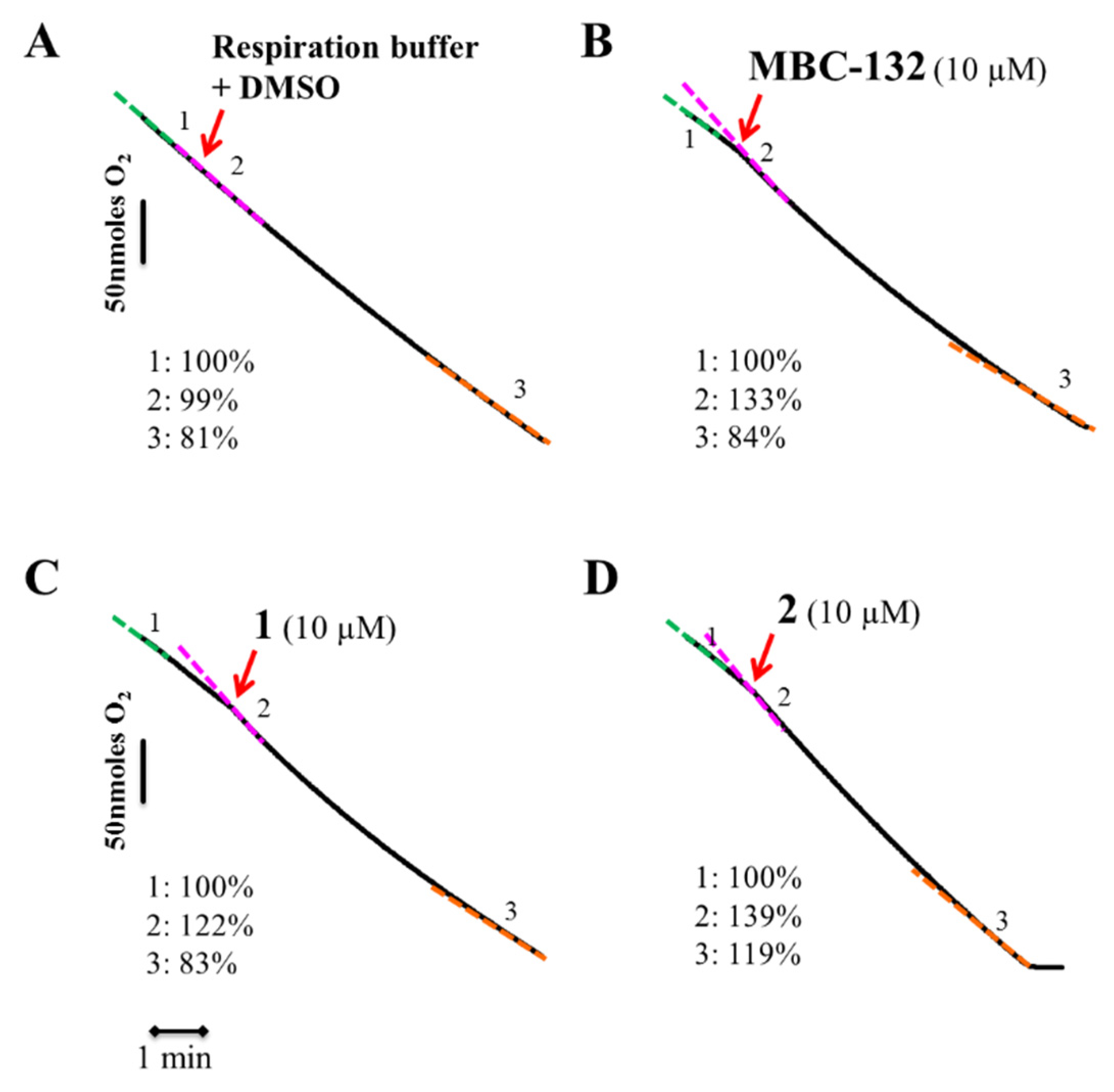
3.5.2. Inhibition of Oxygen Consumption
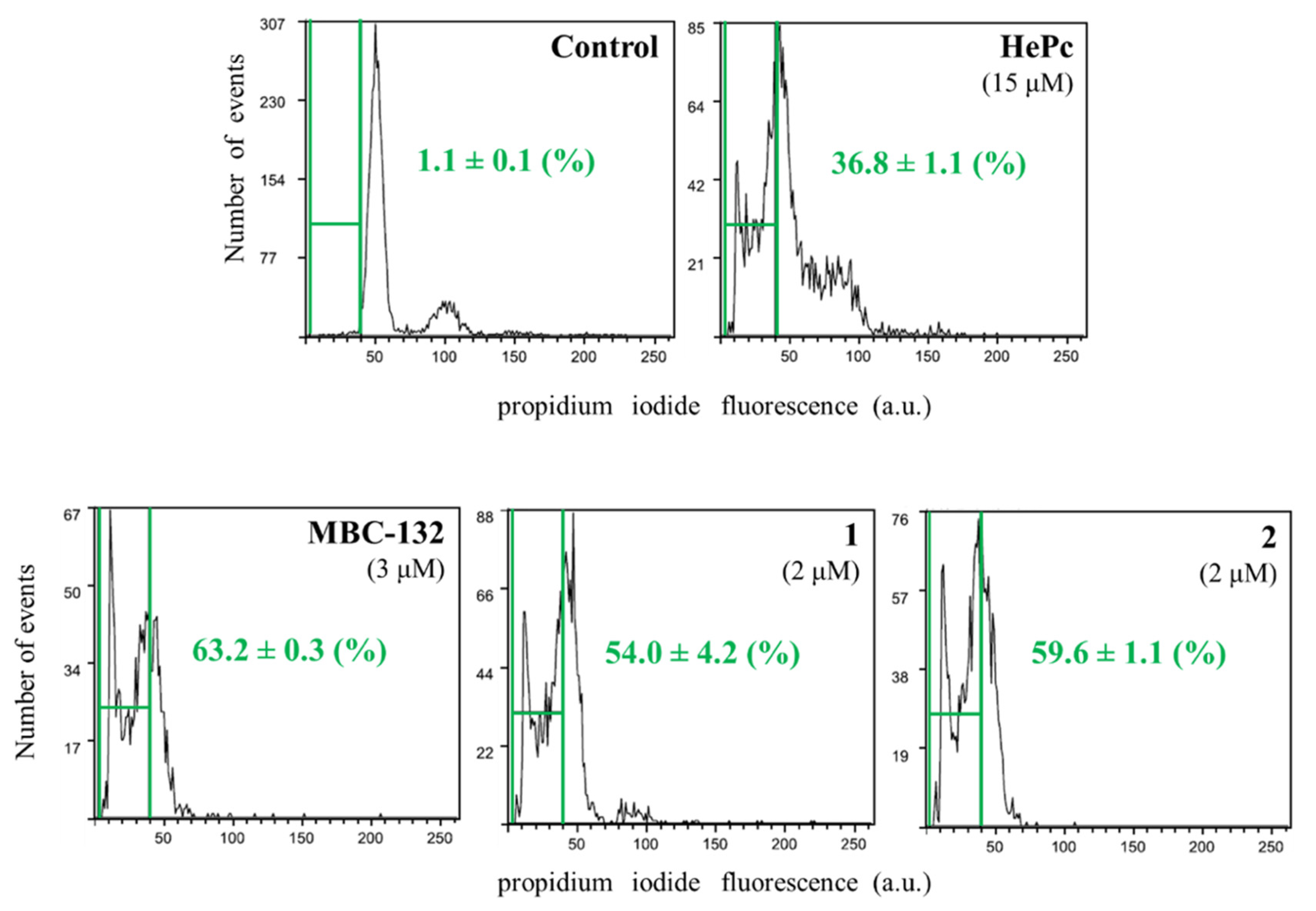
3.5.3. Induction of Programmed Cell Death in L. donovani Promastigotes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Booth, M. Climate change and the neglected tropical diseases. Adv. Parasitol. 2018, 100, 39–126. [Google Scholar] [CrossRef]
- Alves, F.; Bilbe, G.; Blesson, S.; Goyal, V.; Monnerat, S.; Mowbray, C.; Ouattara, G.M.; Pécoul, B.; Rijal, S.; Rode, J.; et al. Recent development of visceral leishmaniasis treatments: Successes, pitfalls, and perspectives. Clin. Microbiol. Rev. 2018, 31, e00048-18. [Google Scholar] [CrossRef] [Green Version]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Sundar, S.; Singh, A. Chemotherapeutics of visceral leishmaniasis: Present and future developments. Parasitology 2018, 145, 481–489. [Google Scholar] [CrossRef]
- Braga, S.S. Multi-target drugs active against leishmaniasis: A paradigm of drug repurposing. Eur. J. Med. Chem. 2019, 183, 111660. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Chico, L.K.; Van Eldik, L.J.; Watterson, D.M. Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discov. 2009, 8, 892–909. [Google Scholar] [CrossRef] [Green Version]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase mutations in human disease: Interpreting genotype-phenotype relationships. Nat. Rev. Gen. 2010, 11, 60–74. [Google Scholar] [CrossRef]
- Kini, S.G.; Garg, V.; Prasanna, S.; Rajappan, R.; Mubeen, M. Protein kinases as drug targets in human and animal diseases. Curr. Enzym. Inhib. 2017, 13, 99–106. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Borba, J.V.B.; Silva, A.C.; Ramos, P.I.P.; Grazzia, N.; Miguel, D.C.; Muratov, E.N.; Furnham, N.; Andrade, C.H. Unveiling the kinomes of Leishmania infantum and L. braziliensis empowers the discovery of new kinase targets and antileishmanial compounds. Comput. Struct. Biotechnol. J. 2019, 17, 352–361. [Google Scholar] [CrossRef]
