
C9orf72 Hexanucleotide Repeat Expansion-Related Neuropathology Is Attenuated by Nasal Rifampicin in Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Behavioral Tests
2.3. Immunohistochemical Analysis
2.4. Rifampicin Treatment
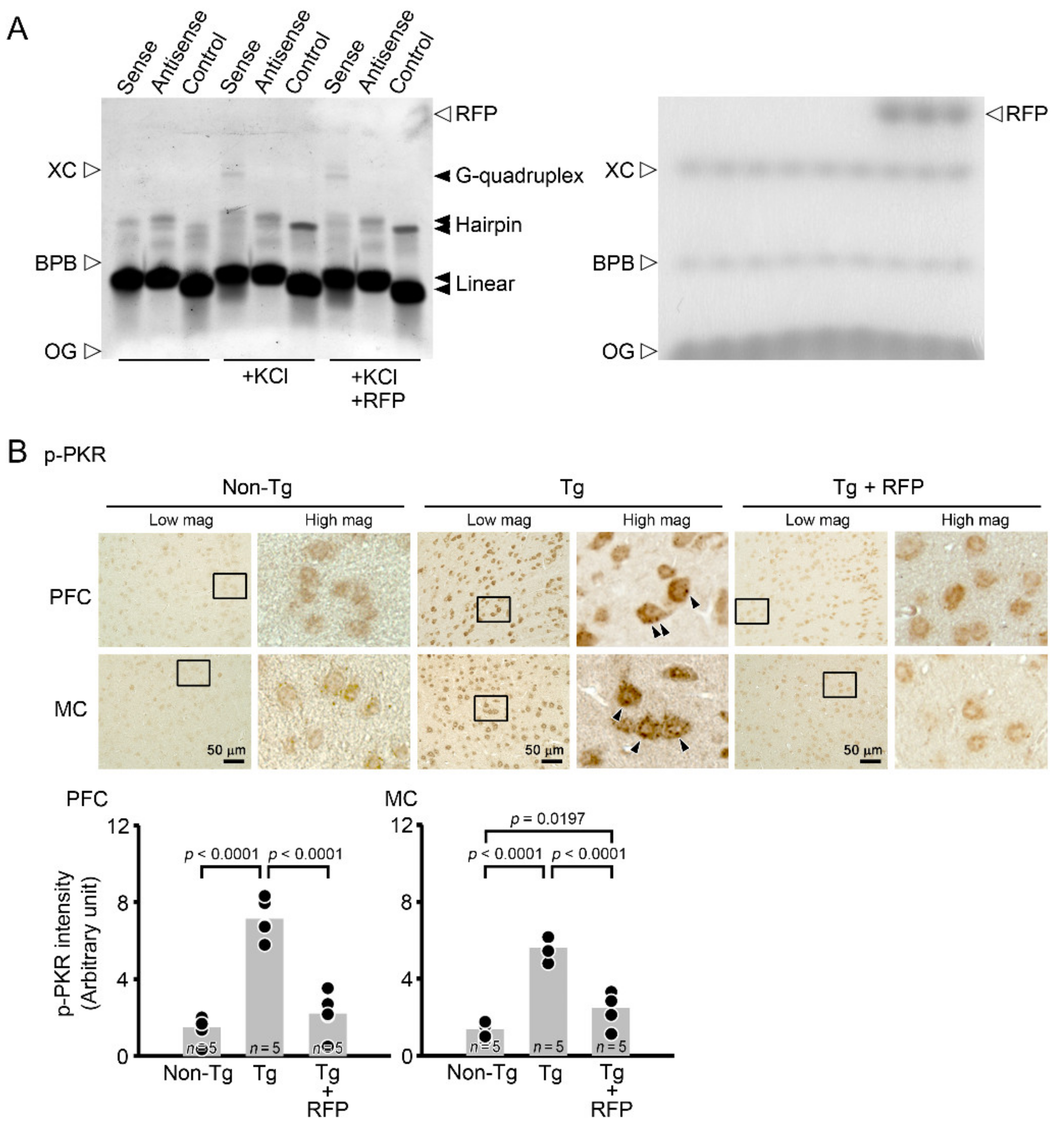
2.5. Gel Shift Assay for G-Quadruplex
2.6. Immunohistochemistry for Phosphorylated PKR
2.7. Statistical Analysis
3. Results
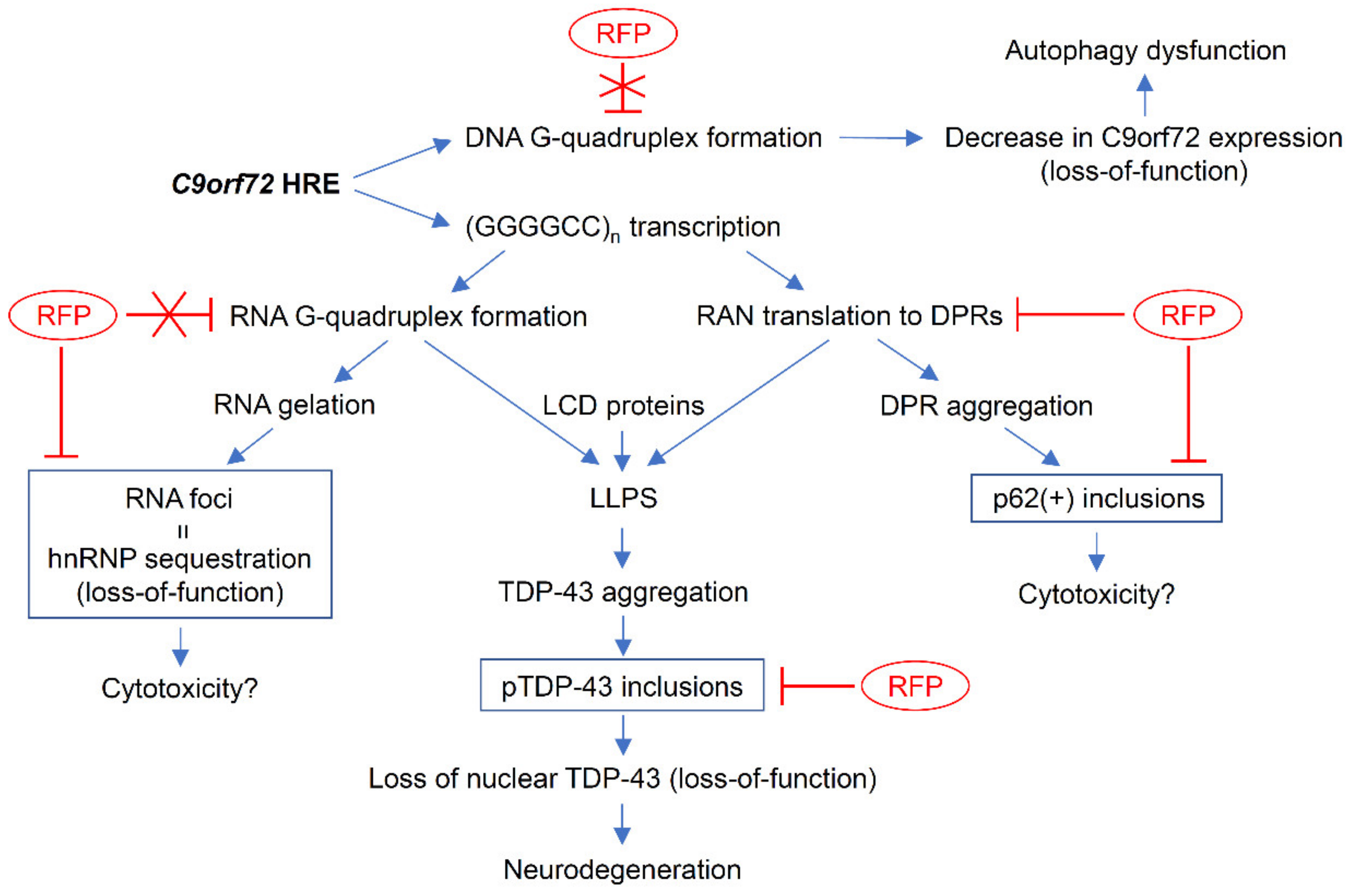
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edbauer, D.; Haass, C. An amyloid-like cascade hypothesis for C9orf72 ALS/FTD. Curr. Opin. Neurobiol. 2016, 36, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L. Four-stranded nucleic acids: Structure, function and targeting of G-quadruplexes. Chem. Soc. Rev. 2008, 37, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Beckers, J.; Tharkeshwar, A.K.; Damme, P.V. C9orf72 ALS-FTD: Recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy 2021, 17, 3306–3322. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.H.; Hung, S.T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.; Shaw, P.G.; Kim, M.S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef]
- Conlon, E.G.; Lu, L.; Sharma, A.; Yamazaki, T.; Tang, T.; Shneider, N.A.; Manley, J.L. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 2016, 5, e17820. [Google Scholar] [CrossRef]
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Stalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef] [Green Version]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Highley, J.R.; Dickman, M.J.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.A.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- McEachin, Z.T.; Parameswaran, J.; Raj, N.; Bassell, G.J.; Jiang, J. RNA-mediated toxicity in C9orf72 ALS and FTD. Neurobiol. Dis. 2020, 145, 105055. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; Dejesus-Hernandez, M.; Blitterswijk, M.M.V.; Jansen-West, K.; Paul, J.W., III; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Broeckhoven, C.V.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.; Caulfield, T.; Daughrity, L.; Dunmore, J.H. Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.M.; Schludi, M.H.; Zee, J.V.D.