- Baker, N.; Catta-Preta, C.M.C.; Neish, R.; Sadlova, J.; Powell, B.; Alves-Ferreira, E.V.C.; Geoghegan, V.; Carnielli, J.B.T.; Newling, K.; Hughes, C.; et al. Systematic functional analysis of Leishmania protein kinases identifies regulators of differentiation or survival. Nat. Commun. 2021, 12, 1244. [Google Scholar] [CrossRef]
- Parsons, M.; Worthey, E.A.; Ward, P.N.; Mottram, J.C. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genom. 2005, 6, 127. [Google Scholar] [CrossRef] [Green Version]
- Garg, M.; Goyal, N. MAPK1 of Leishmania donovani modulates antimony susceptibility by downregulating P-glycoprotein efflux pumps. Antimicrob. Agents Chemother. 2015, 59, 3853–3863. [Google Scholar] [CrossRef] [Green Version]
- Wiese, M. A mitogen-activated protein (MAP) kinase homologue of Leishmania mexicana is essential for parasite survival in the infected host. EMBO J. 1998, 17, 2619–2628. [Google Scholar] [CrossRef] [Green Version]
- Zylbersztejn, A.M.B.; De Morais, C.G.V.; Lima, A.K.C.; Souza, J.E.D.O.; Lopes, A.H.; Da-Silva, S.A.G.; Silva-Neto, M.A.C.; Dutra, P.M.L. CK2 secreted by Leishmania braziliensis mediates macrophage association invasion: A comparative study between virulent and avirulent promastigotes. BioMed Res. Int. 2015, 2015, 167323. [Google Scholar] [CrossRef] [Green Version]
- Merritt, C.; Silva, L.E.; Tanner, A.L.; Stuart, K.; Pollastri, M.P. Kinases as druggable targets in trypanosomatid protozoan parasites. Chem. Rev. 2014, 114, 11280–11304. [Google Scholar] [CrossRef] [Green Version]
- Dacher, M.; Morales, M.A.; Pescher, P.; Leclercq, O.; Rachidi, N.; Prina, E.; Cayla, M.; Descoteaux, A.; Späth, G.F. Probing druggability and biological function of essential proteins in Leishmania combining facilitated null mutant and plasmid shuffle analyses. Mol. Microbiol. 2014, 93, 146–166. [Google Scholar] [CrossRef]
- Wyllie, S.; Thomas, M.; Patterson, S.; Crouch, S.; De Rycker, M.; Lowe, R.; Gresham, S.; Urbaniak, M.D.; Otto, T.D.; Stojanovski, L.; et al. Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis. Nature 2018, 560, 192–197. [Google Scholar] [CrossRef]
- Dichiara, M.; Marrazzo, A.; Prezzavento, O.; Collina, S.; Rescifina, A.; Amata, E. Repurposing of human kinase inhibitors in neglected protozoan diseases. ChemMedChem 2017, 12, 1235–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirado-Duarte, D.; Marín-Villa, M.; Ochoa, R.; Blandón-Fuentes, G.; Soares, M.J.; Robledo, S.M.; Varela-Miranda, R.E. The Akt-like kinase of Leishmania panamensis: As a new molecular target for drug discovery. Acta Trop. 2018, 177, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Chhajer, R.; Bhattacharyya, A.; Didwania, N.; Shadab, M.; Das, N.; Palit, P.; Vaidya, T.; Ali, N. Leishmania donovani Aurora kinase: A promising therapeutic target against visceral leishmaniasis. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 1973–1988. [Google Scholar] [CrossRef] [PubMed]
- Ojo, K.K.; Gillespie, J.R.; Riechers, A.J.; Napuli, A.J.; Verlinde, C.L.; Buckner, F.S.; Gelb, M.H.; Domostoj, M.M.; Wells, S.J.; Scheer, A.; et al. Glycogen synthase kinase 3 is a potential drug target for African trypanosomiasis therapy. Antimicrob. Agents Chemother. 2008, 52, 3710–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xingi, E.; Smirlis, D.; Myrianthopoulos, V.; Magiatis, P.; Grant, K.M.; Meijer, L.; Mikros, E.; Skaltsounis, A.L.; Soteriadou, K. 6-Br-5methylindirubin-3′oxime (5-Me-6-BIO) targeting the leishmanial glycogen synthase kinase-3 (GSK-3) short form affects cell-cycle progression and induces apoptosis-like death: Exploitation of GSK-3 for treating leishmaniasis. Int. J. Parasitol. 2009, 39, 1289–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Bhavanasi, D.; Klein, P.S. Wnt signaling in normal and malignant stem cells. Curr. Stem Cell Rep. 2016, 2, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and associated signaling pathways involved in cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef]
- Roca, C.; Campillo, N.E. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2016–2019). Expert Opin. Ther. Pat. 2020, 30, 863–872. [Google Scholar] [CrossRef]
- Ojo, K.K.; Arakaki, T.L.; Napuli, A.J.; Inampudi, K.K.; Keyloun, K.R.; Zhang, L.; Hol, W.G.J.; Verlinde, C.L.M.J.; Merritt, E.A.; Van Voorhis, W.C. Structure determination of glycogen synthase kinase-3 from Leishmania major and comparative inhibitor structure-activity relationships with Trypanosoma brucei GSK-3. Mol. Biochem. Parasitol. 