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.M.; Rollinson, S.; Robinson, A.; Callister, J.B.; Thompson, J.C.; Snowden, J.S.; Gendron, T.; Petrucelli, L.; Masuda-Suzukake, M.; Hasegawa, M.; et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 2013, 1, 68. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Frick, P.; Neumann, M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 2014, 127, 347–357. [Google Scholar] [CrossRef]
- Fay, M.M.; Anderson, P.J.; Ivanov, P. ALS/FTD-Associated C9ORF72 Repeat RNA Promotes Phase Transitions In Vitro and in Cells. Cell Rep. 2017, 21, 3573–3584. [Google Scholar] [CrossRef] [Green Version]
- Jafarinia, H.; Giessen, E.V.D.; Onck, P.R. Phase Separation of Toxic Dipeptide Repeat Proteins Related to C9orf72 ALS/FTD. Biophys. J. 2020, 119, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Smikle, R.; Reid, M.J.; Mizielinska, S. Altered Phase Separation and Cellular Impact in C9orf72-Linked ALS/FTD. Front. Cell. Neurosci. 2021, 15, 664151. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courchaine, E.M.; Lu, A.; Neugebauer, K.M. Droplet organelles? EMBO J. 2016, 35, 1603–1612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, B.; Hartmann, H.; May, S.; Möhl, C.; Ederle, H.; Michaelsen, M.; Schludi, M.H.; Dormann, D.; Edbauer, D. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 2017, 26, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Fang, Y.S.; Tsai, K.J.; Chang, Y.J.; Kao, P.; Woods, R.; Kuo, P.H.; Wu, C.C.; Liao, J.Y.; Chou, S.C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Tusi, S.K.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Ono, K.; Sakai, A.; Yamashita, M.; Mizuguchi, M.; Klein, W.L.; Yamada, M.; Mori, H.; Tomiyama, T. Rifampicin is a candidate preventive medicine against amyloid-beta and tau oligomers. Brain 2016, 139, 1568–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umeda, T.; Hatanaka, Y.; Sakai, A.; Tomiyama, T. Nasal Rifampicin Improves Cognition in a Mouse Model of Dementia with Lewy Bodies by Reducing alpha-Synuclein Oligomers. Int. J. Mol. Sci. 2021, 22, 8453. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Mordes, D.A.; Morrison, B.M.; Ament, X.H.; Cantrell, C.; Mok, J.; Eggan, P.; Xue, C.; Wang, J.Y.; Eggan, K.; Rothstein, J.D. Absence of Survival and Motor Deficits in 500 Repeat C9ORF72 BAC Mice. Neuron 2020, 108, 775–783. [Google Scholar] [CrossRef]