2011, 176, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efstathiou, A.; Gaboriaud-Kolar, N.; Smirlis, D.; Myrianthopoulos, V.; Vougogiannopoulou, K.; Alexandratos, A.; Kritsanida, M.; Mikros, E.; Soteriadou, K.; Skaltsounis, A.L. An inhibitor-driven study for enhancing the selectivity of indirubin derivatives towards leishmanial Glycogen Synthase Kinase-3 over leishmanial cdc2-related protein kinase 3. Parasites Vectors 2014, 7, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efstathiou, A.; Smirlis, D. Leishmania protein kinases: Important regulators of the parasite life cycle and molecular targets for treating leishmaniasis. Microorganisms 2021, 9, 691. [Google Scholar] [CrossRef] [PubMed]
- Martínez de Iturrate, P.; Sebastián-Pérez, V.; Nácher-Vázquez, M.; Tremper, C.S.; Smirlis, D.; Martín, J.; Martínez, A.; Campillo, N.E.; Rivas, L.; Gil, C. Towards discovery of new leishmanicidal scaffolds able to inhibit Leishmania GSK-3. J. Enzym. Inhib. Med. Chem. 2020, 35, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittner, S.; Temtsin, G.; Sasson, Y. Synthesis of N-quinonyl carbamates via 2-chloro-3-isocyanato-1, 4-naphthoquinone. Synthesis 2000, 2000, 1084–1086. [Google Scholar] [CrossRef]
- Husu, B.; Kafka, S.; Kadunc, Z.; Tišler, M. Amination of naphthoquinones with azidotrimethylsilane. Mon. Chem. 1988, 119, 215–222. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Schrödinger Release 2015-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2015.
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R. Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comp.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger Release 2015-4: Maestro; Schrödinger, LLC: New York, NY, USA, 2015.
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtke, P.; Bidon-Chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radoux, C.J.; Olsson, T.S.; Pitt, W.R.; Groom, C.R.; Blundell, T.L. Identifying interactions that determine fragment binding at protein hotspots. J. Med. Chem. 2016, 59, 4314–4325. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-1: Glide; Schrödinger, LLC: New York, NY, USA, 2017.