- Nguyen, L.; Laboissonniere, L.A.; Guo, S.; Pilotto, F.; Scheidegger, O.; Oestmann, A.; Hammond, J.W.; Li, H.; Hyysalo, A.; Peltola, R.; et al. Survival and Motor Phenotypes in FVB C9-500 ALS/FTD BAC Transgenic Mice Reproduced by Multiple Labs. Neuron 2020, 108, 784–796. [Google Scholar] [CrossRef]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef]
- Fang, P.; Kazmi, S.A.; Jameson, K.G.; Hsiao, E.Y. The Microbiome as a Modifier of Neurodegenerative Disease Risk. Cell Host Microbe 2020, 28, 201–222. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Brandner, S. Invited Review: The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 2020, 46, 522–545. [Google Scholar] [CrossRef] [Green Version]
- Wells, C.; Brennan, S.; Keon, M.; Ooi, L. The role of amyloid oligomers in neurodegenerative pathologies. Int. J. Biol. Macromol. 2021, 181, 582–604. [Google Scholar] [CrossRef]
- Jain, A.; Vale, R.D. RNA phase transitions in repeat expansion disorders. Nature 2017, 546, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72-ALS/FTD. Cell Rep. 2016, 17, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.R.; Patani, R.; Sidle, K. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016, 96, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umeda, T.; Uekado, R.; Shigemori, K.; Eguchi, H.; Tomiyama, T. Nasal Rifampicin Halts the Progression of Tauopathy by Inhibiting Tau Oligomer Propagation in Alzheimer Brain Extract-Injected Mice. Biomedicines 2022, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Tanaka, A.; Sakai, A.; Yamamoto, A.; Sakane, T.; Tomiyama, T. Intranasal rifampicin for Alzheimer’s disease prevention. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 304–313. [Google Scholar] [CrossRef]
- Umeda, T.; Sakai, A.; Shigemori, K.; Yokota, A.; Kumagai, T.; Tomiyama, T. Oligomer-Targeting Prevention of Neurodegenerative Dementia by Intranasal Rifampicin and Resveratrol Combination—A Preclinical Study in Model Mice. Front. Neurosci. 2021, 15, 763476. [Google Scholar] [CrossRef]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 589–607. [Google Scholar] [CrossRef]