- Sebastián-Pérez, V.; Roca, C.; Awale, M.; Reymond, J.L.; Martinez, A.; Gil, C.; Campillo, N.E. Medicinal and Biological Chemistry (MBC) library: An efficient source of new hits. J. Chem. Inform. Model. 2017, 57, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodríguez-Muela, N.; Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.E.; Perez-Castillo, A.M.; et al. Subtly modulating glycogen synthase kinase 3 β: Allosteric inhibitor development and their potential for the treatment of chronic diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef]
- Luque-Ortega, J.R.; Rivas, L. Characterization of the leishmanicidal activity of antimicrobial peptides. Methods Mol. Biol. 2010, 618, 393–420. [Google Scholar] [CrossRef]
- Zuo, X.; Djordjevic, J.T.; Oei, J.B.; Desmarini, D.; Schibeci, S.D.; Jolliffe, K.A.; Sorrell, T.C. Miltefosine induces apoptosis-like cell death in yeast via Cox9p in cytochrome c oxidase. Mol. Pharmacol. 2011, 80, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Domínguez, J.M.; Fuertes, A.; Orozco, L.; Monte-Millán, M.D.; Delgado, E.; Medina, M. Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib. J. Biol. Chem. 2012, 287, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Palomo, V.; Soteras, I.; Perez, D.I.; Perez, C.; Gil, C.; Campillo, N.E.; Martinez, A. Exploring the binding sites of glycogen synthase kinase 3. identification and characterization of allosteric modulation cavities. J. Med. Chem. 2011, 54, 8461–8470. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Hou, X.; Jacob, C. 1,4-naphthoquinones: From oxidative damage to cellular and inter-cellular signaling. Molecules 2014, 19, 14902–14918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silakari, P.; Priyanka; Piplani, P. p-Benzoquinone as a privileged scaffold of pharmacological significance: A review. Mini-Rev. Med. Chem. 2020, 20, 1586–1609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, G.; Xu, S.; Song, Y. Recent advances of quinones as a privileged structure in drug discovery. Eur. J. Med. Chem. 2021, 223, 113632. [Google Scholar] [CrossRef]
- Ferreira, V.F.; de Carvalho, A.S.; Ferreira, P.G.; Lima, C.G.S.; da Silva, F.C. Quinone-based drugs: An important class of molecules in medicinal chemistry. Med. Chem. 2021, 17, 1073–1085. [Google Scholar] [CrossRef]
- Schroeder, R.L.; Goyal, N.; Bratton, M.; Townley, I.; Pham, N.A.; Tram, P.; Stone, T.; Geathers, J.; Nguyen, K.; Sridhar, J. Identification of quinones as novel PIM1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3187–3191. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.J.; Choi, C.K.; Lee, C.S.; Park, M.H.; Tian, X.; Kim, N.D.; Lee, K.I.; Choi, J.K.; Ahn, J.H.; Shin, E.Y.; et al. Small molecules that allosterically inhibit p21-activated kinase activity by binding to the regulatory p21-binding domain. Exp. Mol. Med. 2016, 48, e229. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.J.; Ko, I.L.; Lee, C.L.; Hu, H.C.; Chang, F.R.; Wu, Y.C.; Leu, Y.L.; Wu, C.C.; Lin, C.Y.; Pan, C.Y.; et al. Targeting allosteric site of Akt by 5,7-dimethoxy-1,4-phenanthrenequinone suppresses neutrophilic inflammation. EBioMedicine 2019, 40, 528–540. [Google Scholar] [CrossRef] [Green Version]
- Bolton, J.L.; Dunlap, T. Formation and biological targets of quinones: Cytotoxic versus cytoprotective effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- Pinto, A.V.; De Castro, S.L. The trypanocidal activity of naphthoquinones: A review. Molecules 2009, 14, 4570–4590. [Google Scholar] [CrossRef]
- Lizzi, F.; Veronesi, G.; Belluti, F.; Bergamini, C.; López-Sánchez, A.; Kaiser, M.; Brun, R.; Krauth-Siegel, R.L.; Hall, D.G.; Rivas, L.; et al. Conjugation of quinones with natural polyamines: Toward an expanded antitrypanosomatid profile. J. Med. Chem. 2012, 55, 10490–10500. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Ueda, K.; Sakaguchi, K.; Tokuda, H.; Iida, A. One-Pot synthesis of benzo [f] indole-4, 9-diones from 1, 4-naphthoquinones and terminal acetylenes. Chem. Pharm. Bull. 2011, 59, 1289–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josey, B.J.; Inks, E.S.; Wen, X.; Chou, C.J. Structure–activity relationship study of vitamin K derivatives yields highly potent neuroprotective agents. J. Med. Chem. 2013, 56, 1007–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, A.A.; Duboise, S.M.; Eperon, S.; Rivas, L.; Hodgkinson, V.; Traub-Cseko, Y.; McMahon-Pratt, D. Developmental life cycle of Leishmania—Cultivation and characterization of cultured eextracellular amastigotes. J. Eukaryot. Microbiol. 1993, 40, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Werstuck, G.H. Macrophage function and the role of GSK3. Int. J. Mol. Sci. 2021, 22, 2206. [Google Scholar] [CrossRef]
- Hoffmeister, L.; Diekmann, M.; Brand, K.; Huber, R. GSK3: A kinase balancing promotion and resolution of inflammation. Cells 2020, 9, 820. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Vieyra, R.; Silva-García, O.; Gómez-García, A.; Gutiérrez-Castellanos, S.; Álvarez-Aguilar, C.; Baizabal-Aguirre, V.M. Glycogen synthase kinase 3β modulates the inflammatory response activated by bacteria, viruses, and parasites. Front. Immunol. 2021, 12, 675751. [Google Scholar] [CrossRef]
- Nandan, D.; Camargo de Oliveira, C.; Moeenrezakhanlou, A.; Lopez, M.; Silverman, J.M.; Subek, J.; Reiner, N.E. Myeloid cell IL-10 production in response to Leishmania involves inactivation of glycogen synthase kinase-3β downstream of phosphatidylinositol-3 kinase. J. Immunol. 2012, 188, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Paul, J.; Naskar, K.; Chowdhury, S.; Chakraborti, T.; De, T. TLR mediated GSK3β activation suppresses CREB mediated IL-10 production to induce a protective immune response against murine visceral leishmaniasis. Biochimie 2014, 107, 235–246. [Google Scholar] [CrossRef]