- Chintalaphani, S.R.; Pineda, S.S.; Deveson, I.W.; Kumar, K.R. An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics. Acta Neuropathol. Commun. 2021, 9, 98. [Google Scholar] [CrossRef]
- Silva, A.; Almeida, A.V.D.; Macedo-Ribeiro, S. Polyglutamine expansion diseases: More than simple repeats. J. Struct. Biol. 2018, 201, 139–154. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W.; Bosch, L.V.D. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 2020, 39, e101112. [Google Scholar] [CrossRef] [PubMed]
- Bañez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN Translation in Huntington Disease. Neuron 2015, 88, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.C.; Buescher, J.L.; Oatis, B.; Funk, J.A.; Nash, A.J.; Carrier, R.L.; Hoyt, K.R. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci. Lett. 2007, 411, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoux, I.; Willam, M.; Griesche, N.; Krummeich, J.; Watari, H.; Offermann, N.; Weber, S.; Dey, P.N.; Chen, C.; Monteiro, O.; et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. Elife 2018, 7, e38744. [Google Scholar] [CrossRef]
- Bassez, G.; Audureau, E.; Hogrel, J.Y.; Arrouasse, R.; Baghdoyan, S.; Bhugaloo, H.; Gourlay-Chu, M.L.; Corvoisier, P.L.; Peschanski, M. Improved mobility with metformin in patients with myotonic dystrophy type 1: A randomized controlled trial. Brain 2018, 141, 2855–2865. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatanaka, Y.; Umeda, T.; Shigemori, K.; Takeuchi, T.; Nagai, Y.; Tomiyama, T. C9orf72 Hexanucleotide Repeat Expansion-Related Neuropathology Is Attenuated by Nasal Rifampicin in Mice. Biomedicines 2022, 10, 1080. https://doi.org/10.3390/biomedicines10051080
Hatanaka Y, Umeda T, Shigemori K, Takeuchi T, Nagai Y, Tomiyama T. C9orf72 Hexanucleotide Repeat Expansion-Related Neuropathology Is Attenuated by Nasal Rifampicin in Mice. Biomedicines. 2022; 10(5):1080. https://doi.org/10.3390/biomedicines10051080
Chicago/Turabian StyleHatanaka, Yukari, Tomohiro Umeda, Keiko Shigemori, Toshihide Takeuchi, Yoshitaka Nagai, and Takami Tomiyama. 2022. "C9orf72 Hexanucleotide Repeat Expansion-Related Neuropathology Is Attenuated by Nasal Rifampicin in Mice" Biomedicines 10, no. 5: 1080. https://doi.org/10.3390/biomedicines10051080