- Ribeiro, G.A.; Cunha-Júnior, E.F.; Pinheiro, R.O.; da-Silva, S.A.G.; Canto-Cavalheiro, M.M.; da Silva, A.J.M.; Costa, P.R.R.; Netto, C.D.; Melo, R.C.N.; Almeida-Amaral, E.E.; et al. LQB-118, an orally active pterocarpanquinone, induces selective oxidative stress and apoptosis in Leishmania amazonensis. J. Antimicrob. Chemother. 2013, 68, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Prati, F.; Bergamini, C.; Molina, M.T.; Falchi, F.; Cavalli, A.; Kaiser, M.; Brun, R.; Fato, R.; Bolognesi, M.L. 2-Phenoxy-1,4-naphthoquinones: From a multitarget antitrypanosomal to a potential antitumor profile. J. Med. Chem. 2015, 58, 6422–6434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tielens, A.G.; van Hellemond, J.J. Surprising variety in energy metabolism within Trypanosomatidae. Trends Parasitol. 2009, 25, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.; Forquer, I.; Boitz, J.; Soysa, R.; Elya, C.; Fulwiler, A.; Nilsen, A.; Polley, T.; Riscoe, M.K.; Ullman, B. Targeting the cytochrome bc 1 complex of Leishmania parasites for discovery of novel drugs. Antimicrob. Agents Chemother. 2016, 60, 4972–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, L.; Luque-Ortega, J.R.; Manzano, J.I.; Castanys, S.; Rivas, L.; Gamarro, F. Tafenoquine, an antiplasmodial 8-aminoquinoline, targets Leishmania respiratory complex III and induces apoptosis. Antimicrob. Agents Chemother. 2010, 54, 5344–5351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, L.; Luque-Ortega, J.R.; López-Martín, C.; Castanys, S.; Rivas, L.; Gamarro, F. The 8-aminoquinoline analogue sitamaquine causes oxidative stress in Leishmania donovani promastigotes by targeting succinate dehydrogenase. Antimicrob. Agents Chemother. 2011, 55, 4204–4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basmaciyan, L.; Azas, N.; Casanova, M. Different apoptosis pathways in Leishmania parasites. Cell Death Discov. 2018, 4, 90. [Google Scholar] [CrossRef]
- Prati, F.; Uliassi, E.; Bolognesi, M.L. Two diseases, one approach: Multitarget drug discovery in Alzheimer’s and neglected tropical diseases. MedChemComm 2014, 5, 853–861. [Google Scholar] [CrossRef]
- Dorlo, T.P.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef]





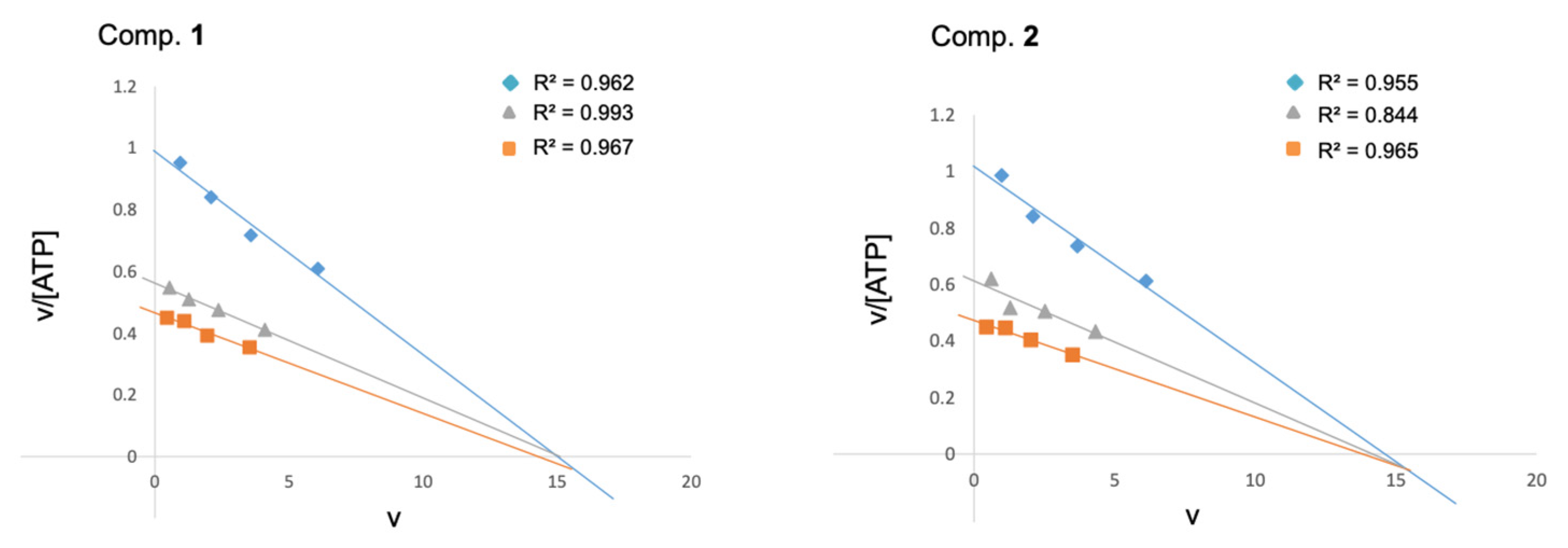
 ), 2.5 (
), 2.5 (  ) and 5 (
) and 5 (  ) μM, respectively. The phospho-glycogen synthase peptide-2 (GS2) was used as a substrate at a fixed concentration of 25 μM. Each kinetic point was made by duplicate, and represented as the mean of two independent experiments.
), 2.5 ( ) and 5 ( ) μM, respectively. The phospho-glycogen synthase peptide-2 (GS2) was used as a substrate at a fixed concentration of 25 μM. Each kinetic point was made by duplicate, and represented as the mean of two independent experiments.
) μM, respectively. The phospho-glycogen synthase peptide-2 (GS2) was used as a substrate at a fixed concentration of 25 μM. Each kinetic point was made by duplicate, and represented as the mean of two independent experiments.
), 2.5 ( ) and 5 ( ) μM, respectively. The phospho-glycogen synthase peptide-2 (GS2) was used as a substrate at a fixed concentration of 25 μM. Each kinetic point was made by duplicate, and represented as the mean of two independent experiments.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Chemical Structure | LdGSK-3s b IC50 (μM) | L. donovani Promastigotes IC50 (μM) | L. pifanoi Axenic Amastigotes IC50 (μM) | MPM c IC50 (μM) | Selectivity Index (SI) d |
|---|---|---|---|---|---|---|
| MBC-10 |  | ~10 e | 10.5 ± 1.2 | 11.2 ± 2.5 | ND | ND |
| MBC-132 |  | 2.5 ± 0.1 | 1.51 ± 0.02 | 0.51 ± 0.01 | 2.6 ± 0.0 | 5.1 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Comp. | R | X | LdGSK-3s b IC50 (μM) | L. infantum Promastigotes IC50 (μM) | L. pifanoi Axenic Amastigotes IC50 (μM) | MPM c IC50 (μM) | Selectivity Index (SI) d |
| MBC-132 |  | Cl | 2.5 ± 0.1 | 1.51 ± 0.02 | 0.51 ± 0.04 | 2.6 ± 0.0 | 5.1 |
| 1 |  | Cl | 3.7 ± 0.3 | 1.54 ± 0.01 | 0.15 ± 0.40 | 3.1 ± 0.1 | 20.6 |
| 2 |  | Cl | 2.5 ± 0.2 | 1.47 ± 0.02 | 0.4 ± 0.2 | 4.51 ± 0.30 | 11.2 |
| 3 |  | Cl | 2.8 ± 0.1 | 4.9 ± 0.2 | 4.7 ± 0.8 | 9.8 ± 0.2 | 2.1 |
| 4 |  | Cl | 4.1 ± 0.1 | 2.9 ± 0.2 | 3.7 ± 1.1 | 7.4 ± 0.8 | 2.0 |
| 5 |  | Cl | 4.5 ± 0.6 | 3.8 ± 1.9 | 3.6 ± 0.6 | 8.1 ± 1.0 | 2.3 |
| 6 |  | Cl | >10 | 19.1 ± 1.2 | >25 | - | - |
| 7 |  | Cl | >10 | 16.9 ± 0.4 | >25 | - | - |
| 8 |  | Cl | >10 | 19.8 ± 3.6 | 17.8 ± 1.1 | 17.0 ± 0.3 | 1.0 |
| 9 |  | Cl | >10 | >25 | >25 | - | - |
| 10 |  | Cl | >10 | >50 | >50 | - | - |
| 11 |  | Cl | >10 | >25 | >25 | - | - |
| 12 |  | Cl | >10 | >50 | >50 | - | - |
| 13 |  | Cl | >10 | >25 | >25 | - | - |
| 14 |  | Cl | >10 | 1.4 ± 0.4 | 2.1 ± 0.6 | 1.3 ± 0.3 | 0.6 |
| 16 |  | H | >10 | 4.1 ± 0.8 | 1.7 ± 0.2 | 3.8 ± 0.3 | 2.2 |
| 17 |  | H | >10 | 4.5 ± 0.7 | 1.7 ± 0.1 | 4.2 ± 0.2 | 2.5 |
| 18 |  | H | >10 | 4.5 ± 0.5 | 1.2 ± 0.1 | 4.0 ± 0.2 | 3.3 |
| 19 |  | H | >10 | 5.9 ± 1.8 | 1.8 ± 0.3 | 4.3 ± 0.3 | 2.4 |
| 20 |  | H | >10 | 7.1 ± 2.1 | 3.2 ± 0.1 | 7.5 ± 0.6 | 2.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebastián-Pérez, V.; Martínez de Iturrate, P.; Nácher-Vázquez, M.; Nóvoa, L.; Pérez, C.; Campillo, N.E.; Gil, C.; Rivas, L. Naphthoquinone as a New Chemical Scaffold for Leishmanicidal Inhibitors of Leishmania GSK-3. Biomedicines 2022, 10, 1136. https://doi.org/10.3390/biomedicines10051136
Sebastián-Pérez V, Martínez de Iturrate P, Nácher-Vázquez M, Nóvoa L, Pérez C, Campillo NE, Gil C, Rivas L. Naphthoquinone as a New Chemical Scaffold for Leishmanicidal Inhibitors of Leishmania GSK-3. Biomedicines. 2022; 10(5):1136. https://doi.org/10.3390/biomedicines10051136
Chicago/Turabian StyleSebastián-Pérez, Victor, Paula Martínez de Iturrate, Montserrat Nácher-Vázquez, Luis Nóvoa, Concepción Pérez, Nuria E. Campillo, Carmen Gil, and Luis Rivas. 2022. "Naphthoquinone as a New Chemical Scaffold for Leishmanicidal Inhibitors of Leishmania GSK-3" Biomedicines 10, no. 5: 1136. https://doi.org/10.3390/biomedicines10051136